GB 29687-2013 食品安全国家标准 水产品中阿苯达唑及其代谢物多残留的测定 高效液相色谱法
用新柱子做了阿苯达唑的样品,然后走空白和溶剂空白,发现阿苯达唑的峰很高。请问大家,做阿苯达唑是不是容易在柱子上残留?还是其他原因?如果是柱子残留,什么柱子不易残留?
最近做阿苯达唑,有几个问题想请教下:1.取样量问题:精密称取适量(约相当于阿苯达唑20mg),这个取样量不是20mg,因为阿苯达唑药片里含有其他辅料,所以取样量应该比20mg多,对吗?2.试剂问题:用乙醇稀释至刻度,这个乙醇是无水乙醇吗?3.结果偏低,是什么影响的?
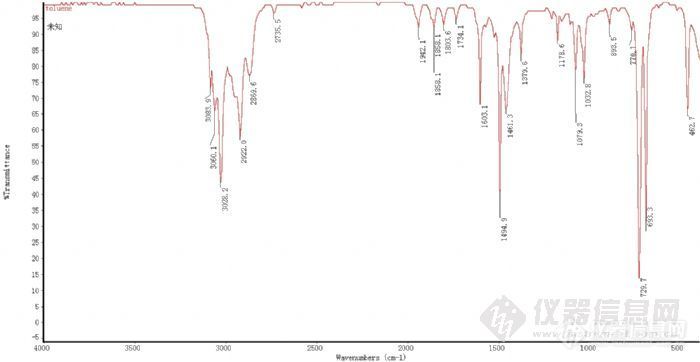
今天拿到一张苯标准品、一张甲苯标准品的IR谱图。想请问一下,里面的吸收峰都是什么振动引起的呢?峰该怎样归属呢?谢谢大家~这张是苯的标准图:http://ng1.17img.cn/bbsfiles/images/2011/06/201106211618_300779_1905813_3.jpg这张是甲苯的标准图:http://ng1.17img.cn/bbsfiles/images/2011/06/201106211618_300780_1905813_3.jpg
用761-2008标准柱子是30*0.25*0.25跟标准的一样ECD检测器,标准品配成0.04微克/毫升检测出的结果只在2分钟左右有个很小的峰试进样正己烷,也在2分钟左右有个小的波动如果联苯菊酯标准品出的峰是溶剂峰的话,为什么联苯菊酯本身不出峰?柱子是新的,没有老化,有影响么?联苯菊酯的浓度是不是过小?
阿伐那非杂质是阿伐那非的同分异构体或相关化合物,其纯度、含量和杂质情况对阿伐那非的药效和安全性有重要影响。在药物研发和生产过程中,需要使用标准品来检测和鉴定阿伐那非及其杂质的性质和含量。COTO标准品是一种高纯度的标准物质,用于测定阿伐那非及其杂质的纯度、含量和化学性质。通过与COTO标准品进行对比和分析,可以确定阿伐那非及其杂质的结构、组成和含量,从而保证阿伐那非的质量和安全性。在药物研发和生产过程中,COTO标准品的使用非常重要。它可以提供可靠的参照物,用于质量控制、药物分析和化学计量学研究。通过使用COTO标准品,可以确保阿伐那非及其杂质的准确性和可靠性,为药物的安全性和有效性提供保障。总的来说,COTO标准品在阿伐那非杂质的研究和控制中具有重要作用。通过使用COTO标准品,可以更好地了解阿伐那非及其杂质的性质和含量,从而确保药物的安全和有效性。同时,也需要加强生产过程中的管理和监督,加强质量标准和监管措施的执行力度,确保药物质量和安全。
看到国标GB/T 14677-1993空[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质[/color][/url]量 甲苯、二甲苯、苯乙烯的测定 [url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法中,做苯的标准曲线时要用二硫化碳做溶剂,我看到有人也用甲醇做溶剂,溶剂的不同会有影响吗? 另外在这个标准里面,不管是标样还是被测气体样品,都用采样管来处理然后通过加热解析进入色谱。我们这里装置简陋,可以在做标准曲线的时候,直接向气化室进配好的标准溶液,在做被测气体的时候,直接通过六通阀进气样吗,然后根据被测气体出峰的面积在标准曲线上找对应的含量吗? 希望了解的人回答,万分感谢!很感谢大家的回答,但还是没有我想要的答案,抛开国标不说,如果我用甲醇或者二硫化碳为溶剂配置了一定浓度的苯溶液,假设为1mg/mL,然后用微量注射器取1、2、3、4、……uL进样,这样苯的进样质量就是1、2、3、4、……ug,以进样质量为横坐标,峰面积为纵坐标作图。然后在后面测空气样品时(直接空气进样),可否将前面的图作为标准曲线用,通过测得的峰面积得到进样中苯的质量,然后根据进样体积得到苯的浓度?写的有点长,感谢大家耐心阅读并作出回答。
2011年2月15日,新西兰食品安全局(NZFSA)公布了2011年食品农化物最大残留限量标准。此次涉及的农化物包括阿维菌素、乙酰甲胺磷、阿苯达唑、氯氨吡啶酸、双甲脒、氨基三唑、阿莫西林、氨苄青霉素、安普罗铵、阿泊拉霉素、艾维激素、阿扎康唑、甲基谷硫磷、三唑锡、嘧菌酯、巴喹普林等256种化学物质,涵盖的食品范围包括鳄梨、猕猴桃、梨果、草莓;牛脂肪、牛肝脏、牛肉、绵羊脂肪、绵羊肾、绵羊肝、绵羊肉等126种,其中涉及最多的三类食品依次为梨果(43种),葡萄(34种),马铃薯(32种)。此次规定了食品中氯霉素的最大残留限量标准为0.0003mg/kg,是被允许的最大残留限量值中最低的。
请问哪里能买到拌种灵和苯醚甲环唑的标准品呢?
色谱柱DB23和DB-5都试过,都没有标准品峰进样口温度230,检测器温度250,柱温:60℃,以10℃/min程序升温至120℃,恒温1min,以10℃/min程序升温至200℃,恒温2min,进样量1uL;标准品为1uL苯乙醇溶于2ml乙酸乙酯溶剂峰正常,相同条件下苯乙酮标准品也有峰。为什么1-苯乙醇标准品不出峰???
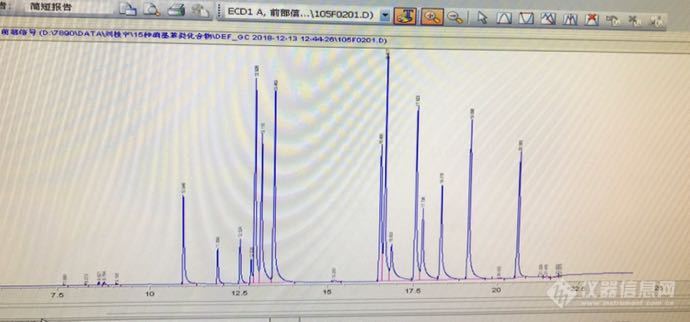
做HJ 648 水质中硝基苯类化合物的检测,15种标准品的色谱峰拖尾,DB-1701柱子,30×0.32×0.25,进样口250°,检测器300°,柱子流量1ml,初温50°保持2min.,以每分钟10°升到200°,保持1min.,再以每分钟12°升到250°,保持2min.,换过非极性的柱子OV-101,分离效果更差,请问这里有没有做过这个标准的老师指导一下。[img=,690,322]https://ng1.17img.cn/bbsfiles/images/2018/12/201812140954328263_5265_1620184_3.png[/img]
看到国标GB/T 14677-1993空[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质[/color][/url]量 甲苯、二甲苯、苯乙烯的测定 [url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法中,做苯的标准曲线时要用二硫化碳做溶剂,我看到有人也用甲醇做溶剂,溶剂的不同会有影响吗? 另外在这个标准里面,不管是标样还是被测气体样品,都用采样管来处理然后通过加热解析进入色谱。我们这里装置简陋,可以在做标准曲线的时候,直接向气化室进配好的标准溶液,在做被测气体的时候,直接通过六通阀进气样吗,然后根据被测气体出峰的面积在标准曲线上找对应的含量吗? 希望了解的人回答,万分感谢!
做胶粘剂中苯、甲苯、二甲苯,卤代烃等的含量,所用苯、甲苯、二甲苯,卤代烃是否为标准品?还是色谱纯的试剂即可??谢谢!!!

[font=宋体] 阿戈美拉汀杂质是在阿戈美拉汀的生产或保存过程中产生的非目标化合物。这些杂质可能会影响阿戈美拉汀的纯度和药效。阿戈美拉汀[/font][font=宋体]在临床上[/font][font=宋体][font=宋体]是一种治疗抑郁症的药物,属于褪黑素受体激动剂和[/font][font=Calibri]5-[/font][font=宋体]羟色胺受体拮抗剂。[/font][/font][font=宋体][font=宋体] 阿戈美拉汀杂质有多种类型,每一种都具有不同的化学特性,如[/font][font=Calibri]CAS[/font][font=宋体]号、分子式、分子量等。例如,阿戈美拉汀杂质[/font][font=Calibri]7-Desmethyl-3-hydroxyagomelatine[/font][font=宋体]([/font][font=Calibri]3-Hydroxy-7-desmethyl agomelatine[/font][font=宋体])是[/font][font=Calibri]Agomelatine[/font][font=宋体]的代谢产物,其[/font][font=Calibri]CAS[/font][font=宋体]号为[/font][font=Calibri]166526-99-4[/font][font=宋体],纯度为[/font][font=Calibri]98%[/font][font=宋体],具有特定的化学结构和性质。另一种阿戈美拉汀杂质是[/font][font=Calibri]AgoMelatine DiMer Urea[/font][font=宋体],其[/font][font=Calibri]CAS[/font][font=宋体]号为[/font][font=Calibri]185421-27-6[/font][font=宋体]。[/font][font=Calibri]CATO[/font][font=宋体]标准品提供的阿戈美拉汀全套的杂质[/font][/font][font=宋体],[/font][font=宋体]这些杂质对于药物的纯度和稳定性研究至关重要,也是药物研发过程中不可或缺的一部分[/font][font=宋体]。[img=,606,514]https://ng1.17img.cn/bbsfiles/images/2024/02/202402182106267012_9724_6381607_3.png!w606x514.jpg[/img][/font][font=宋体][color=#05073b][back=#fdfdfe] 广州[/back][/color][/font][font='Segoe UI'][color=#05073b][back=#fdfdfe]佳途科技[/back][/color][/font][font=宋体][color=#05073b][back=#fdfdfe]股份有限公司[/back][/color][/font][font='Segoe UI'][color=#05073b][back=#fdfdfe]深知药物研发与质量控制的重要性[/back][/color][/font][font=宋体][font=宋体],[/font][font=Calibri]CATO[/font][font=宋体]标准品厂家,提供阿戈美拉汀全套[/font][/font][font=宋体]的[/font][font=宋体]杂质,为客户提供更加精准、可靠的分析标准品,助力药物研发事业的快速发展[/font][font=宋体],[/font][font=宋体]以满足客户在药物研发和质量控制方面的需求。[/font]
最近用液相色谱测定非食品添加物质十二烷基苯磺钠,发现从国标中心购买的十二烷基苯磺酸钠标准溶液居然有四个峰,怎么回事呀?有谁作过吗?到底哪个峰是十二烷基苯磺酸钠呢?烦请高手指点一二!

大神们,求助:为什么直接进苯的标准品峰形很好,但是热解析进样是拖尾的?第一张热解析进样,第二张直接进样甲醇中苯,http://ng1.17img.cn/bbsfiles/images/2016/12/201612281625_02_2196181_3.jpghttp://ng1.17img.cn/bbsfiles/images/2016/12/201612281626_01_2196181_3.jpg
配制邻苯二甲酸酯类标准品时,购买的标准品是纯品,但是也是液体的,100mg,配制的时候,一次全部清洗到100ml棕色容量瓶中,标准品溶液的浓度是多少?是1mg/ml吗?还是要用减量法称量一下具体的含量。谢谢!我用正己烷溶解定容的,保存的时候直接放在-10°的冰箱冷冻室保存,可以吗?说什么的都有,如果是称量的话,这样也不好吧,做邻苯二甲酸盐的来说说吧
这几天用岛津的碳18分析氨基酸,用的异硫氰酸苯酯为衍生剂。 用水代替氨基酸标准品,会出现很多杂峰,所以想应该是衍生过程引进的。衍生的方法是 标准品+三乙胺乙腈溶液+异硫氰酸苯酯乙腈溶液 反应一个小时候用正己烷萃取各位大虾有更好的衍生方法么?本实验室没有氮吹仪,没法吹干哦
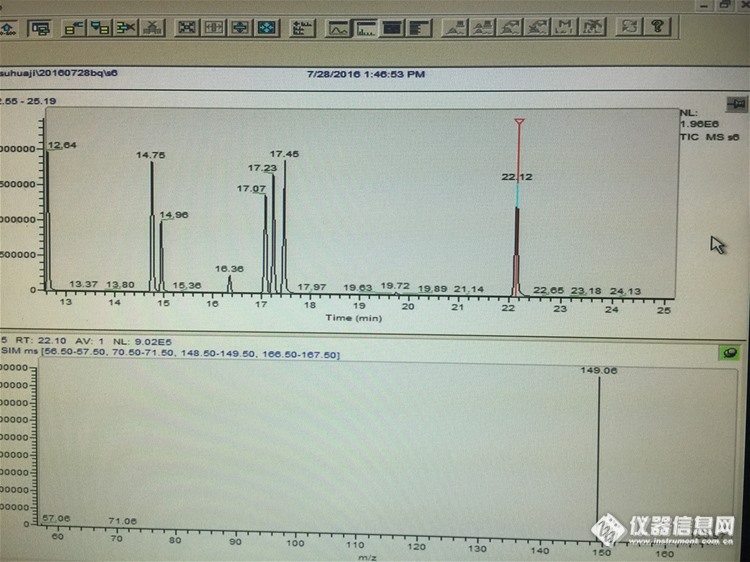
按照国标21928做16种邻苯二甲酸酯类塑化剂的标准品,买的标准品是AccuStandard的,下面的是做出来的图(只截取了后面的一部分),纵观做的几次标曲,前面的14种线性都还好,有的可以达到三个9,可是最后两种DNOP和DNP每次做的线性都好差,问问各位,是不是我做的哪里出了问题,还是大家也有出现这种情况? 还有就是图中那个22.12的峰,保留时间差不多,按理说应该是DNP的峰,可是丰度比和标准上的57:149:71:167(100:94:48:13)明显对不上,所以一直怀疑它到底是不是DNP,如果不是的话又一直都能做出来又是什么鬼。求各位帮忙解答一下 求大神帮忙解答一下http://ng1.17img.cn/bbsfiles/images/2016/07/201607290928_602477_3064284_3.jpg
大家好,我做苯磺隆标样的时候,为什么只出溶剂峰,而没有样品峰呢?条件是按参考文献来的,标准品浓度为0.1,0.5,1.0,2.0,3.0ug/ml,最后还用母液10ug/ml进样也是不出样品峰。
做甜蜜素的时候标准品不出峰了,仪器条件和前处理方法都是之前做过的,年前的时候还是好的。然后最近标准品都不出峰了,溶剂峰是正常的。换了新的标准品还是一样。想问下大家 这是什么原因导致的呢?谢谢
各位老师,请教一个GCMS检测邻苯二甲酸酯的问题,检测标准品时,每个峰前都会有一个小峰,采购的100ppm原标是用甲醇配制的,校准曲线是用丙酮稀释的,大家有遇到过这个现象吗?是甲醇丙酮溶剂汽化体积不同造成的吗?谢谢。
流动相是乙腈和0.1%磷酸水,空白溶剂是50%甲醇水对照品为6种标准品的混标:分别跑了初始5%,10%,20%,到50%盐的梯度,倒过来的梯度也跑了,都是只有3分钟前出一个大峰和多个连在一起的小峰。还分别跑了20%,30%,40%,50%,60%,70%,80%,90%乙腈的等度都跑了 除了等度50%的在3分钟后有出峰,其他的都在3分钟前出大峰和一些小峰。随着乙腈增加,峰是有微小的前移变化,但是都在3分钟前出峰。文献中查到的标准品色谱条件,分别有乙腈从5%10%20%逐步梯度到50%, 跑30分钟,出峰时间都适中在10~20分钟左右。然后昨天也另外完全按其中一个单标国家药典(甲醇:水=25:75)的方法(除了溶剂不同,药典是乙醇,我们是50%甲醇水)跑了一针,也是只有2分钟左右的空白溶剂相同的大峰。多种方法跑的空白溶剂,出的大峰,是在2.4分钟左右出现。看了出峰的柱效,没有超过3000的,大多在1000左右。这是色谱柱的问题吗,还是流动相ph的影响,会影响那么大吗,怎么调都分不开,像色谱柱完全没有保留能力。
找来找去都是PVC塑料或是纺织品,有没有涂料中邻苯二甲酸酯检测方面的标准阿?
最近在做塑料制品中的18种多环芳烃,5ug/mL浓度(为了确定保留时间所以浓度较高)的峰形都很好。配制了20ng/mL~400ng/mL共6个浓度点的标准溶液,出来的谱图无一例外表现出:除了前面3个化合物外,其余15种都拖尾严重,挨着的2个或3个化合物以一个峰宽很大的峰出来,而且保留时间都比5ug/mL时延迟。奇怪的是,我做了中间浓度点的加标实验,样液中的一系列化合物峰形都很正常。标准物质用甲苯配制,塑料制品用甲苯提取。 也就是说,标准物质在溶剂中拖尾严重,在样液中峰形正常。这是怎么回事? 我老化了柱子还是如此。望高人指点。
标准品里有50多种混合物,方法什么的都没变更过,以前是出峰的,最近几次做,苯乙烯出了一个特别小的峰,其他物质出峰都挺好的,线性也不错。手动调谐做了捡漏,没有漏气问题。不知再在哪些地方做检查?
在配置邻苯二甲酸脂的标准品的过程中,用正己烷配置的一定浓度标准品比如1.0ppm 在常温下放置一周,体积并无变化.为何上机分析时与一起配置的相同浓度(1.0ppm存储于冰箱中)峰面积相差几倍?(检测仪器重复性很好)
做苯标准曲线色谱峰出的不是很好(苯峰几乎没有),会是活性炭管吸附能力不好吗
各位有没有做ISO 14389:2014 纺织品邻苯二甲酸酯测试的?这十种邻苯的混合标准品是自己配置的吗?有没有现成的混标储备液可以购买?(10项邻苯,包括:DINP、DEHP 、DNOP、DIDP、BBP、DBP、DPP、DIHP、DIBP、DMEP。)
最近根据:居住区大气中苯、甲苯和二甲苯卫生检验标准方法气相色谱法(GB 11737一89)做苯\甲苯\二甲苯;标准最低管浓度0.005μg/mL,结果一个峰也没有出来,怀疑是仪器灵敏度不够?我是瓦里安3800 DB-FFAP柱,色谱条件没问题,是自己原来做车间空气苯类的条件,在这基础上还把分流调成1:5.(原来是1:15).但是不出峰,怎么办啊????







 我要推广仪器
我要推广仪器
 下载APP
下载APP