[em06] 四氢呋喃、二氧六环、吡啶、甲苯 照残留溶剂测定法(附录Ⅷ P第三法)试验。精密称取苯适量,加甲醇制成每1 ml中约含60μg的溶液,作为内标溶液。精密称取四氢呋喃、二氧六环、吡啶、甲苯适量,加甲醇制成每1ml中各含720μg、380μg、200μg和890μg的溶液,作为对照贮备溶液;精密量取对照贮备溶液1ml与内标溶液1ml,置10ml量瓶中,加水稀释至刻度,摇匀,作为对照溶液。精密称取本品1.0g,置10ml量瓶中,加内标溶液1ml,加水溶解并稀释至刻度,摇匀,作为供试品溶液。用二乙烯基-乙基乙烯苯型高分子小球作为固定相,柱温190℃,依法测定。残留溶剂含量应符合规定。我让色谱公司按这个要求做了不锈钢柱子(柱填料:401有机载体(二乙烯基苯/乙基乙烯苯共聚体)60-80目),可是不出峰,后来把柱寄回去了,现在又寄给我的柱子(柱填料:10%PEG-20M CHROMOSORB PAW-DMCS 80-100目),峰是有了,可是分不开,我做[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]的氮气4圈,空气4.2圈,氢气4.5圈,后我又把氮气开到3圈,还是这个样子.是怎么回事呢,请高手赐教.谢谢!!!
[font=SimSun, STSong, &]降甜剂即2-(4-甲氧基苯氧基)-丙酸钠,这种香料的实际作用只是使别人口感上觉得甜度下降了还是说它跟糖有了化学反应造成了糖的转化?[/font]
有谁知道气相色谱测定3-甲氧基苯甲腈的方法?毛细管柱的
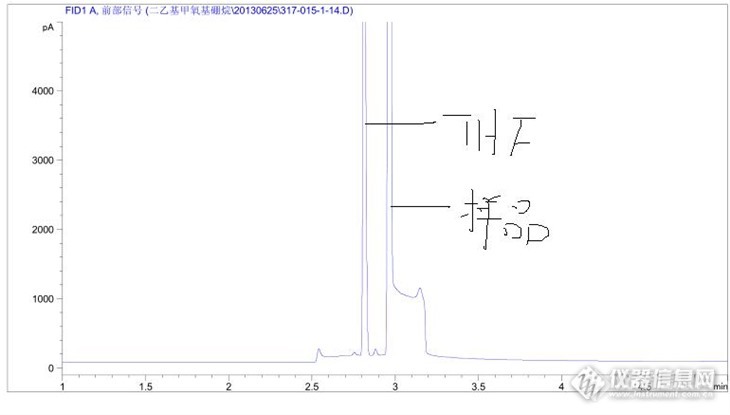
二乙基甲氧基硼烷的THF(四氢呋喃)溶液,有少量甲醇和乙醚。FID检测器HP-5的色谱柱 80℃1分钟 10℃/min 升至180℃ 2分钟以前做的挺好,但是最近成这样了,是什么原因啊?换了新柱子还是这样。http://ng1.17img.cn/bbsfiles/images/2013/07/201307051012_449530_2260586_3.jpg这是以前做的http://ng1.17img.cn/bbsfiles/images/2013/07/201307051012_449531_2260586_3.jpg这是现在的
大家谁有对甲氧基苯乙醇,麻烦打个质谱图让我看看,我现在在做一个反应,产物质谱图不确定,谢谢!
我用CPSC方法测试邻苯,走的是买的质控样品,如果按照标准上总体积为四氢呋喃体积加正己烷体积,计算结果超过标准值50%,如果只计算正己烷体积,则在标准值范围内,请问这个总体积计算是否要把四氢呋喃体积计算在内。
由于手上没有2,3,4-三甲氧基乙酰苯(2,3,4,-trimethoxyacetophenone,C11H14O4)的对照品,所以想求助大家有没有它的光谱图,有的请发一份上来好吗?谢谢大家帮忙啦,
求2,3苯并呋喃测试方法,仪器参数
求购咨询苯基三甲氧基硅烷与辛基三乙氧基硅烷的色度检测标准和仪器,或者告诉下哪里有权威的检测机构也好的。谢谢!
甜味抑制剂"2-(4-甲氧基苯氧基)-丙酸钠"如何检测?用HPLC可以检测,用GC或GCMS如何检测,如何衍生化?先酸化吗?在衍生化?
谁手上有2,4,5-三甲氧基苯甲醛质量标准?邦邦忙好吗?
各位好,目前小弟正在做呋喃苯烯酸钠的检测,不知道怎么回事流动相,空白都有污染,导致添加回收,以及基质标准曲线不成线性,根本无法校正,请教各位。
化妆品中呋喃香豆素类(三甲沙林、8-甲氧基补骨脂素、5-甲氧基补骨脂素)和欧前胡内酯的检测方法
气相测3.5-二甲氧基苯甲酸甲酯要用什么标准物做外标
各位大侠好,请问有谁知道三甲氧基苯甲醛这种物质用HPLC检测时,紫外吸收波长是多少,以及使用什么流动相呢?小女子在此谢过了!http://simg.instrument.com.cn/bbs/images/brow/em09511.gif
我的样品分子量在2000以下,流动相用进口的四氢呋喃。每次新开封一瓶四氢呋喃后,我会倒一部分四氢呋喃到一个棕色瓶中,用于平时试样的配制。但我发现,几天后棕色瓶中的四氢呋喃就氧化了,氧化物的分子量也在2000以下,出峰正好对我的试样形成干扰了。请教大家有什么好的防止四氢呋喃氧化的方法?还是我每天制样时,必须从流动相那个瓶中抽取四氢呋喃,才能减少这种干扰?
[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]测3.5-二甲氧基苯甲酸甲酯要用什么标准物做外标
[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]测3.5-二甲氧基苯甲酸甲酯要用什么标准物做外标
求呋喃能力验证过的大神支个招。实验室正在做呋喃的测量审核,因为前两次能力验证的实测值都大于120%,没过,一直在纠结,我们是不是衍生试剂2硝基苯甲醛加大4倍,还是PH调不对。按照农业部783号公告做的。昨天做的最后一步去脂肪加入定容液加正己烷的时候,只有考核样乳化了,其他质控样完好。白做了,总共7天时间。如果这次再不过真的要被骂了。求各位大神支个招。
请告诉2-甲基-4-甲氧基-二苯胺的液相色谱分析方法
小弟做了好几次2-溴,3,3‘-二甲氧基联苯的氢谱,都发现甲基有三重峰,做何解?谢了!
仪器走了10ppm 的,3,3二甲氧基联苯胺走的不好,啥情况呢?[img=,690,517]https://ng1.17img.cn/bbsfiles/images/2024/01/202401131427369784_1355_3971926_3.png[/img][img=,690,517]https://ng1.17img.cn/bbsfiles/images/2024/01/202401131427375863_7057_3971926_3.png[/img]
【求助】2-乙氧基苯甲脒(伐地那非(vardenafil)合成的中间体)各种文献上都查不到分析方法,有会的大虾吗?
FID使用时相关问题,我是在乙醇里加入等体积四氢呋喃、甲苯、氟苯浓度大概在百分之一,出峰顺序是四氢呋喃、乙醇、氟苯、甲苯,提高柱温以后,乙醇的含量减少了是怎么回事?相同样品,同样的注射量,恒温程序走的,当提升温度把柱温由60变到80,乙醇的积分面积百分数从90%下降到80%是因为什么,请各位大神解惑
正在做硝基呋喃扩项,机器是Waters的TQ-S,第一次衍生的时候,是把内标和标样分开衍生的,用的农业部781公告,具体操作是硝基呋喃四种标样(Dr.E的固体标样),用乙腈稀释,母液100ug/mL,吸10uL50mL离心管,加入4mL水,再加0.5mL的盐酸,再加150微升0.1mol/l的邻硝基苯甲醛,涡旋振荡之后放入37℃恒温培养箱,16h后加磷酸氢二钾,然后用NaoH调pH。第一次衍生的时候,AHD和SEM的内标没有响应。后来重新配标液,继续衍生的时候,出了2-NP-2SCA的所有物质都出峰了,响应都很好。然后再配SEM单标衍生,已经配到1000-10000ppb,进0.1微升,都没任何响应。前后已经做了10天了,求各位大神指点。图如下,求版主指点!
请教:乙烯基甲醚、丙烯醛、二甲氧基二氢吡喃、戊二醛这几种物质可否有[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]检测?用什么样的柱子和检测条件?
用微波处理化妆品,测定其中的重金属(Cr、Hg、Pb...),化妆品的成分是对甲氧基苯甲酸,消解后澄清,用水定容出现絮状沉淀。请教出现絮状沉淀原因及正确的前处理方法。
求乙酰氯,二溴丁烷,对甲氧基苯胺,硫代乙酸钾,溴苯含量的分析方法,那个大哥大姐知道的帮帮忙
四氢呋喃纯化时蒸馏要氮气保护吗?我最近在做四氢呋喃的纯化,是用二苯甲酮、钠回流的,之前做好先用氢氧化钾干燥。等体系变墨绿色的时候就可以蒸馏出来了。我用干燥塔隔绝湿气,这时候要用氮气保护码 四氢呋喃有被氧化的机会。请给位高手指教!!!

请教各位老师:采用四氢呋喃超声萃取邻苯二甲酸酯进样测定数据与实际差距较大,是哪块步骤出了问题?0.3g样品+10ml四氢呋喃超声30-60min,加入20ml正己烷静置,聚四氟乙烯膜过滤。样品为邻苯浓度1000ppm的PVC,实测只有5ppm,定量方法为外标法,同一方法参数对5ppm、10ppm等浓度标液进行检测无异常。[img=,690,920]https://ng1.17img.cn/bbsfiles/images/2023/07/202307161725554321_1260_5881265_3.jpg!w690x920.jpg[/img][img=,690,517]https://ng1.17img.cn/bbsfiles/images/2023/07/202307161725556394_1136_5881265_3.jpg!w690x517.jpg[/img]







 我要推广仪器
我要推广仪器
 下载APP
下载APP