急需3MCPD(3-单氯-1,2-丙二醇)标准品或者含量90%以上的纯品联系电话021-58487988-6210联系人 程先生
急需采购聚丙二醇(PPG)GPC系列标准品Poly(propylene glycol),有谁知道在哪里可以买到,忘告之。多谢!!!
求购二甘醇标准品(现货)
如题!公司有个客户的文件里面提到要用DPG(Dipropilene glycol)做标准品,同事觉得用丙二醇可以替代使用,不知道坛子里有没有人用前者做标准品的啊,能否给点意见不??不甚感激!
苯酚-硫酸法是一种常用的检测粗多糖含量的方法,其原理是苯酚-硫酸试剂可与游离的寡糖、多糖中的己糖、糖醛酸起显色反应,在480-490 nm处有最大吸收值,吸收值与糖含量呈线性关系。此法是先用标准品多糖制作标准曲线后,再通过多糖的显色反应测定吸光度,然后根据其在曲线上的位置推算出多糖的浓度从而推算其含量。此法操作简单、快速、灵敏、重复性好,对每种多糖仅需制作一条标准曲线[1]。目前大家研究较多的、生物活性较高的一些真菌多糖,如香菇多糖、灵芝多糖、姬松茸多糖、猴头菇多糖、灰树花多糖等[2],在结构上大多是以β-(1→3)、β-(1→4)或β-(1→6)糖苷键连接的葡聚糖,另外,分子量也一般分布在十几万到几十万之间。因此,由北京卫生防疫站建立,经中国预防科学院营养与食品卫生研究所验证的《粗多糖含量的测定方法》中建议使用50万分子量的葡聚糖作为标准品[3]。为行业内粗多糖含量的测定统一了标准,使各企业之间多糖类产品更具有可比性。燕麦β-葡聚糖是一种β-(1→3)-(1→4)键接的线性葡聚糖,在结构、粘度等其他物理性质上与常见的植物和真菌多糖很相似,适合作为植物、真菌来源多糖含量测定的标准品。但由于多糖纯化困难,市面上不少葡聚糖纯度较低,不适合作为标准品。下面,我们来比较两种不同纯度的燕麦β-葡聚糖产品作为多糖标准品的区别。1 材料与方法1.1 实验材料高纯度燕麦β-葡聚糖PS-Con-Ⅰ由武汉百特纯大分子科技有限公司提供,纯度大于97%(其中,另外3%主要是结合水),低纯度燕麦β-葡聚糖由某食品研究所提供,纯度约50%,苯酚、浓硫酸均为化学纯。1.2 实验方法样品溶解:高纯度燕麦β-葡聚糖经70℃水浴,15min后完全溶解。低纯度燕麦β-葡聚糖70℃水浴,30min后仍有不溶物,升高溶解温度至90℃后继续溶解30min,仍有少量不溶物,过滤。溶液配制:配制0.1mg/ml葡聚糖标准溶液,50mg/ml苯酚溶液备用。标准曲线的制作:精密吸取葡聚糖标准液0.10,0.40, 0.80,1.20,1.60,2.00ml(分别相当于葡聚糖0.01,0.04,0.08,0.12,0.16,0.20mg),补充水至2.0mL,加入苯酚溶液1.0ml,混匀,再加入浓硫酸5ml,混匀,沸水浴2分钟,混匀,冷却后用分光光度计在485nm波长处以试剂空白溶液为参比,测定吸光度值(A),以A为横坐标,葡聚糖含量C为纵坐标绘制标准曲线。2 结果与分析2.1 样品溶解高纯度燕麦β-葡聚糖溶解速度较快,溶液澄清透明,说明此产品溶解性良好。低纯度燕麦β-葡聚糖难以溶解,且溶解1h后仍有不溶物存在,说明此产品溶解性差,杂质较多。 2.2 标准曲线下表为两种标准品分别配制不同葡聚糖浓度(含量)反应后得到的吸光值:葡聚糖含量(mg)0.010.040.080.120.162.00高纯度标样吸光值0.0530.0800.2000.2620.3530.450低纯度标样吸光值0.0010.0550.1130.1730.2400.320通过数据处理,得到标准曲线如下:高纯度燕麦β-葡聚糖 C=0.4657A-0.0068 (R=0.9955)低纯度燕麦β-葡聚糖 C=0.609A+0.0101(R=0.9985)比较这两个标准曲线发现,当待测样品吸光值一定,使用低纯度葡聚糖作为标准品得到的标准曲线计算葡聚糖含量值时,明显高于高纯度标准品。究其原因,低纯度葡聚糖所含杂质较多,在作为标准品时,部分杂质不能溶解,却计入了标准品葡聚糖总量,因此,使得结果偏高。另外,即使溶解的物质中,也有可能存在部分不能参加反应的蛋白等杂质,同样会造成结果偏高。由以上数据和分析可以得出,测定粗多糖含量不能使用低纯度葡聚糖作为标准品,应尽量选用高纯度葡聚糖标准品,按照国家建议方法和行业标准进行检测,这样才能保证各企业多糖系列产品在含量和纯度上的可比性,有利于规范企业行为和保健品市场。参考文献[1] 胡居吾,范青生,肖小年. 粗多糖测定方法的研究. 江西食品工业. 2005, 1[2] 李明元. 真菌粗多糖测定方法的研究. 食品研究与开发. 2007, 5[3] 粗多糖的测定方法. 北京卫生防疫站建立,经中国预防科学院营养与食品卫生研究所验证. 食品伙伴网[em0805]
二氯甲烷从不同浓度的有机溶机水溶液(DMF, 甲醇,酸,碱)中萃取标准品,计算回收率,不知道大家有没有什么经验分享一下,由于水中有有机溶剂,不知道萃取效率能达到多少?有没有这方面资料可以参考呢?谢谢
职业卫生甲硫醇的标准品是抽甲硫醇气体溶解到二氯甲烷中,这个甲硫醇气体是指买来的标气还是溶液挥发的?我们买的标准品是10%丙二醇的甲硫醇溶液,这个该怎么配呢,是密封挥发溶解到二氯甲烷,还是直接稀释到二氯甲烷中,哪位可以解答下谢谢
[color=red]【由于该附件或图片违规,已被版主删除】[/color][img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=54454]GB11311-91品容器及包装材料用聚对苯二甲酸乙二醇酯成型品卫生标准[/url]
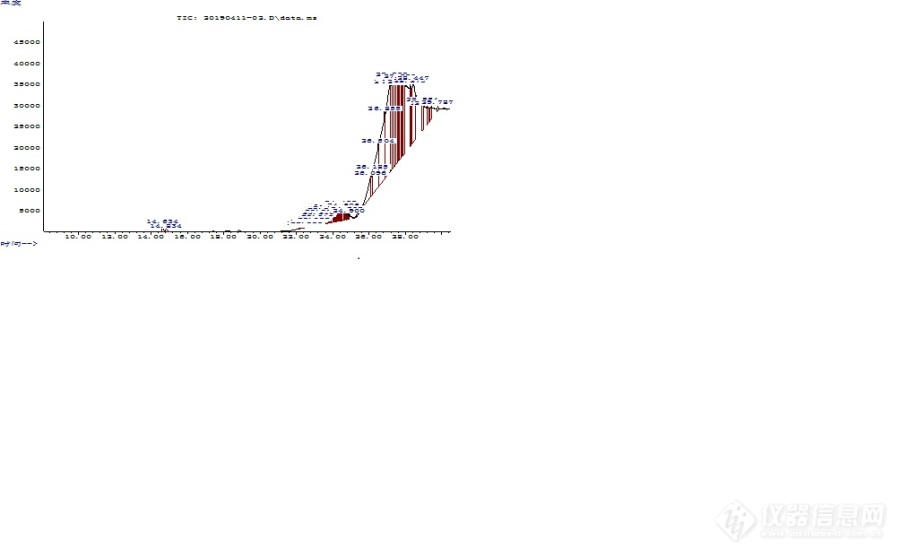
最近在开展食品中3-氯-1,2-丙二醇方法摸索,在对标样进行衍生,并作标准曲线的时候发现有如下问题,请各位大神不吝指教。1、选取5个点作为3-氯-1,2-丙二醇标准样品,质量分别为0.0286ug、0.143ug、0.286ug、0.572ug、1.144ug,在作标准曲线时,在每个标样中分别加入等量的D5-3-氯-1,2-丙二醇,其质量为0.222ug,上述标样中均加入正己烷定容至2mL,然后按标准要求加入40uL的七氟丁酰基咪唑进行衍生反应,后续的步骤严格按标准要求执行;2、衍生结束后,上[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质[/color][/url]进行测定,这时候问题出现了,按照上述的内标加入量,D5-3-氯-1,2-丙二醇在各标样的中的量应该是相同的,按理论来说,其在谱图中的峰面积也应该是大致相同的,最起码不会有太大的差别,但实验结果却是内标的峰面积会随着标样浓度的增加而不断的增大,其具体的数值为38845、86170、185376、412975、887632;反观标样3-氯-1,2-丙二醇的峰面积,其实测值与标样浓度值对应增长,成线性关系,具体测定值分别为26801、64667、138047、300758、640761。如果按照外标作标准曲线的话,线性是没问题的,但如果按内标作曲线的话,作出来的曲线压根就没法使用,可以说完全不成线性,这就是我请请教各位同行的问题,你们在用内标作这个项目的时候也会遇到这个问题吗?如果没有,那么你们的内标面积都能保持一致吗?为了各直观的说明这个问题,我把我做的5个标准点谱图按浓度从低到高一并附上以作参考,其中前面的峰为内标,后面的峰为3-氯-1,2-丙二醇。[img=,690,425]https://ng1.17img.cn/bbsfiles/images/2019/04/201904151752487962_2765_1640909_3.jpg!w690x425.jpg[/img][img=,690,425]https://ng1.17img.cn/bbsfiles/images/2019/04/201904151752589614_3231_1640909_3.jpg!w690x425.jpg[/img][img=,690,425]https://ng1.17img.cn/bbsfiles/images/2019/04/201904151753062214_3758_1640909_3.jpg!w690x425.jpg[/img][img=,690,425]https://ng1.17img.cn/bbsfiles/images/2019/04/201904151753121534_1266_1640909_3.jpg!w690x425.jpg[/img][img=,690,425]https://ng1.17img.cn/bbsfiles/images/2019/04/201904151753188554_5523_1640909_3.jpg!w690x425.jpg[/img]
GB 29698-2013 食品安全国家标准 奶及奶制品中17β-雌二醇、雌三醇、炔雌醇多残留的测定 气相色谱-质谱法
看了一些认证过的标准品证书,标准品是经过纯品采用称量法使用基体溶液经过认证,申请GBW(E)的。我想问一下实验室啥情况用纯品,啥时候用认证过的标准品。有没有老师有正己烷中的邻苯二甲酸二异丁酯的证书啊,那个厂家的都行,以前领导买的是甲醇中的邻苯二甲酸二异丁酯,现在这个证书用不了。
我看国标中之要求纯度大于95%,但是并没有要求其他的。我可以用试剂代替么?主要不确定度达到要求,比如甲醇,我必须买甲醇标准品还是用色谱纯甲醇代替呢?

在分析技术的园地里发现许多朋友提到有关标准品的各种问题。其实我想主要的就是很少有人把这个题目讲解的透彻一点,让大家都能够一次彻底了解。自己在美国工作了30余年,也一直在分析的领域上,所以决定花时间把它写下来与大家共享。也算是抛砖引玉,能够给大家一个思考的机会,同时大家也互相交流,相得益彰。闲话少说,言归正传。分析的方法分成两种,一种就是绝对的方法,是不需要标准品,当然使用这种方法必须使用相当纯度的化合物。分析的方法譬如元素分析,DSC,NMR, 滴定,电化学等等,这些方法都可以用来决定化合物的纯度。如何取得相当纯度的化合物,一般我们就用色谱,首先肯定色谱的纯度(其实是均匀度homogeneity),当然在注射相当量的前提下(要能察觉到0.1%的杂质),只能看见一个主峰,也就是说均匀度接近100%(根据峰面积计算)。此时我们拿这个接近100%均匀度的化合物来定性,我们必须确认其专有性(specificity,属性)的存在,然后再定量得出此化合物的纯度。这里说的纯度是指化学组成分而言(composition)。因为这些方法没有办法测出金属盐类,或非金属盐类。因此我们要有另外的方法来决定离子的存在与其含量。当然还有其它成分如水分,有机溶剂等等。当我们把所有的成分定出来之后,总和就是我们这个化合物的组成成分。有了这些数据,自然我们可以求得物质平衡。所以,任何标准品的订定都必须经过这个程序。另外一种分析的方法就是相对的方法。顾名思义,就是需要标准品来对比。除了上述的绝对的方法外,其它的方法都是相对的。看看我们做色谱,就是一个最好的例子。我们需要标准品来定性及定量我们所要分析的样品。我以前曾经开发过新药(New Molecular Entity),标准品就需要自己来定了。经过色谱的分离,纯化干燥后,进行了如上段所叙述的定性,定量工作,我们取得了化学纯度。这个标准品,我们把它叫做原始标准品(primaryreference standard)。如果说化学纯度是95%,也就是说每次我秤了1毫克,里面含有此化合物0.95毫克。因为这种原始标准品,制备很繁杂而且获得的量也不大(在一定时间内),所以没有必要每次都使用这种原始标准品来进行色谱或其它相对分析方法分析样品。因此我们可以拿我们的化学原料药(一般就是工艺开发后的产品)来订为二级标准品(secondaryreference standard)。有人把它说成工作标准品,其实是有商酌余地的。工作标准品也可以是原始标准品,我们不需费力,可以采购而来。我们所要的做的工作,就是把这个二级标准品做色谱分析,然后用原始标准品来做工作标准品,完成定性,定量。如此,我们可以根据峰面积,及原始标准品的相应比(responsefactor = peak area / concentration)求出二级标准品的含量。以后我们做色谱的时候,就可以把这个订好的二级标准品拿来做我们的工作标准品来分析我们的样品。前面提到原始标准品的由来。可以自己定,当然也可以购买。在美国就是美国药典(USP),它不是一个官方机构。但是只要有官方承认的标准品,自然就去买,不管贵贱。如果太贵,那就买一次,然后用来做原始标准品,定出我们自己的二级标准品。一般美国药典的标准品都有规定干燥的程序。在执行干燥完成后,如果没有表明纯度,那就是100%。当下一个批次原始标准品发行的时候,前一批次自动失效。也就是有效期完全看新的批次何时出现。这样也避免了稳定性的考察。当然我们可以拿这个原始标准,来考察我们二级标准品的稳定性。国内的情形我不太清楚,我相信只要官方机构认可的标准品,都可以直接使用的。一般最常用的化学原料药的美国药典标准品,200毫克在200美元左右。如果我们使用微量天平,每次用量1.000毫克,那就可以每次使用原始标准品来做色谱,就避免再去设定二级标准品的麻烦了。另外我想大家都知道,当我们定量的时候,同一个标准品,我们必须秤两次,配置两个不同浓度的工作液。这样可以确认两个工作液是否配置正确。关于到底应该进样几针。我一向的做法就是第一个进样六针,做为系统适应度(system suitability)的检查,其它的任何样品,一律三针。有的人说两针就可以了。我要提醒的是两点决定一条线,得出的统计结论那是数学公式计算出来的。三点才能决定一个面,也就是高斯的分配图(GaussianDistribution)。所以至少在统计学上三点比两点得出的结果要好。又有人说了,如果我一个样品,要走60分钟,那不是完蛋了。首先,是否有必要要走60分钟。我认为最理想的方法是在30-45分钟(当然看功力了),这是指对杂质的分析,必须走梯度。我曾经用5厘米的柱子,梯度,30分钟分出17个化合物,包刮两个主要成分,两个抗氧剂,其它的就是杂质,分解物。如果走等梯度,一般是不超过10分钟为原则。这样就没有问题了。如果真的要走60分钟,那就必须选择好的仪器。并不是所有HPLC的仪器都可以无忧的走完全部样品。当然,柱子也十分重要。扯远了,以后有时间再分开来讨论。
收到二乙二醇 又名二甘醇 要求测定纯度,这个有标准方法吗? DB-WAX 和RTX-5 哪个做好?
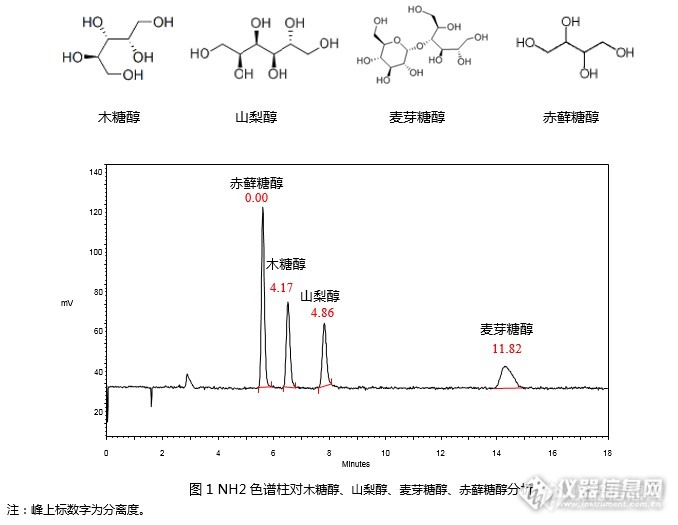
[align=center]GB 5009.279-2016 食品安全国家标准 食品中木糖醇、山梨醇、麦芽糖醇、赤藓糖醇的测定-NQAD[/align]《GB 5009.279-2016 食品安全国家标准食品中木糖醇、山梨醇、麦芽糖醇、赤藓糖醇的测定》第二法中推荐使用蒸发光散射检测器对4种糖醇进行检测。本实验室使用资生堂高灵敏度气溶胶通用型检测器NQAD对该项目进行检测。使用资生堂氨基柱CAPCELL PAK NH2 UG80 S5 4.6 mm i.d. × 250 mm(GQAD 05507)依据国标方法进行分析,可以实现4种糖醇的良好分析(见图1)。[img=,678,525]http://ng1.17img.cn/bbsfiles/images/2017/08/201708030937_01_2222981_3.png[/img][img=,611,257]http://ng1.17img.cn/bbsfiles/images/2017/08/201708030937_02_2222981_3.png[/img]进一步对标准曲线进行绘制,依据国家标准,以峰面积为纵坐标,标准工作液浓度为横坐标,以赤藓糖醇浓度为0.14 mg/mL, 0.21 mg/mL, 0.28 mg/mL, 0.35 mg/mL, 0.42 mg/mL, 0.49 mg/mL,木糖醇、山梨醇、麦芽糖醇浓度为0.10 mg/mL, 0.15 mg/mL, 0.20 mg/mL, 0.25 mg/mL, 0.30 mg/mL, 0.35 mg/mL的混合系列标准工作液,进行标准曲线绘制。如图2~5所示,赤藓糖醇在0.14 mg/mL~0.49 mg/mL浓度范围内,木糖醇、山梨醇、麦芽糖醇在0.1 mg/mL~0.35 mg/mL浓度范围内线性良好,相关系数R[sup]2[/sup]均在0.99以上。[img=,534,330]http://ng1.17img.cn/bbsfiles/images/2017/08/201708030938_01_2222981_3.png[/img][img=,573,327]http://ng1.17img.cn/bbsfiles/images/2017/08/201708030938_02_2222981_3.png[/img][img=,573,326]http://ng1.17img.cn/bbsfiles/images/2017/08/201708030938_03_2222981_3.png[/img][img=,556,342]http://ng1.17img.cn/bbsfiles/images/2017/08/201708030938_04_2222981_3.png[/img]注:图中色谱峰线条不平滑是由于图像在复制过程中解像度问题引起的。
最近发现,我把标准品用甲醇溶解并稀释后进质谱,得到的结果有很多杂质,但是标准品厂商提供的纯度又在94%以上,所以想搞清楚标准品厂商是如何计算标准品的纯度的?说明一下,我所用的标准品是Dr.E公司的
2010年9月从中国计量院购买了一组甲醇标准品(溶于乙醇)证书显示浓度为1毫克/毫升,有效期一年最近刚好打算参加此比对,其中有甲醇项目,问下诸位此甲醇标准品是否可以使用?谢谢
请教聚乙二醇、二乙二醇的分析方法或标准,哪位知道的大侠告诉下哦,谢谢。。。
转自中国色谱网,作者:cation一个分析方法要选择使用内标或外标法来定量,并没有一定的规定,所以这里就尝试来分析一下, 如何找到一个合适的定量方式, 怎么去选择一个内标准品 .........内标准品的选择 传统上在教科书中都会很模糊的告诉学生内标准的选择方式,例如说要选安定性好 与分析物性质要相近 在分析的基质中不能出现等,现在我对这些个" 选择方式"没有太大的兴趣,因为这个大家都知道,那现在就从另一个角度的来看看内标准品的选择还有哪些需要注意的.1. 只选一个内标准品? 当然, 如果你的分析目标物就只有一个, 在正常的状况下,内标准"应该"也只会有一个才对! 但是如果你的分析是多成份的,那就必须十分小心地看待在一个分析方法内标准品的选择, 如果分析物的在层析图中是平均分布在各处, 那你就必须看看你的检测方法中是否有规定内标物与分析物之间的滞留时间差范围是多少,依此规定来选择内标,但是如果并没有规定,最好也是选择一个以上的内标来使用,因为即使化学性质不会差太多,但在沸点方面却会有满大的差异, 内标与分析物的沸点差异过大,在GC的注射口中就无法把因为discrimibation所造成的误差校正回来. 如果你的分析物是性质相差颇大的 (例如说同时含有醇,酸...),那别怀疑一定是要使用一个以上的内标准品,如果多个物种再加上多成份,那就很复杂了. 最低的限度也要依照分析物的沸点高低来使用多个不同的内标准品. 在美国环保署的检验方法USEPA 8270C,是一个检测半挥发性的污染物的规范,前后列了不下一百种的分析物,就使用了六个不同的内标准品作为校正的依据,来照顾到各个不同沸点的分析物!! 滥用药物分析大概是最严谨的了,即使是结构性质极相近的分析物,例如morphine和codeine,amphetamine和methamphetamine,在分析时为求准确,都是以各自的D同位素取代的标准品作为内标.2.基质中一定不能存在? 这个问题当然是肯定的, 不然定量结果会很不稳定或者是很凄惨. 但是有些时候你根本不知道哪些东西在分析样品的基质中不会存在! 这时候怎么办? 找以往的文献看看别人是用甚么,这是一个方法, 但要注意的是文献不一定就是对的! 使用分析物的氢同位素(D)取代物,是最妥当的,但问题是价格昂贵,而且不是每一种分析物的氢同位素(D)取代物都有贩售,当然,如果本钱够,你可以去订购专门替你合成氢同位素(D)取代物. 如果要自己选, 那就必须了解一下分析物的成分了. 以前曾经替人定性和定量过一阵子海水鱼中所含不饱和脂肪酸,这鱼中不饱和脂肪酸的碳数都是奇数(或是偶数, 我已经忘了), 所以内标就使用了几个偶数(或奇数)的不饱和脂肪酸, 像这种内标物绝不可能出现在分析物中,且性质十分相近,所以定量的结果一般误差都不会太大! 如果完全无法确定怎办? 有时候就是赌一赌啰.........举例来说:如果你的分析物结构中含有氯的话,就可以寻找一个化合物,而这个化合物是把其中的氯换成氟或溴,例如你可以使用2-氟联苯或2-溴联苯来当作分析2-氯联苯的内标准品. 以环境分析为例,通常在自然界中的氟及溴化物并不多, 在EPA的方法中,就经常把结构相似的氟及溴化物添加入样品中,来做为分析含氯化合物的QC样品.所以找氟及溴来取代氯原子的化合物来作为内标,基本上还算是很安全的. 如果实在连上述转换一个基团的内标都找不到,那至少在选择时一定要找同一类的,分析酸就找酸当内标,分析醇就找醇当内标,直链接构分析物就不要找一个环状的化合物来当内标,分子量不要相差太大,这算是最基本的要求!!3.内标和分析物的滞留时间必须接近? 这个说法原则上没错,因为如果滞留时间接近,它们的物理或者是化学的性质在某种程度上是接近的,所以有些分析方法会有这样的规定. 但在某些特殊的状况下却出了问题, 事实上还真的碰过, 结果在更换到第三个内标准品后, 它的RT远离分析物, 反而解决了定量误差的问题!! 这个案例以后有空在来聊聊. 4.内标的浓度应该是多少? 除了某些分析方法会规定必须加入多少浓度的内标外,其它的就只能靠分析员自己来决定了. 以我为例, 通常会先决定一个分析方法的检量线范围,然后开始配制不同浓度的内标准品来注射入仪器中,分析完后选出一个大概是检量线最高浓度的波峰高度三分之二左右的浓度,作为该项分析的内标浓度. 这样的选择方式,可以兼顾到高低浓度的需要,一般来说,内标浓度过高除了会增加成本之外,对于低浓度的校正是会产某些程度的误差. 在某些分析方法中会强制妳使用内标法而不是外标法或线性回归的方式来定量,但是如果你的基质很复杂,一针打进仪器中,结果大大小小的出来一堆波峰, 这个时候如果你又不是使用质谱作为侦测器的话,就必须考虑加大内标准品的使用浓度, 来降低内标准品的可能发生的积分误差!!5.内标准怎么配制? 一但决定了添加内标准品的浓度,就可以开始配制分析时所用的内标准品标准溶液,相对于测试时的小量, 分析员必须先估计妳在这一批的分析样品有多少, 然后计算整个分析需要添加入多少的量,例如说妳需要大概100mL的内标准品标准溶液,这时就必须再加多30%以上,一次就配好整个分析要用的量! 分装或者是整瓶放到-20度的冰箱中存放,免得做到一半发觉内标准品标准溶液用完了,再重新配制一次,如果你是分析的新手, 搞不好前后两次配得不一样浓度,那就会很凄惨了…..有些领域的分析,很重视这种内标波峰的积分值是否是有一定的再现性!!讲完了内标准品, 下次就可以来看看内标法和外标法了………..

大家看一下丙二醇的离子碎片怎么变成这样了,真是奇怪呢。标准的45应该是基峰,现在反而是61了。http://ng1.17img.cn/bbsfiles/images/2011/03/201103252106_285276_2194847_3.jpg
谈谈内标准品(内标物质)传统上在教科书中都会很模糊的告诉学生内标准的选择方式,例如说要选安定性好 与分析物性质要相近 在分析的基质中不能出现等,现在我对这些个" 选择方式"没有太大的兴趣,因为这个大家都知道,那现在就从另一个角度的来看看内标准品的选择还有哪些需要注意的.1. 只选一个内标准品?当然, 如果你的分析目标物就只有一个, 在正常的状况下,内标准"应该"也只会有一个才对! 但是如果你的分析是多成份的,那就必须十分小心地看待在一个分析方法内标准品的选择, 如果分析物的在层析图中是平均分布在各处, 那你就必须看看你的检测方法中是否有规定内标物与分析物之间的滞留时间差范围是多少,依此规定来选择内标,但是如果并没有规定,最好也是选择一个以上的内标来使用,因为即使化学性质不会差太多,但在沸点方面却会有满大的差异, 内标与分析物的沸点差异过大,在GC的注射口中就无法把因为discrimibation(分辨)所造成的误差校正回来.如果你的分析物是性质相差颇大的 (例如说同时含有醇,酸...),那别怀疑一定是要使用一个以上的内标准品,如果多个物种再加上多成份,那就很复杂了. 最低的限度也要依照分析物的沸点高低来使用多个不同的内标准品. 在美国环保署的检验方法USEPA 8270C,是一个检测半挥发性的污染物的规范,前后列了不下一百种的分析物,就使用了六个不同的内标准品作为校正的依据,来照顾到各个不同沸点的分析物!!滥用药物分析大概是最严谨的了,即使是结构性质极相近的分析物,例如morphine和codeine,amphetamine和methamphetamine,在分析时为求准确,都是以各自的D同位素取代的标准品作为内标.2.基质中一定不能存在?这个问题当然是肯定的, 不然定量结果会很不稳定或者是很凄惨. 但是有些时候你根本不知道哪些东西在分析样品的基质中不会存在! 这时候怎么办? 找以往的文献看看别人是用甚么,这是一个方法, 但要注意的是文献不一定就是对的! 使用分析物的氢同位素(D)取代物,是最妥当的,但问题是价格昂贵,而且不是每一种分析物的氢同位素(D)取代物都有贩售,当然,如果本钱够,你可以去订购专门替你合成氢同位素(D)取代物. 如果要自己选, 那就必须了解一下分析物的成分了.以前曾经替人定性和定量过一阵子海水鱼中所含不饱和脂肪酸,这鱼中不饱和脂肪酸的碳数都是奇数(或是偶数, 我已经忘了), 所以内标就使用了几个偶数(或奇数)的不饱和脂肪酸, 像这种内标物绝不可能出现在分析物中,且性质十分相近,所以定量的结果一般误差都不会太大! 如果完全无法确定怎办? 有时候就是赌一赌啰.........举例来说:如果你的分析物结构中含有氯的话,就可以寻找一个化合物,而这个化合物是把其中的氯换成氟或溴,例如你可以使用2-氟联苯或2-溴联苯来当作分析2-氯联苯的内标准品. 以环境分析为例,通常在自然界中的氟及溴化物并不多, 在EPA的方法中,就经常把结构相似的氟及溴化物添加入样品中,来做为分析含氯化合物的QC样品.所以找氟及溴来取代氯原子的化合物来作为内标,基本上还算是很安全的.如果实在连上述转换一个基团的内标都找不到,那至少在选择时一定要找同一类的,分析酸就找酸当内标,分析醇就找醇当内标,直链接构分析物就不要找一个环状的化合物来当内标,分子量不要相差太大,这算是最基本的要求!!3.内标和分析物的滞留时间必须接近?这个说法原则上没错,因为如果滞留时间接近,它们的物理或者是化学的性质在某种程度上是接近的,所以有些分析方法会有这样的规定. 但在某些特殊的状况下却出了问题, 事实上还真的碰过, 结果在更换到第三个内标准品后, 它的RT远离分析物, 反而解决了定量误差的问题!! 这个案例以后有空在来聊聊. 4.内标的浓度应该是多少?除了某些分析方法会规定必须加入多少浓度的内标外,其它的就只能靠分析员自己来决定了. 以我为例, 通常会先决定一个分析方法的检量线范围,然后开始配制不同浓度的内标准品来注射入仪器中,分析完后选出一个大概是检量线最高浓度的波峰高度三分之二左右的浓度,作为该项分析的内标浓度. 这样的选择方式,可以兼顾到高低浓度的需要,一般来说,内标浓度过高除了会增加成本之外,对于低浓度的校正是会产某些程度的误差.在某些分析方法中会强制你使用内标法而不是外标法或线性回归的方式来定量,但是如果你的基质很复杂,一针打进仪器中,结果大大小小的出来一堆波峰, 这个时候如果你又不是使用质谱作为侦测器的话,就必须考虑加大内标准品的使用浓度, 来降低内标准品的可能发生的积分误差!!5.内标准怎么配制?一旦决定了添加内标准品的浓度,就可以开始配制分析时所用的内标准品标准溶液,相对于测试时的小量, 分析员必须先估计你在这一批的分析样品有多少, 然后计算整个分析需要添加入多少的量,例如说妳需要大概100mL的内标准品标准溶液,这时就必须再加多30%以上,一次就配好整个分析要用的量! 分装或者是整瓶放到-20度的冰箱中存放,免得做到一半发觉内标准品标准溶液用完了,再重新配制一次,如果你是分析的新手, 搞不好前后两次配得不一样浓度,那就会很凄惨了…..有些领域的分析,很重视这种内标波峰的积分值是否是有一定的再现性!!来源于食品伙伴网。
如题。一直以来困扰我呐。为什么药厂的客户总是钟情于中检所,药典的标准品呢?有次一个药厂的客户因为中检所的产品断货,和我们买了一大批Dr.的罗红霉素,结果说是不合适,测下来的纯度不够,我们和供应商反映,最终无果。药厂的贴友多说说,必须使用药典系统的产品么?
绘制标准曲线所用的标准品必须是99%的纯度吗?90%纯度的行不行?为什么?
http://www.greenherbs.com.cn/bbs/dispbbs.asp?boardid=2&Id=7671652001 盐酸四氢唑林 Tetrahydrozoline Hydrochloride 对照品/标准品1651621 δ-9-四氢大麻酚 Delta-9-Tetrahydrocannabinol 对照品/标准品1651009 盐酸四环素 Tetracycline Hydrochloride 对照品/标准品1650006 盐酸羟丁卡因 Tetracaine Hydrochloride 对照品/标准品1649007 丙酸睾酮 CIII Testosterone Propionate CIII 对照品/标准品1648004 庚酸睾酮 CIII Testosterone Enanthate CIII 对照品/标准品1647001 环戊丙酸睾酮CIII Testosterone Cypionate CIII 对照品/标准品1646009 睾酮 CIII Testosterone CIII 对照品/标准品1645006 睾内酯CIII Testolactone CIII 对照品/标准品1644003 萜品醇 Terpin Hydrate 对照品/标准品1643929 特非那定杂质B Terfenadine Related Compound B 对照品/标准品1643907 特非那定杂质A Terfenadine Related Compound A 对照品/标准品1643805 特非那定 Terfenadine 对照品/标准品1643703 特康唑 Terconazole 对照品/标准品1643510 特布他林杂质A Terbutaline Related Compound A 对照品/标准品1643500 硫酸特布他林 Terbutaline Sulfate 对照品/标准品1643496 盐酸特比萘酚 Terbinafine Hydrochloride 对照品/标准品1643485 特拉唑嗪杂质C Terazosin Related Compound C 对照品/标准品1643474 特拉唑嗪杂质B Terazosin Related Compound B 对照品/标准品1643463 特拉唑嗪杂质A Terazosin Related Compound A 对照品/标准品1643452 盐酸特拉唑嗪 Terazosin Hydrochloride 对照品/标准品1643408 替马西泮 CIV Temazepam CIV 对照品/标准品1643394 他唑巴坦杂质 A Tazobactam Related Compound A 对照品/标准品1643383 他唑巴坦;泰唑巴坦 Tazobactam 对照品/标准品1643361 牛磺酸 Taurine 对照品/标准品1643340 酒石酸 Tartaric Acid 对照品/标准品1643328 鞣酸 Tannic Acid 对照品/标准品1643306 枸橼酸他莫昔芬 Tamoxifen Citrate 对照品/标准品1643281 消旋盐酸坦洛新 Racemic Tamsulosin Hydrochloride 对照品/标准品1643260 盐酸坦洛新 Tamsulosin Hydrochloride 对照品/标准品1642904 塔格;塔格糖 Tagatose 对照品/标准品1642813 他克莫司杂质A Tacrolimus Related Compound A 对照品/标准品1642802 他克莫司 Tacrolimus 对照品/标准品1642700 盐酸他克林 Tacrine Hydrochloride 对照品/标准品1642507 舒洛芬 Suprofen 对照品/标准品1642256 琥珀酸舒马曲坦相关杂质 Sumatriptan Succinate Related Impurities 对照品/标准品1642223 琥珀酸舒马普坦杂质C Sumatriptan Succinate Related Compound C 对照品/标准品1642212 琥珀酸舒马普坦杂质A Sumatriptan Succinate Related Compound A 对照品/标准品1642201 琥珀酸舒马普坦 Sumatriptan Succinate 对照品/标准品1642154 舒马普坦 Sumatriptan 对照品/标准品1642100 舒利苯酮 Sulisobenzone 对照品/标准品1642019 舒林酸杂质A Sulindac Related Compound A 对照品/标准品1642008 舒林酸 Sulindac 对照品/标准品1639003 乙酰磺胺异恶唑 Sulfisoxazole Acetyl 对照品/标准品1638000 磺胺异恶唑 Sulfisoxazole 对照品/标准品1637008 磺吡酮 Sulfinpyrazone 对照品/标准品1636504 酞磺胺噻唑 Sulfathiazole 对照品/标准品1636005 柳氮磺吡啶 Sulfasalazine 对照品/标准品1635228 磺胺喹沙啉杂质A Sulfaquinoxaline Related Compound A 对照品/标准品1635206 磺胺喹沙啉 Sulfaquinoxaline 对照品/标准品1635002 磺胺吡啶熔点标准品 Sulfapyridine Melting Point Standard 对照品/标准品1634000 磺胺吡啶 Sulfapyridine 对照品/标准品1633506 氨苯磺酸 (磺胺酸 ) Sulfanilic Acid 对照品/标准品1633007 磺胺熔点标准品 Sulfanilamide Melting Point Standard 对照品/标准品1632004 磺胺 Sulfanilamide 对照品/标准品1631500 磺胺甲恶唑 N4- 葡胺 Sulfamethoxazole N4-glucoside 对照品/标准品1631001 磺胺甲恶唑 Sulfamethoxazole 对照品/标准品1630009 磺胺甲二唑 Sulfamethizole 对照品/标准品1629000 磺胺二甲嘧啶 Sulfamethazine 对照品/标准品1628007 磺胺甲嘧啶 Sulfamerazine 对照品/标准品1626500 磺胺多辛 Sulfadoxine 对照品/标准品1626001 磺胺地索辛 Sulfadimethoxine 对照品/标准品1625009 磺胺嘧啶 Sulfadiazine 对照品/标准品1624505 磺胺氯达嗪 Sulfachlorpyridazine 对照品/标准品1624006 磺胺醋酰钠 Sulfacetamide Sodium 对照品/标准品1623808 磺胺醋酰 Sulfacetamide 对照品/标准品1623706 磺胺苯酰 Sulfabenzamide 对照品/标准品1623681 硝酸硫康唑 Sulconazole Nitrate 对照品/标准品1623670 舒巴坦 Sulbactam 对照品/标准品1623648 枸橼酸舒芬太尼 CII Sufentanil Citrate CII 对照品/标准品1623637 蔗糖 Sucrose 对照品/标准品1623626 蔗糖素(三氯蔗糖) Sucralose 对照品/标准品1623615 蔗糖八乙酸酯; 蔗糖八醋酸酯 Sucrose Octaacetate 对照品/标准品1623604 氯琥珀酰单胆碱 Succinylmonocholine Chloride 对照品/标准品1623502 氯琥珀胆碱 Succinylcholine Chloride 对照品/标准品1623003 硫酸链霉素 Streptomycin Sulfate 对照品/标准品1622408 甜菊糖 Stevioside 对照品/标准品1622000 硬脂醇 Stearyl Alcohol 对照品/标准品1621507 硬脂酰聚氧甘油酯 Stearoyl Polyoxyglycerides 对照品/标准品1621008 硬脂酸 Stearic Acid 对照品/标准品1620220 司他夫定系统适用性实验用混合物 Stavudine System Suitability Mixture 对照品/标准品1620209 司他夫定;3'-脱氧-2',3'-双脱氢胸苷 Stavudine 对照品/标准品1620005 司坦唑醇 CIII Stanozolol CIII 对照品/标准品1619505 角鲨烯 Squalane 对照品/标准品1619017 螺内酯杂质A Spironolactone Related Compound A 对照品/标准品1619006 螺内酯 Spironolactone 对照品/标准品1618003 盐酸大观霉素 Spectinomyc
实验室最近要做个外标的标准曲线,但是标准品到处都缺货,只有纯度是≥98或者99%的试剂级的。试剂级大概就相当于国内的化学纯CP级吧?这能用么?做标准品的一般是看纯度还是要看级别的呀?
苯酚-硫酸法是一种常用的检测粗多糖含量的方法,其原理是苯酚-硫酸试剂可与游离的寡糖、多糖中的己糖、糖醛酸起显色反应,在480-490 nm处有最大吸收值,吸收值与糖含量呈线性关系。此法是先用标准品多糖制作标准曲线后,再通过多糖的显色反应测定吸光度,然后根据其在曲线上的位置推算出多糖的浓度从而推算其含量。此法操作简单、快速、灵敏、重复性好,对每种多糖仅需制作一条标准曲线[1]。目前大家研究较多的、生物活性较高的一些真菌多糖,如香菇多糖、灵芝多糖、姬松茸多糖、猴头菇多糖、灰树花多糖等[2],在结构上大多是以β-(1→3)、β-(1→4)或β-(1→6)糖苷键连接的葡聚糖,另外,分子量也一般分布在十几万到几十万之间。因此,由北京卫生防疫站建立,经中国预防科学院营养与食品卫生研究所验证的《粗多糖含量的测定方法》中建议使用50万分子量的葡聚糖作为标准品[3]。为行业内粗多糖含量的测定统一了标准,使各企业之间多糖类产品更具有可比性。燕麦β-葡聚糖是一种β-(1→3)-(1→4)键接的线性葡聚糖,在结构、粘度等其他物理性质上与常见的植物和真菌多糖很相似,适合作为植物、真菌来源多糖含量测定的标准品。但由于多糖纯化困难,市面上不少葡聚糖纯度较低,不适合作为标准品。下面,我们来比较两种不同纯度的燕麦β-葡聚糖产品作为多糖标准品的区别。1 材料与方法1.1 实验材料高纯度燕麦β-葡聚糖PS-Con-Ⅰ由武汉百特纯大分子科技有限公司提供,纯度大于97%(其中,另外3%主要是结合水),低纯度燕麦β-葡聚糖由某食品研究所提供,纯度约50%,苯酚、浓硫酸均为化学纯。1.2 实验方法样品溶解:高纯度燕麦β-葡聚糖经70℃水浴,15min后完全溶解。低纯度燕麦β-葡聚糖70℃水浴,30min后仍有不溶物,升高溶解温度至90℃后继续溶解30min,仍有少量不溶物,过滤。溶液配制:配制0.1mg/ml葡聚糖标准溶液,50mg/ml苯酚溶液备用。标准曲线的制作:精密吸取葡聚糖标准液0.10,0.40, 0.80,1.20,1.60,2.00ml(分别相当于葡聚糖0.01,0.04,0.08,0.12,0.16,0.20mg),补充水至2.0mL,加入苯酚溶液1.0ml,混匀,再加入浓硫酸5ml,混匀,沸水浴2分钟,混匀,冷却后用分光光度计在485nm波长处以试剂空白溶液为参比,测定吸光度值(A),以A为横坐标,葡聚糖含量C为纵坐标绘制标准曲线。2 结果与分析2.1 样品溶解高纯度燕麦β-葡聚糖溶解速度较快,溶液澄清透明,说明此产品溶解性良好。低纯度燕麦β-葡聚糖难以溶解,且溶解1h后仍有不溶物存在,说明此产品溶解性差,杂质较多。 2.2 标准曲线下表为两种标准品分别配制不同葡聚糖浓度(含量)反应后得到的吸光值:葡聚糖含量(mg)0.010.040.080.120.162.00高纯度标样吸光值0.0530.0800.2000.2620.3530.450低纯度标样吸光值0.0010.0550.1130.1730.2400.320通过数据处理,得到标准曲线如下:高纯度燕麦β-葡聚糖 C=0.4657A-0.0068 (R=0.9955)低纯度燕麦β-葡聚糖 C=0.609A+0.0101(R=0.9985)比较这两个标准曲线发现,当待测样品吸光值一定,使用低纯度葡聚糖作为标准品得到的标准曲线计算葡聚糖含量值时,明显高于高纯度标准品。究其原因,低纯度葡聚糖所含杂质较多,在作为标准品时,部分杂质不能溶解,却计入了标准品葡聚糖总量,因此,使得结果偏高。另外,即使溶解的物质中,也有可能存在部分不能参加反应的蛋白等杂质,同样会造成结果偏高。由以上数据和分析可以得出,测定粗多糖含量不能使用低纯度葡聚糖作为标准品,应尽量选用高纯度葡聚糖标准品,按照国家建议方法和行业标准进行检测,这样才能保证各企业多糖系列产品在含量和纯度上的可比性,有利于规范企业行为和保健品市场。参考文献[1] 胡居吾,范青生,肖小年. 粗多糖测定方法的研究. 江西食品工业. 2005, 1[2] 李明元. 真菌粗多糖测定方法的研究. 食品研究与开发. 2007, 5[3] 粗多糖的测定方法. 北京卫生防疫站建立,经中国预防科学院营养与食品卫生研究所验证. 食品伙伴网
色谱柱DB23和DB-5都试过,都没有标准品峰进样口温度230,检测器温度250,柱温:60℃,以10℃/min程序升温至120℃,恒温1min,以10℃/min程序升温至200℃,恒温2min,进样量1uL;标准品为1uL苯乙醇溶于2ml乙酸乙酯溶剂峰正常,相同条件下苯乙酮标准品也有峰。为什么1-苯乙醇标准品不出峰???
哪位知道国产的异丁醇,异戊醇标准品哪有?
感谢哪位大侠分享下HG/T 4134-2010 工业聚乙二醇PEG标准 万分感激
求购:国产三唑醇标准品,哪位可以提供点信息?谢谢![em61] [em61]







 我要推广仪器
我要推广仪器
 下载APP
下载APP