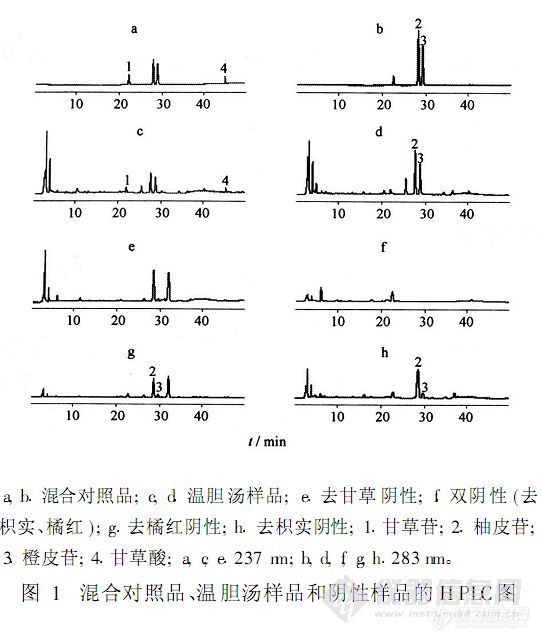
【作者】 许栋明; 程可建;【Author】 XU Dongming,CHENG Kejian(1.Science and Technology Innovation of Small and Mid-sized Enterprise Fund Management Center,Ministry of Science and Technology,Beijing 10038,China;2.Bescholor Research Center,Peking University,Beijing 100084,China)【机构】 科技部科技型中小企业技术创新基金管理中心; 北大世佳研究中心;【摘要】 目的:建立HPLC同时测定温胆汤中甘草苷、柚皮苷、橙皮苷和甘草酸含量的方法。方法:DIKMA Diamonsil(2)-C18柱(4.6 mm×250 mm,5μm);流动相乙腈(A)-0.1%磷酸溶液(B),线性梯度洗脱;检测波长237,283 nm;柱温25℃;流速1.0 mL.min-1;进样量10μL。结果:甘草苷、柚皮苷、橙皮苷和甘草酸铵的进样量与峰面积,分别在0.019 9~0.119(r=0.999 7),0.180~1.08(r=0.999 7),0.146~0.873(r=0.999 8),0.0393~0.236μg(r=0.999 7)呈良好的线性关系;平均加样回收率依次为97.7%,97.7%,97.1%,98.5%,RSD 1.4%,2.0%,2.0%,1.9%。结论:该方法快速,简便,重复性好,适合于同时测定温胆汤样品中甘草苷、柚皮苷、橙皮苷和甘草酸的含量。 更多还原http://ng1.17img.cn/bbsfiles/images/2012/08/201208131329_383466_2379123_3.jpg
如题,请问有什么关于新橙皮苷或者橙皮苷含量测定的国标吗
HPLC-DAD分析酸浆中木犀草素及木犀草素-7-β-D-葡萄糖甙成分酸浆(拉丁文名:Physali alkekengi L.)又名红菇娘、挂金灯、戈力、灯笼草、灯笼果、洛神珠、泡泡草、鬼灯等北方称为菇蔫儿、姑娘儿,以果实供食用。化学成分含酸浆苦素A(Physalin A)、酸浆苦素B、酸浆苦素C、木犀草素(Luteolin)及木犀草素-7-β-D-葡萄糖甙。果实含枸橼酸、草酸、维生素C、酸浆红色素(physalien)、酸浆醇(physanol)A,B。花萼含α胡萝卜素、酸浆黄质(physoxanthin)及叶黄素等,种子油的不皂化物中分得多种4α-甲基甾醇,主要为禾本甾醇(gramisterol)和钝叶醇(obtusifoliol)及4种新甾体。此外尚含多种4-脱甲基甾醇,如胆甾醇和24-乙基胆甾醇等。还含有多种三萜3β-一元醇,其中环木菠萝烷醇(cycloartanol)35%,环木菠萝烯醇(cycloartenol)27%、羊毛脂-8-烯-3β-醇(lanost-8-en-3β-ol)。木犀草素(luteolin)是一种天然黄酮类化合物,存在于多种植物中,具有抗炎、抗肿瘤、抗过敏等方面的作用。化学是如下:http://ng1.17img.cn/bbsfiles/images/2016/08/201608311303_607620_2217446_3.jpg目前,国内传统中药有效成分的提取方法普遍存在提取率低、杂质清除率不高、生产周期过长、能耗高、溶剂用量大等缺点。随着中药现代化进程的不断深入,许多现代高新技术不断地被应用到中药有效成分的提取和分离,使得中药有效成分的提取更高效和简便。超声-微波协同萃取技术直接将超声振动与开放式微波两种作用方式相结合,充分利用超声波振动的空化作用以及微波的高能作用,实现了低温常压条件环境下,对固体样品进行快速、高效、可靠的预处理,与常规提取方法相比,超声-微波协同萃取技术具有快速、节能、节省溶剂、污染小等优点。本实验应用超声-微波协同萃取法提取酸浆中的木犀草素及木犀草素-7-β-D-葡萄糖甙,采用高效液相-二极管阵列检测法(HPLC-DAD)测定提取物中木犀草素及木犀草素-7-β-D-葡萄糖甙的含量,药材中二者成分的含量分别为:1.200mg/g 和0.43mg/g,二个峰,木犀草素-7-β-D-葡萄糖甙峰位置分别为:221nm,270nm,木犀草素峰位置分别为:226nm,276nm,由于木犀草素-7-β-D-葡萄糖甙比木犀草素多了一个 β-D-吡喃葡萄糖基团,天麻素二个峰位置都发生了蓝移,样品中二个峰的光谱图与标准品二个峰的光谱图相同,可以进一步确定酸浆中含有木犀草素及木犀草素-7-β-D-葡萄糖甙。主要仪器与试剂主要仪器Agilent1100型四元梯度高效液相色谱仪(美国 Agilent 公司)Agilent TC-C18(ODS)色谱柱(5μm,4.6×250mm,美国 Agilent 公司)CW-2000 超声-微波协同萃取仪(新拓微波溶样测试技术有限公司)DJ-10A 型倾倒式粉碎机(上海隆拓仪器设备有限公司)RE-52AA 型旋转蒸发仪(河南巩义仪器厂)LXJ-IIB 型低速大容量多管离心机(上海安亭科学仪器厂)试剂木犀草素(中检所,含量98%;)木犀草素-7-β-D-葡萄糖甙(中检所,含量98%;)酸浆全草(采于黑龙江)除甲醇、乙腈为色谱纯(国药集团化学试剂有限公司),其余试剂除专门提到外,均为分析醇,实验用水为二次蒸馏水。实验方法供试品溶液的制备 精密称取酸浆粉末1.0g,置于超声-微波萃取仪玻璃容器中,加入50mL70%甲醇,开启超声微波,控制在恒温50℃下提取40min,萃取3次,合并提取液,浓缩至近干,残渣加入甲醇溶解,转移至10mL 量瓶中,加甲醇稀释至刻度,摇匀,过0.45μm 的微孔滤膜,取续滤液,即得。提取条件的考察溶剂的选择:精密称取酸浆粉末1.0g,置于超声-微波萃取仪玻璃容器中,分别用水、70%甲醇、70%乙醇溶液超声-微波协同萃取40min(n=3),萃取3次,合并提取液,浓缩至近干,残渣加入甲醇溶解,转移至10mL 量瓶中,加甲醇稀释至刻度,摇匀,过0.45μm的微孔滤膜,取续滤液,HPLC 测定萃取率。溶剂体积分数的选择:分别用体积分数为40%、50%、60%、70%、80%、90%和纯甲醇溶液超声-微波协同萃取30min(n=3),方法同上。溶剂用量的选择:分别用10mL、20mL、50mL、80mL、100mL70%甲醇提取,方法同上。提取时间的选择:分别用70%甲醇超声-微波协同萃取20min、30min、40min、50min、60min(n=3),方法同上。提取温度的选择:分别在40、45、50、55、60℃下用70%甲醇超声-微波协同萃取40min,方法同上。对照品溶液的制备 分别精密称取常温减压干燥12h 的木犀草素及木犀草素-7-β-D-葡萄糖甙对照品适量,加甲醇配制成木犀草素-7-β-D-葡萄糖甙为200μg/mL、木犀草素为100μg/mL 的混合对照品溶液,冷藏备用。色谱条件 色谱柱:Agilent TC-C18柱(5μm,4.6×250mm);流动相:A-0.1%乙酸水溶液;B-甲醇,线性梯度洗脱:0~30 min,3%~5% B;30~35 min,5%~20%B;35~40min,20%~20%B;检测波长:270nm;流速:1mL/min;柱温:30℃;进样量:20μL。结果与讨论提取条件的优化结果溶剂的优化结果:分别用水、70%甲醇、70%乙醇溶液超声-微波协同萃取30min(n=3),结果表明70%甲醇提取木犀草素-7-β-D-葡萄糖甙的量较高,而木犀草素的量差异不明显,因此选择70%甲醇提取。溶剂体积分数的优化结果:分别用体积分数为40%、50%、60%、70%、80%、90%和纯甲醇溶液超声-微波协同萃取30min(n=3),结果表明,在甲醇体积分数70%时,木犀草素-7-β-D-葡萄糖甙和木犀草素的提取率随着甲醇浓度的增加而增加;但当甲醇体积分数在70%以上时,木犀草素葡萄糖甙的提取率呈现下降趋势,木犀草素没有明显的变化。木犀草素葡萄糖甙属于一种苷,分子量小,极性较大,当甲醇体积分数过高时,溶液极性降低,使得极性较强的木犀草素葡萄糖甙不易溶出,而木犀草素极性相对木犀草素葡萄糖甙小,影响不明显,因此实验选择70%甲醇作为提取溶剂。溶剂用量的优化结果:分别用10mL、20mL、50mL、80mL、100mL70%甲醇提取,结果表明溶剂体积在50mL时木犀草素葡萄糖甙和木犀草素的提取率最高,之后随着溶剂用量的增加,木犀草素葡萄糖甙和木犀草素的提取率趋于稳定,因此溶剂用量选用50mL 进行提取 。提取时间的优化结果:分别用70%甲醇超声-微波协同萃取20min、30min、40min、50min、60min(n=3),结果表明超声-微波协同萃取时间从20~40min的过程中木犀草素葡萄糖甙和木犀草素的提取率逐渐增加;而提取时间超过40min之后,提取率反而逐渐下降。超声-微波协同萃取时间太长,植物中大量细胞细胞破碎,使得大量粘性物质等进入提取液,溶剂杂质增多、粘度增大,影响了有效成分的溶出,有效成分含量反而减少,因此选择提取时间为40min。提取温度的优化结果:分别在40、45、50、55、60℃下用70%甲醇超声-微波协同萃取40min,实验表明,提取温度在50~60℃的范围内,木犀草素葡萄糖甙和木犀草素的提取率没有明显差异,考虑到温度太高容易破坏活性成分,因此选择提取温度为50℃。流动相的考察在实验过程中,流动相首先考察了甲醇-水、乙腈-水等度洗脱对酸浆超声-微波协同萃取样品溶液进行分离,乙腈-水作为流动相时,出峰较快,不能较好地把木犀草素葡萄糖甙和木犀草素与其他杂质成分分离;甲醇-水作为流动相时,出现峰形拖尾现象,分离效果不理想。为改善上述现象,改用0.1%乙酸代替水并采用梯度洗脱,经过反复筛选之后,最终确定流动相组成为 A -0.1%乙酸水溶液, B -甲醇,洗脱程序为0~30 min , 3%~5% B;30~35 min ,5%~20% B ;35~40 min 20%~3% B,木犀草素葡萄糖甙和木犀草素和其他杂质成分能够很好的分离,得到较理想的色谱图。对照品溶液和酸浆萃取样品的HPLC-DAD 分析下图分别显示了在上述的色谱条件下,采用 DAD 进行检测得到的两种混合对照品及酸浆萃取样品的 HPLC 分离色谱图。图1色谱图中木犀草素葡萄糖甙和木犀草素的保留时间分别为18.74min, 26.87min,根据保留时间判断,图2中的 a、b 色谱峰分别初步鉴定为木犀草素葡萄糖甙和木犀草素。图3、4分别显示了混合对照品和酸浆萃取物中保留时间18.74min, 26.87min 的色谱峰进行 DAD 检测后得到的光谱图,木犀草素葡萄糖甙和木犀草素 UV 光谱图形状相似,出现 二个峰,木犀草素葡萄糖甙峰位置分别为:221nm,270nm,木犀草素峰位置分别为:226nm,276nm,由于木犀草素葡萄糖甙比木犀草素多了一个 β-D-吡喃葡萄糖基团,木犀草素葡萄糖甙二个峰位置都发生了蓝移,样品中二个峰的光谱图与
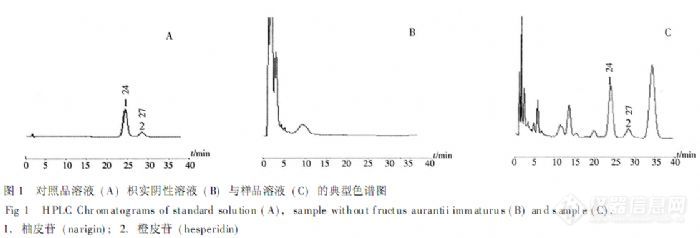
最近检测枳壳 中的新橙皮苷和柚皮苷 因为之前公司没有自己开发方法,所以按照10版药典的方法,检测的结果是:对照品的峰都可以 但是样品出现的两个峰 出峰时间和对照品的不一样,都提前了三分钟,样品和标准品混合之后,出现了四个峰。请问高人 是怎么回事,如何解决。实验条件:乙腈:水=20:80 PH=3.0 柱温25 流速1.0 自动进样 10ul 色谱柱waters150*4.6*5 样品处理:称取定量样品加50ML甲醇,称定,加热回流1.5h,放冷,补足重量,过滤,取10ml滤液置于25ml 加甲醇定容。
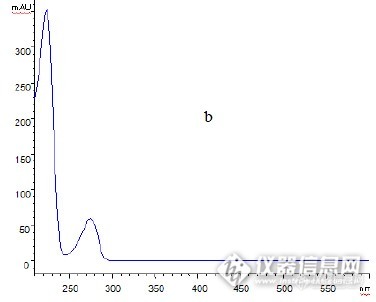
作者:李仁秋 (云南省昆明市儿童医院, 云南 昆明 650011)摘 要:目的: 建立高效液相色谱法测定新止咳合剂中橙皮苷的含量。方法: 采用 Dikma 钻石 C18 色谱柱( 4. 6mm @250mm, 5Lm) ; 流动相为甲醇) 0. 2% 磷酸溶液( 38: 62) ; 检测波长为 283nm,流速为 1. 0ml/ min, 柱温为 30 e 。结果: 橙皮苷进样量在 0. 01~ 2. 0Lg ( r= 0. 999 97)范围内与峰面积线性关系良好,平均加样回收率为 99. 12%, RSD= 0. 84%( n= 9) 。结论:本法简便、 快速、 专属性强、 重现性好;可作为新止咳合剂的质量控制方法。关键词:橙皮苷; 高效液相色谱法;新止咳合剂; 含量测定http://ng1.17img.cn/bbsfiles/images/2012/07/201207161641_377910_2379123_3.jpg
药品名称:橙皮苷外文名称:HesperidinCAS号:520-26-3溶解性:易溶于吡啶、氢氧化钠溶液,溶于二甲基甲酰胺,微溶于甲醇和热冰醋酸,极微溶于乙醚,丙酮、氯仿和苯。该品1g溶于50L水。无臭、无味。植物来源:芸香科柑桔属植物甜橙、柠檬。以下为使用资生堂色谱柱对橙皮苷检测得到的谱图,请参考。http://ng1.17img.cn/bbsfiles/images/2016/12/201612151447_02_2222981_3.jpgA. 供试品溶液 B. 对照品溶液【色谱条件】色谱柱:CAPCELL PAK C18 S5; 4.6 mm i.d.×150 mm流动相:0.1%磷酸溶液/乙腈/甲醇=74/14/12流 速 : 1.0mL/min温 度 : 25 °C检 测 : UV284nm进样量:10μL注:文献中液相方法与《中国药典(2015)》中小儿至宝丸项下橙皮苷分析方法一致。如图,对照品溶液有较好峰形,供试品溶液中的橙皮苷峰能与其他杂质峰获得较好分离,分离度大于1.50。
药用金莲花研究进展 金莲花为毛茛科金莲花属(Trollius L.)金莲花亚科金莲花族(Trollieae),别名有旱金莲花,旱地莲,金芙蓉,旱金莲,金梅草,亚洲金莲花,金疙瘩等。在清代吴其浚所著的《植物名实图考》中,误将毛茛科金莲花的名称套在了南美原产的“Tropae-lam majus”(金莲花科金莲花)上。此后以讹传讹,以致后代不少植物文本和文献也沿用了吴氏的错谬,把毛茛科金莲花和金莲花科金莲花相互混淆。2004年,李万方从形态、习性和药用价值等方面对这两种金莲花进行了区分,为金莲花的正确表述提供了依据。据统计,世界上金莲花属植物约有25种,主要分布在北半球温带及寒温带山区,其中约有16种出自中国,分布在山西、河北、内蒙古、山西、东北、四川、云南、台湾等省区,以山西、河北、内蒙古产量最大。现代医学研究表明,金莲花属植物多具有抗炎、抗病毒作用,在医药领域的应用前景非常广阔。以下就金莲花属植物的研究进展进行综合性的介绍。1 化学成分 金莲花属植物中的化学成分主要由黄酮类化合物、生物碱、有机酸、挥发油等。关于金莲花植物的报道主要集中在黄酮类化合物。20世纪80年代,从金莲花中分离到牡荆苷和荭草苷两种黄酮类化合物,90年代从长瓣金莲花中也分离出这两种物质,另外还分离出了牡荆素-2"-O-β-D-吡喃木糖苷(vitexin-2"-O-β-D-pyranxyloside)、荭草素-2"-O-β-D-吡喃木糖苷(orietin-2"-O-β-D-pyranxyloside)等四种碳苷黄酮。近年,黄文哲等又从短瓣金莲花中分离到一种氧苷黄酮--槲皮素-3-O-新橙皮糖苷。玄参黄酮、白杨黄酮、木犀草素、槲皮素、日本椴苷、及鼠尾草素等也在短瓣金莲花中被发现。2004年,宋冬梅等还从阿尔泰金莲花中分离到一种甲氧基黄酮--柯伊利素,这是首次从金莲花属植物中得到该物质。 从
最近要用HPLC测橙皮苷,谁有这方面的资料发上来,讨论一下。谁知道哪有卖橙皮苷标准品的在北京?

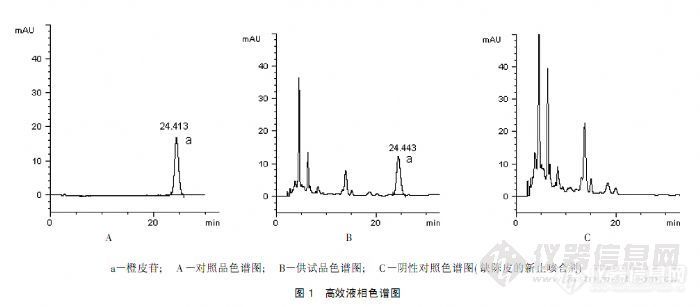
作者:蔡俊安(;河南百年康鑫药业有限公司;)摘要:目的建立香砂和中丸中橙皮苷的含量测定方法。方法采用高效液相色谱法,Diamonsil ODS1 C18色谱柱,以甲醇-醋酸-水(35∶4∶61)为流动相,流速为1.0 mL/min,检测波长为283 nm。结果橙皮苷在0.42~8.4μg范围内呈良好线性,回归方程为Y=830 125.1X+5 849.875,r=0.999 8,平均加样回收率为99.35%,RSD=0.68%(n=6)。结论本方法简便、准确、专属性强,测定结果重复性好。谱图:http://ng1.17img.cn/bbsfiles/images/2012/08/201208061429_381892_1606903_3.jpg
有做过的吧?还用从橙汁中提取橙皮苷吗?如果用的话,用什么方法提取,如果不用,直接检测就可以吗?谢谢。
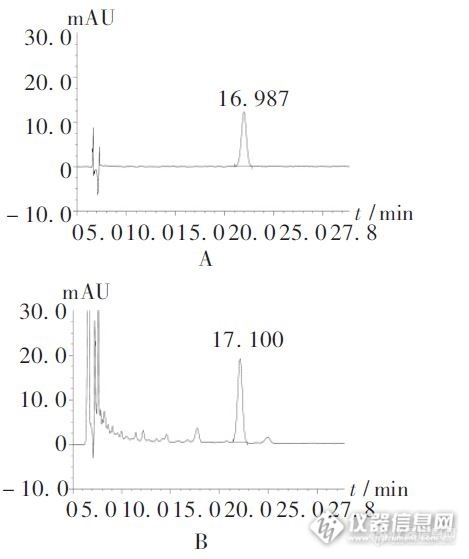
【作者】 庞小雄; 梁松庆; 钟兆健; 关敏婷;【机构】 广东药学院; 广东药学院 广东广州510006; 广东广州510006; 广东广州510006广药学院2007届药学专业本科毕业生;【摘要】 目的:测定拈痛丸中橙皮苷的含量。方法:采用反相高效液相色谱法,Diamonsil-C18柱,甲醇-水(43∶57)为流动相,流速为1.0 ml/min,检测波长283 nm,柱温30℃。结果:橙皮苷平均回收率为98.4%,RSD为2.14%(n=6)。结论:该法可用于拈痛丸中橙皮苷的含量测定。 更多还原【关键词】 拈痛丸; 橙皮苷; 反相高效液相色谱法; 含量测定;
我在做陈皮含量的时候,橙皮苷对照品用甲醇不溶解,请问谁可以指点一下?
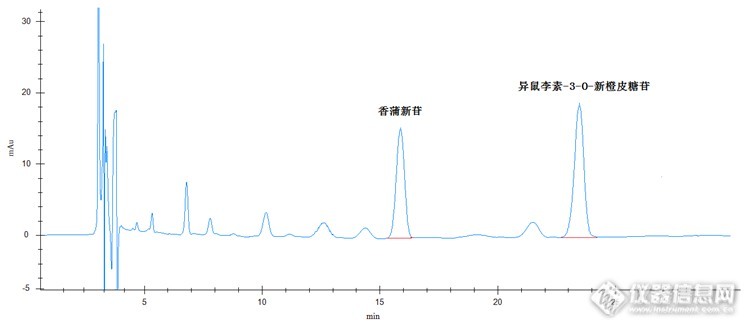
检测蒲黄药材快速、准确出结果 蒲黄药材具有止血,化瘀,通淋等药物功效。常用于治疗吐血,衄血,咯血,崩漏,外伤出血,经闭痛经,跌伤肿痛,血淋涩痛等疾病效果较好,属于名贵药材。 蒲黄里的药物成分很多,主要有异鼠李素-3-O-新橙皮糖苷、香蒲新苷。下面我们就针对这两种成分进行实验部分原理 取适量蒲黄药材,加纯甲醇溶解,超声波提取,经进样器进样,色谱柱分离,紫外检测器检测,保留时间定性,峰面积定量计算。仪器及试剂仪器:高效液相色谱仪(紫外检测器+柱温箱),超声波清洗仪,溶剂过滤器,针筒式过滤器,电子天平试剂:甲醇(色谱纯),乙腈(色谱纯),磷酸溶液(分析纯),超纯水样品制备 对照品溶液的制备:准确称取异鼠李素-3-O-新橙皮糖苷、香蒲新苷对照品各2.5mg于50ml容量瓶中,加甲醇至刻度,配制成浓度均为50μg/ml的对照品溶液,备用。 供试品溶液的制备:准确称取本品约0.5g,置具塞锥形瓶中,精密加入50ml甲醇后,称定重量并记录,冷浸12小时后超声波超声30min,放冷,再次称定重量,用甲醇补足减少的重量,摇匀,滤过,待测。色谱条件检测器:紫外检测器色谱柱:普通C18,4.6 X 250mm,50μm流动相:乙腈:0.05%磷酸溶液=15:85(V:V)检测波长:254nm进样量20μl柱温:室温对照品色谱图: http://ng1.17img.cn/bbsfiles/images/2015/07/201507301828_558136_2536753_3.png供试品色谱图: http://ng1.17img.cn/bbsfiles/images/2015/07/201507301828_558137_2536753_3.png 从以上色谱图我们可以看出样品出峰时间较晚,峰形也较差。下面我们换用一根天津博纳艾杰尔科技有限公司生产的耐酸性色谱柱,效果我们请看色谱图。对照品色谱图:http://ng1.17img.cn/bbsfiles/images/2015/07/201507301828_558138_2536753_3.png 供试品色谱图: http://ng1.17img.cn/bbsfiles/images/2015/07/201507301829_558139_2536753_3.png 换了这根色谱柱,色谱图的峰形好了很多,保留时间也明显有所提前,但保留时间还是有点晚。下面我们把流动相比例调整了下,调整为乙腈:0.05%磷酸溶液=22:78(V:V),效果接着往下看。对照品色谱图: http://ng1.17img.cn/bbsfiles/images/2015/07/201507301829_558140_2536753_3.png供试品色谱图: http://ng1.17img.cn/bbsfiles/images/2015/07/201507301829_558141_2536753_3.png 当然保留时间还可以再缩短些(通过提高色谱柱温度,或提高高压泵流速,或换用更高效更短的色谱柱),但供试品中异鼠李素-3-O-新橙皮糖苷的附近有干扰物,为了保证分离度,这个分析应该时间比较合理(一定要根据样品情况而定,如果样品很纯净,对被测物没有干扰,保留时间当然是越短越好),尽量不要再缩短了,保证分离度是第一位的。 检测蒲黄药材的这个方法到现在已经比较完美。但有几点事项还需注意。1. 样
016年8月24日,加拿大卫生部发布公告,批准甜菊糖苷作为甜味剂在食品中使用。加拿大卫生部食品理事会对甜菊糖苷的安全性进行了详细的评价,并未发现存在安全性风险。新修订的《允许添加的甜味剂列表》中甜菊糖苷的使用范围和最大限量见下表。该修订自8月23日生效。 甜味剂 食品类别 添加限量 甜菊糖苷(包含以下任何一种甜菊糖苷,莱鲍迪甙A,莱鲍迪甙B,莱鲍迪甙C,莱鲍迪甙D,莱鲍迪甙女,莱鲍迪甙男,杜尔可甙A,甜茶苷等) (1) 桌面甜味剂 (1)按照GMP (2)早餐谷物;糖果釉料的休闲食品;坚果涂抹酱;花生涂抹酱;加糖调味料或涂层的混合物休闲食品;非标准巧克力糖果;非标准巧克力糖果涂料;非标准水果利差;非标准果泥;非标准沙拉酱;非标准酱汁;非标准餐桌糖浆 (2) 0.035% (以甜菊糖计算) (3)非标准化的饮料浓缩物;非标准饮料;非标准混合饮料 (3) 0.02% 消费的饮料中(以甜菊糖计算) (4)烘烤混合;灌装混合;馅料;摘心混合;浇头;非标准化的烘焙制品;非标准甜点混合物;非标准甜点; 酸奶 (4) 0.035%消费产品(以甜菊糖计算) (5)口气清新剂的产品; 口香糖 (5) 0.35%(以甜菊糖计算) (6)未标准化调味品 (6) 0.013%(以甜菊糖计算) (7)标准化糖果(除了非标准化的巧克力糖果);非标准糖果涂层(除了非标准化的巧克力糖果涂层) (7) 0.07%(以甜菊糖计算) (8)代餐棒;补充营养棒 (8) 0.02% (以甜菊糖计算) 相关附件:
【作者】 金艺; 张红霞; 许海燕; 赵怀清;【Author】 JIN Yi,ZHANG Hong-xia,XU Hai-yan,ZHAO Huai-qing(School of Pharmacy,Shenyang Pharmaceutical University,Shenyang 110016)【机构】 沈阳药科大学药学院; 沈阳药科大学药学院 沈阳110016; 沈阳110016;【摘要】 目的建立HPLC法测定胃力片中柚皮苷和橙皮苷的含量。方法采用HPLC法,色谱柱为Diamonsil C18(4.6mm×150mm,5μm),以甲醇-1%冰醋酸水溶液(32∶68,v/v)为流动相,流速为1.0mL.min-1,检测波长为283nm。结果柚皮苷与橙皮苷线性范围分别为14.0~126.0μg.mL-1(r=0.9997)、2.88~25.92μg.mL-1(r=0.9997),平均回收率分别为96.4%、97.8%。结论该方法简便准确、重现性好、可为胃力片质量控制提供依据。http://ng1.17img.cn/bbsfiles/images/2012/08/201208061313_381840_2379123_3.jpg
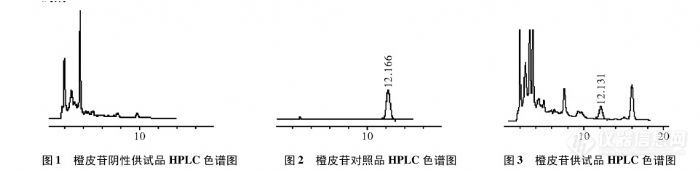
按照药典第一部中检测陈皮中橙皮苷的含量时发现,橙皮苷微溶于甲醇溶液,成浑浊状态,且峰面积较小为一千多,不全溶解是否影响到峰面积的大小?如果采取超声等方法使其溶解是否会有影响?想请教一下此项检测项目的有效方法?
据欧盟网站消息,3月24日欧盟委员会发布(EU)2016/441号,修订(EC)No1333/2008号法规附录II,批准甜菊糖苷作为甜味剂用于芥末。 甜菊糖苷是一种无热量甜味剂,用于芥末可替代蔗糖,因此可延长芥末的货架期,增加微生物稳定性,还可为产品增加风味。 欧盟委员会认为,芥末中添加甜菊糖苷不会对人体健康构成影响,因此批准其作为芥末添加剂,将限量定为120mg/kg。 新条例自发布后第20天起生效。
[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=72759]健胃片中橙皮苷的含量[/url]

10,抽取5个版友);中奖名单:mengzhaocheng(注册ID:mengzhaocheng)夏天的雪(注册ID:bingwang228)WUYUWUQIU(注册ID:wulin321)捌道巴拉巴巴巴(注册ID:v3082413)莫名其妙(注册ID:moyueqiu)http://ng1.17img.cn/bbsfiles/images/2017/02/201702271518_01_1610895_3.jpghttp://ng1.17img.cn/bbsfiles/images/2017/02/201702271518_02_1610895_3.jpg【注意事项】同样的答案,每人只能发一次PS:该贴浏览权限为“回贴仅作者和自己可见”,回复的版友仅能看到版主的题目及自己的回答内容,无法看到其他版友的回复内容。下午3点之后解除,即可看到正确答案、获奖情况及所有版友的回复内容。=======================================================================RP-HPLC法测定番石榴叶中桑黄素来苏糖苷和桑黄素阿拉伯糖苷的含量方法:HPLC基质:药品应用编号:102953固定相:Diamonsil C18(2)色谱柱/前处理小柱:Diamonsil 5μm C18(2), 250 x 4.6mm色谱条件:色谱柱:Diamonsil C18 250 mm× 4.6 mm, 5μm(Cat#:99603) 流动相: 甲醇-0.1% 磷酸溶液( 35: 65) 流速: 1.0 mL/min 柱温: 室温 进样量: 10 μL 检测器: UV 355 nm文章出处:中国实验方剂学杂志 2009, 15(6):21-23关键字:反相高效液相色谱, 番石榴叶, 桑黄素来苏糖苷, 桑黄素阿拉伯糖苷, Diamonsil C18, 钻石二代, 含量测定谱图:摘要:目的:建立番石榴叶中桑黄素来苏糖苷和桑黄素阿拉伯糖苷的含量测定方法。方法:用Diamonsil C18色谱柱(250mm×4.6mm,5μm),以甲醇-0.1%磷酸溶液(35∶65)为流动相,流速为1.0mL·min-1,检测波长为355nm。结果:桑黄素来苏糖苷在0.06132~0.14308μg范围内呈良好的线性关系(r=0.9999);桑黄素阿拉伯糖苷在0.05844~0.13636μg范围内呈良好的线性关系(r=1),平均回收率分别为98.6%和100.7%,RSD分别为1.0%和1.7%。结论:本方法对番石榴叶质量标准的制定具有很好的参考价值。http://www.dikma.com.cn/Public/Uploads/images/108-1.JPG
[size=15px][color=#595959]由于生活方式和饮食习惯的改变,[/color][/size][b][size=15px][color=#595959]糖尿病[/color][/size][/b][size=15px][color=#595959]已成为影响人类健康的第三大原因,仅次于心[/color][/size][b][size=15px][color=#595959]血管[/color][/size][/b][size=15px][color=#595959]疾病和[/color][/size][b][size=15px][color=#595959]恶性肿瘤[/color][/size][/b][size=15px][color=#595959]。[b]创面愈合受损[/b]是糖尿病的重要继发性并发症,通常导致肢体丧失和残疾。目前,缺乏可行的解决方案来[/color][/size][b][size=15px][color=#595959]管理[/color][/size][/b][size=15px][color=#595959]糖尿病创面(DW)。[/color][/size] [b][size=15px][color=#595959]铁死亡[/color][/size][/b][size=15px][color=#595959]是一种独特的程序性细胞死亡形式,不同于细胞凋亡和[/color][/size][b][size=15px][color=#595959]坏死[/color][/size][/b][size=15px][color=#595959],其特征是细胞内铁超载和铁依赖性脂质过氧化产物的积累。在大鼠DW模型中发现了明显的铁死亡相关变化,并证明局部应用[b]铁死亡抑制剂[/b](铁抑素-1)可加速DW的愈合。DW患者葡萄糖代谢降低,导致持续[/color][/size][size=15px][color=#595959]高血糖[/color][/size][size=15px][color=#595959]水平。长期持续的高血糖不仅会导致活性氧(ROS)的过量产生和脂质过氧化的激活,但也会损害铁代谢途径,导致游离铁水平升高,从而诱导氧化应激和铁死亡,导致细胞[/color][/size][b][size=15px][color=#595959]功能障碍[/color][/size][/b][size=15px][color=#595959]和死亡。此外,先前的研究已经证实了另一种铁死亡抑制剂去铁胺(DFO)在促进DW愈合方面的有效性。这些研究结果共同强调了[b]铁下垂在导致DW延迟愈合的病理机制中的重要作用[/b]。[/color][/size] [b][size=15px][color=#595959]橙皮素(HST)[/color][/size][/b][size=15px][color=#595959]是一种天然存在于柑橘类水果中的[b]类黄酮化合物[/b],广泛存在于各种传统草药中,如葡萄柚皮、橙皮和陈皮。这些植物材料通常用于中药制剂中。该研究旨在[b]探讨HST减少人脐静脉内皮细胞(HUVECs)铁死亡,促进血管生成和伤口愈合的潜在分子机制[/b]。 [size=15px]利用网络药理学[b]预测HST影响的下游靶点[/b]。采用Western blot (WB)和聚合酶链反应([url=https://insevent.instrument.com.cn/t/jp]PCR[/url])检测铁死亡相关标志物的表达。采用谷胱甘肽/氧化谷胱甘肽(GSH/GSSG)和丙二醛(MDA)检测试剂盒检测细胞内铁死亡相关代谢水平。通过Mitosox染色、FerroOrange染色和JC1染色研究细胞内线粒体状态和铁水平。利用分子对接技术确定HST的潜在下游直接靶点。此外,使用HE染色、Masson染色、[b]免疫[/b]组织化学和多普勒血流动力学评估等各种方法分析创面愈合和创面内的新生血管。[/size][size=15px][/size][font=mp-quote, -apple-system-font, BlinkMacSystemFont, &][/font] [/color][/size][align=center] [/align][size=15px][color=#595959][/color][/size][size=15px][color=#595959][/color][/size][size=15px][color=#595959][/color][/size][size=15px][color=#595959]HST有效抑制ERASTIN刺激的细胞内铁死亡水平升高。此外,观察到[b]HST通过激活SIRT3来实现对铁死亡的抑制[/b]。在糖尿病大鼠创面模型中,HST显著促进创面愈合,降低组织铁死亡水平,与体外研究结果一致。[/color][/size][color=#3573b9]结论[/color][size=15px][color=#595959][/color][/size] [b][size=15px][color=#595959][/color][/size][size=15px][color=#595959][/color][/size][size=15px][color=#595959][/color][/size][size=15px][color=#595959][/color][/size][/b][size=15px][color=#595959][font=&][/font][/color][/size][b][size=15px][color=#595959][/color][/size][/b][size=15px][color=#595959][/color][/size][b][size=15px][color=#595959][/color][/size][/b][size=15px][color=#595959]该研究表明,[b]HST可以通过激活SIRT3来抑制铁死亡的进展,保护HUVECs的生理功能。HST有望作为促进糖尿病创面愈合的天然化合物[/b]。[/color][/size][size=15px][color=#595959][/color][/size][size=15px][color=#595959][/color][/size]
木犀草素(luteolin),别称草木犀、黄示灵等,大多以糖苷的形式广泛存在于多种中药材、天然药用植物[1]及蔬菜[2]中的一种黄酮类化合物,是一种天然色素成分,可以作为食用色素添加于食品中。木犀草素的化学名为3′,4′,5,7-四羟基黄酮(3′,4′,5,7-tetrahydroxyflavone),物理状态为淡黄色结晶状粉末,熔点为330 ℃,包含4个酚羟基,具有弱酸性,可溶于碱性溶液中,因脂溶性高而难溶于水,从而阻碍了其在体内的吸收与利用[3]。木犀草素具有抗炎和抗菌[4-5]、抗氧化[6]、抗肿瘤[7]、神经保护[8]、抑制肺纤维化[9]及肺癌[10-11]和心血管疾病[12]等多种药理作用。由于水溶性差(仅为6.0 mg/L)、生物利用度率低等原因限制了其成药性和临床应用。针对这一问题,近年来许多学者开展了增加木犀草素溶解度的研究,如微球[13]、纳米胶束[14]、金属配合物[15]、自微乳[16]、脂质体[17]等,并明显提高了其生物利用度,这表明木犀草素的肠道渗透性不是限制其生物利用度的关键因素,其属于生物药剂学系统II类药物。因此,采用制剂技术提高木犀草素的溶解性是可以改善其成药性和生物利用度的,将有利于推广其临床应用。然而上述开发的剂型仍存在诸多的缺点,如工艺复杂、载药量低、生物安全性差、成本高等,难以大范围推广应用。近年来,逐步发展成熟的纳米混悬剂[18]作为一种新剂型,与传统纳米制剂相比,它具有载药量高、溶出度高、添加剂用量少、易于放大生产等优点。因此,本实验尝试将难溶性木犀草素制备成纳米混悬剂以提高其水溶性和生物利用度,改善其成药性和临床优势。 为此,本实验首先采用微沉淀-高压匀质法制备口服木犀草素纳米混悬剂(luteolin nano-suspension,LNS),并以纳米粒的粒径、稳定性、多分散性指数(polydispersity index,PDI)、ζ电位等为考察指标,采用单因素考察法筛选LNS的稳定剂和最优药物-稳定剂比;接着,对LNS的理化性质进行考察,并分析其物理状态和体外溶出行为;最后通过大鼠外翻肠模型考察药物在肠道不同部位的吸收转运情况,探索药物在肠道内的吸收速率和最佳部位,预测纳米混悬剂可能存在的体内吸收行为,既可以用于木犀草素口服给药的潜在剂型,也为其进一步加工成其他剂型研究提供基础。 1 仪器与材料 1.1 仪器 ZNCL-BS180型恒温磁力搅拌器,北京市永光明医疗仪器有限公司;AL104-1C型精密分析天平,上海鼎科科学仪器有限公司;NS1001L型高压匀质机,意大利GEA [url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]NIR[/color][/url]o Soavi公司;Nanotrac wave II型激光粒度仪型激光粒度仪,美国麦奇克有限公司;LC3100型高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱仪[/color][/url],安徽皖仪科技股份有限公司;ZWY-103D型恒温振荡仪,上海智诚分析仪器制造有限公司;H1650-W型医用离心机,湖南湘仪实验室仪器开发公司;DZF-6030型真空干燥箱,上海精宏实验设备有限公司。JEOL 2010型透射电子显微镜(TEM),日本JEOL公司。 1.2 试剂 木犀草素原料药,批号JZ19021403,质量分数97.0%,南京狄格尔医药科技有限公司;木犀草素对照品,批号ps1032-0025,HPLC质量分数≥98%,成都普思生物科技有限公司;十二烷基磺酸钠(sodium dodecyl sulfonate,SDS),医药级,河南圣拓实业有限公司;泊洛沙姆188(Poloxamer 188,Pluronic,F68),医药级,西安天正药用辅料有限公司;维生素E聚乙二醇琥珀酸酯(D-α-tocopherol polyethylene glycol 1000 succinate,TPGS),医药级,上海惠诚生物科技有限公司;二甲基亚砜(dimethyl sulfoxide,DMSO),分析纯,天津市德恩试剂有限公司。 1.3 动物 SD大鼠购买于河南省实验动物中心,体质量(200±20)g,合格证号:SCXK(豫)2017-0001。所有动物实验均经过河南大学动物伦理委员会审核批准(HUSOM2019-216)。 2 方法与结果 2.1 LNS的制备 2.1.1 LNS中稳定剂的选择 将40 mg木犀草素原料药超声溶解于1 mL的DMSO中作为有机相,再取等量的稳定剂(SDS、F68、TPGS)溶解于纯水中(作为水相,或称反溶剂相);在室温下,将有机相通过注射器快速注入转速为1 800 r/min的反溶剂相中,继续搅拌10 min,得到预混悬剂;将预混悬剂转移至高压匀质机中,分别以20.0、50.0、80.0 MPa的压力循环匀质5、5、25次,得到LNS。 利用动态光散射仪分别考察LNS的粒径、多分散系数(polydispersity index,PDI)、表面电荷(ζ电位)和稳定性。本实验以不同稳定剂(SDS、F68、TPGS)制备的LNS粒径大小、PDI、ζ电位结果如表1所示。3种稳定剂所制备的粒径均在100~500 nm。以SDS为稳定剂制备的纳米混悬剂粒径最大,以F68为稳定剂制备的纳米混悬剂PDI最大,以TPGS为稳定剂制备的纳米混悬剂ζ电位最大,但是3者没有较大的差异,因此对于预测稳定性来说,上述结果难以判断哪个稳定剂制备的LNS会有良好的贮存稳定性。 因此,本实验又对各种条件的贮存稳定性进行了研究,结果见图1。以SDS、F68为稳定剂制备的纳米混悬剂在1周内粒径呈现持续增长的趋势,而以TPGS为稳定剂制备的LNS粒径未出现明显变动,由此可知,本实验中以TPGS为稳定剂制备的LNS具有较好的物理稳定性。 2.1.2 LNS中药物-稳定剂质量比的筛选 将40 mg的木犀草素原料药超声溶解于1 mL的DMSO中作为有机相,再分别按照木犀草素与TPGS的质量比为1∶2、1∶1、2∶1称取TPGS,溶解于水中,得到反溶剂相;再按上述工艺制备LNS,得到不同药物-稳定剂质量比的LNS。利用动态光散射仪分别考察纳米混悬剂的粒径、分布、ζ电位和稳定性。不同药物-稳定剂比制备的LNS的理化性质研究结果见表2和图2。如表2所示,3种不同药物-稳定剂比制备的LNS的粒径分别为(289.3±6.6)、(210.7±2.0)、(34.6±3.7)nm,3种LNS的PDI接近,1∶2时ζ电位最大,2∶1时ζ电位没测到。虽然药物与稳定剂的质量比为2∶1时,其粒径与1∶2、1∶1时相差较大,但是粒径难以反映稳定性情况。因此,接下来考察了1∶2、1∶1、2∶1 3种不同比例下制备的LNS的稳定性,结果如图2所示。当药物-稳定剂比为2∶1和1∶2时,在2周内粒径变化幅度都较为明显,说明其稳定性表现均极差;而当药物-稳定剂比为1∶1时,制备的纳米混悬剂的粒径基本保持稳定,表明其稳定性较好。因此,本实验最终选用药物-稳定剂比为1∶1。 2.1.3 最优制备处方和方法的确定 依照LNS的稳定剂及药物-稳定剂比的筛选结果,初步确定LNS的最优制备处方与方法如下:将精密称取40 mg的木犀草素原料药超声溶解于1 mL的DMSO中作为有机相;将40 mg TPGS搅动溶解于40 mL纯水中作为水相,将有机相快速注入转速为1 800 r/min的水相中,搅动10 min,得到预混悬剂;将制备的预混悬剂倒入高压匀质机的导入槽中,分别以20.0、50.0、80.0 MPa的压力,分别循环匀质5、5、25次,得到LNS。重复制备3批,以粒径、PDI和ζ电位考察制剂处方和制备工艺的稳定性。 2.2 LNS的表征 2.2.1 粒径、ζ电位及形态分析 将最优处方制备的3批LNS分别通过激光粒度分析仪测定其粒径、PDI、ζ电位,结果LNS的粒径为(209.00±3.24)nm(n=3),PDI都低于0.228±0.013(n=3),粒径分布图见图3;ζ电位值为(?16.80±0.27)mV (n=3),较小的PDI和绝对值较大的ζ电位,意味着LNS可能具有较好的长期稳定性[19]。 再取适量的LNS加蒸馏水稀释到适当倍数后,滴在覆有支持膜的铜网上,自然环境下干燥后,通过TEM观察其形态特征及大小,并成像,结果见图4。LNS呈现均匀分散的球形或椭圆形颗粒,粒径约为180 nm,比动态光散射测定结果较小,这可能是由于TEM样品为干燥品,导致粒子外层亲水部分失水而收缩[20]。 2.2.2 储存稳定性 将制备的LNS分别放在4 ℃和室温环境中,在预定的时间点取样,通过激光粒度分析仪测定其粒径和PDI,连续考察14 d,每个样品平行操作3份,结果见表3。LNS在4 ℃和室温下储存2周后,粒径和PDI稍有增加,但变化范围都较小,说明该LNS的储存稳定性较好。 2.2.3 体外胃肠环境中的稳定性 以pH 1.2和pH 6.8的缓冲溶液模拟胃液和肠液,将制备的LNS分别以1∶1与上述2种缓冲溶液混合,并于37 ℃水浴中放置,在预定的时间点0、2、4、6、8、12、24 h时取样,通过激光粒度分析仪测定其粒径,连续考察24 h,每个样品平行操作3份,结果见表4。在2种37 ℃的缓冲溶液中孵育24 h内,LNS的粒径和PDI几乎无变化,表明LNS在2种环境中能保持稳定,这表示LNS口服给药后,在经胃肠道给药时能保持良好的稳定性,这有利于木犀草素到达肠道后仍以纳米晶存在,从而有利于木犀草素的快速释放而获得较高的生物利用度。 2.2.4 纳米混悬剂的物理状态研究 本实验选用DSC来确定LNS中的木犀草素晶型是否发生了改变,测试样品有木犀草素、TPGS、木犀草素与TPGS的物理混合物和LNS。以空铝盘作为空白对照,分别精密称取3~5 mg的木犀草素、TPGS、物理混合物(木犀草素+TPGS)、LNS干粉放于差式扫描量热分析(differential scanning calorimetry,DSC)仪中,N2流(40 mL/min)保护下,以10 ℃/min升温速度持续升温,升温范围设置为40~600 ℃,记录差式扫描量热分析图谱,所有测试样品重复分析3批,结果见图5。木犀草素和LNS、物理混合物均是结晶,其熔融温度为339.38 ℃,稳定剂对木犀草素的熔融温度基本无影响。这表明LNS中的木犀草素仍处于结晶状态,稳定剂的存在不会改变木犀草素的晶型。在木犀草素和LNS中,在50~150 ℃出现了1个宽峰,这可能是由于药物吸收了水分造成的。 再分别称取适量的木犀草素、TPGS、物理混合物(木犀草素+TPGS)、LNS置于X射线粉末衍射(X-ray powder diffraction,XRPD)仪中,以步进测定方式,散射角扫描范围设为5°~60°,电压设为40 kV,电流为30 mA,结果见图6。由图6可知,木犀草素在19.12、23.20、26.32 ℃有3个衍射峰,衍射峰的峰形较为尖锐,峰值较高,表明木犀草素的晶型为结晶型。稳定剂TPGS在15.72、17.48、22.86、25.60、29.26 ℃有衍射特征峰。制备成纳米混悬剂后,虽然LNS图谱中木犀草素的特征峰有所减弱,但与木犀草素相比,在相应位置特征峰均存在,进一步证实制备成LNS后木犀草素并未显著改变晶型,说明稳定剂的加入不会影响木犀草素的晶型,这与DSC分析的结果一致。 2.3 平衡溶解度与过饱和溶出度测试 为了测定木犀草素的平衡溶解度与木犀草素纳米混悬剂的过饱和溶出度,本实验参考文献方法[21]建立了HPLC法。 2.3.1色谱条件 色谱柱为Sino Chrom ODS-BP色谱柱(250 mm×4.6 mm,5 μm);流动相为甲醇-0.3%磷酸水溶液(60∶40);柱温30 ℃;检测波长350 nm;体积流量1 mL/min;进样量10 μL。 2.3.2对照品溶液的配制 精密称取木犀草素对照品2.50 mg,放入100 mL棕色量瓶中,以适量色谱甲醇使之完全溶解,并定容至刻度线,摇匀得到质量浓度为25 μg/mL的木犀草素对照品储备液。 2.3.3 线性关系考察 采用色谱甲醇稀释成质量浓度分别为0.5、1.0、2.0、5.0、7.0、10.0 μg/mL系列的木犀草素对照品溶液,按“2.3.1”项下色谱条件进行分析,以对照品质量浓度为横坐标(X)、峰面积为纵坐标(Y)进行线性回归,得线性回归方程为Y=44 670 X-2 498.3,R2=0.999 8,结果表明木犀草素在0.5~10.0 μg/mL线性关系良好。 2.3.4 专属性、精密度和准确度考察 在建立的HPLC色谱条件下,木犀草素色谱峰不会受pH 1.2和pH 6.8的溶出介质、稳定剂TPGS、Tyrode液以及肠吸收液中所有成分的干扰(图7),表明本实验所建立的含量测定方法具有较好的专属性,能够满足体外溶出和肠吸收试验中木犀草素的含量测定要求。另外,其精密度实验的RSD为1.2%,高、中、低3个质量浓度的样品加样回收率在99.67%~101.47%,RSD均小2%,符合《中国药典》2020年版的规定。 2.3.5 平衡溶解度的测定 为了测定木犀草素在pH值为1.2、6.8缓冲溶液中的平衡溶解度,取5 mL 2种缓冲溶液各3份于西林瓶中,加入过量的木犀草素,将西林瓶置于恒温振荡箱中,在温度为37℃,转速为75 r/min条件下振荡24 h。取出各样品,3 000 r/min下离心10 min后取上清液,然后用0.2 μm滤膜滤过,取续滤液于进样瓶中,按照“2.3.1”项下色谱条件进样测定,并计算木犀草素的平衡溶解度,结果可知,木犀草素在pH值为1.2、6.8的缓冲溶液中的平衡溶解度分别为(3.83±0.23)、(7.81±0.13)μg/mL。 2.3.6 过饱和溶出度的测定 为了考察LNS体外溶出行为,参照《中国药典》2020年版中桨法进行。具体操作如下:在智能溶出仪中,以500 mL模拟胃液为溶出介质,温度为37℃,桨旋转速度为75 r/min,将30 mL LNS加入溶出介质中,以相同质量浓度的木犀草素乙醇溶液作为对照,二者均平行操作3份。以药物刚接触溶出介质开始计时,分别于5、15、30、60、120、130、150、180、240、360、480 min时取样4 mL,取完样后立即补充4 mL相应的新鲜溶出介质。另外,于120 min取样后,每个溶出杯中分别加入适量的Na3PO4溶液,调节pH值为6.8,以模拟肠液。将所取样品溶液经0.2 μm微孔滤膜滤过,取续滤液置于进样瓶中,照“2.3.1”项下色谱条件测定,计算累积溶出度,结果见图8。为了测定过饱和溶出水平,在整个实验过程中,介质中药物的质量浓度都应保持远远大于药物的饱和溶解度[22]。结果如图8所示,在pH 1.2和pH 6.8时,木犀草素-原料药的过饱和溶出始终低于对应的平衡溶解度,LNS的过饱和溶出始终高于对应的平衡溶解度,说明制剂的过饱和度高;在溶出介质的pH值调为6.8后,过饱和溶出水平明显下降,在150 min后过饱和溶出水平逐渐稳定,说明LNS能维持较高的过饱和溶出水平。 结果表明,LNS较木犀草素原料药具有明显优势,其饱和溶出度约是木犀草素原料药的15倍,过饱和度高并能维持较长时间,可以延缓药物在体内因析出晶体而沉淀的过程,从而使稳定剂在较小用量下也能保证药物分子成溶解态,提高了原料药的溶解度,有利于增加其生物利用度[23]。 2.4 小肠吸收实验 为了探索LNS对木犀草素在胃肠道的吸收部位和吸收速率的影响,采用外翻肠囊法[24]研究LNS在肠道不同肠段的吸收特征,以探究药物在肠道内的最佳吸收部位。 2.4.1 对照品溶液的制备 精密量取“2.3.1”项下相应体积的储备液,置于50 mL棕色量瓶中,用Tyrode液定容至刻度,摇匀,配制出质量浓度为1、2、4、8、16、32、40 μg/mL木犀草素对照品溶液。 2.4.2 线性关系考察 按照“2.3.1”项下色谱条件测定,以木犀草素对照品质量浓度为横坐标(X),峰面积为纵坐标(Y)进行线性回归,得到回归方程为Y=45 475 X-19 575,R2=0.999 6,结果表明木犀草素在1~40 μg/mL线性关系良好。 2.4.3 供试品溶液的制备 大鼠按实验质量浓度随机分为3组,每组4只,实验前12 h禁食,自由饮水。颈椎脱臼处死,打开腹腔,小心分离出小肠,分别截取十二指肠、空肠、回肠、结肠相应肠段各10 cm,用生理盐水冲洗至无内容物流出。将肠段放入37 ℃ Tyrode液中,冲洗,在不损伤肠管的情况下,小心剥离肠表面的脂肪及血管,取出,用滤纸吸干表面水分。 将肠管一端结扎,用光滑的玻璃棒外翻,用Tyrode液冲洗过后,向不同肠段中注入3 mL的空白Tyrode液后将另一端也进行结扎形成囊状的肠管。将肠管放入盛有Tyrode液的烧杯中,实验中始终保持37 ℃的恒温,并不断通入95% O2/5% CO2的混合气体。平衡5 min后,将烧杯中的液体倒出,分别加入不同质量浓度(0.15、0.30、0.60 mg/mL)的木犀草素及LNS药液。以肠囊和药液接触时开始计时,取样时间点分别为15、30、45、60、75、90、105、120 min,每个时间点从肠囊内取样500 μL,同时补充同温同体积的空白Tyrode液。待试验结束后,将各段肠囊置于空白Tyrode液中孵育1 h,以清除掉肠囊及肠组织中残留的药物;随后将上述用于木犀草素和LNS吸收实验的各肠段互换,再按上述操作同法重复试验,以进行自身对照交叉试验的后段实验。取上述肠吸收液,加入甲醇500 μL,超声混匀,15 898×g离心(离心半径6.32 cm)2次,每次15 min,取上清液用0.2 μm滤膜滤过,取续滤液适量即得。 按照“2.3.1”项下色谱条件测定,并计算药物在各时间点的累积吸收量(Q,μg)和药物吸收速率常数[Ka,μg/(mincm2)],结果见图9。 由公式计算不同质量浓度下木犀草素在各个时间点的累积吸收量(Q)。 Q是每个时间点木犀草素的累积吸收量,Ci是每个时间点的实际检测质量浓度,V1是加入肠囊内的空白Tyrode液,V2是每次取样的体积 由图9可知,通过对比2种制剂在各肠段中不同质量浓度的药物吸收情况,可以发现药物的同一时间点的吸收量表现出质量浓度相关性。相同质量浓度下,在各肠段中2制剂组吸收量相比,LNS组的药物累积吸收量显著大于木犀草素溶液组,表明LNS相比于木犀草素溶液能够促进药物在肠道的吸收。 根据小肠内(4个肠段)的Q值,通过线性拟合,由公式Ka=L(斜率)/A(肠管平铺面积)求得吸收速率常数(Ka)和相关系数(R2),结果见表5。2种制剂中木犀草素在肠道的不同部位中的吸收速率大小顺序均为十二指肠>空肠>回肠>结肠,这可能归因于十二指肠和空肠肠段的吸收面积较大;这一结果还表明LNS并没有改变木犀草素在肠道内的主要吸收部位和机制。对比相同质量浓度、相同肠段中2种制剂的吸收情况可以发现,LNS中木犀草素的吸收速率显著高于木犀草素溶液的情况,尤其是十二指肠和空肠中LNS和木犀草素溶液的木犀草素吸收速率差异更加明显,这表明LNS可以增加木犀草素的肠吸收,且十二指肠和空肠是主要吸收部位。 另外,还可以发现2种制剂在每一肠段中的吸收速率都存在显著的质量浓度相关性(P<0.01),但是2种制剂在同一肠段中的吸收速率随质量浓度增加而提高的程度有明显差异,即木犀草素溶液随质量浓度的增加,各肠段中吸收速率增幅增大,而LNS随质量浓度的增加,各肠段中吸收速率增幅减小,这些结果表明2种制剂在各肠段中的吸收均有质量浓度相关性,但其吸收速率与质量浓度之间均存在非线性关系,且仅在Ka<0.052时,木犀草素的肠吸收过程可能只受木犀草素溶解度限制,而不受吸收速度限制。然而,木犀草素的实际口服吸收情况是否符合上述规律以及其具体吸收机制如何,将有待于后期开展体内外吸收途径探索和体内药动学研究来进一步证实。 3 讨论 3.1 稳定剂的选择及药物-稳定剂比的确定 由于不同的稳定剂中化学基团的差异,导致稳定剂与药物微粒之间的分子间作用力以及胶粒间的作用力都有明显差异,所以稳定剂种类会影响到纳米混悬剂的稳定性[25]。因此,本实验首先以粒径和稳定性为考察指标,通过单因素筛选法优化了LNS的稳定剂种类,并确定了以TPGS作为稳定剂能达到较好的预期效果;考虑到稳定剂用量对稳定效果的影响[26],随后本实验又考察了药物-稳定剂比对纳米混悬剂的粒径、稳定性、PDI、ζ电位的影响,最终确定最佳药物-稳定剂比为1∶1。 3.2 LNS体外分析方法的建立及研究 3.2.1 波长的选择 木犀草素对照品与稳定剂TPGS在紫外波长200~800 nm扫描,结果显示木犀草素在207、254、350 nm 3处波长处有强吸收;而TPGS在219、286 nm显示出强吸收,350 nm处没有显示出强吸收。为了排除稳定剂TPGS对木犀草素测定的干扰,选用350 nm作为木犀草素的测定波长。 3.2.2 Tyrode溶液的配制 在木犀草素的肠吸收情况研究中,虽有文献报道了外翻肠囊模型和在体单向肠灌流模型[27-29],但关于木犀草素及其制剂在大鼠不同肠段中的吸收情况鲜有报道,且大多数文献对其吸收情况所提甚少。 本实验采用离体外翻肠囊法,可操作性强、重复性好;能够保留较为完整的肠道组织和黏膜特性,其实验结果与机体药物吸收水平比较接近,具有说服力;但肠外翻肠囊法也存在缺点,如长时间暴露在体外,肠管没有血管和神经的控制,肠黏膜功能和形态会失去作用。因此,本研究为解决这一问题,利用Tyrode培养液改善肠管的存在环境,具体配制方法如下:将NaCl(8.0 g/L)、KCl(0.2 g/L)、CaCl2(0.2 g/L)、NaHCO3(1 g/L)、NaH2PO4(0.05 g/L)、MgCl2(0.1 g/L)、葡萄糖(1.0 g/L),用蒸馏水定容至1 000 mL,稀盐酸调pH值为7.2~7.4,由于CaCl2不好溶解,应在其他无机盐溶解完全后再加入,葡萄糖于临用前再加入。并且在实验过程中连续通入95% O2/5% CO2,保证了在实验期间肠管上肠黏膜的活性。实验证明用该模型了解药物的离体吸收,其结果可靠。 3.3 LNS的过饱和溶出 药物在纳米混悬剂中所处的物理状态关系着其粒径和溶出稳定性,通常无定形药物微粒具有较高的饱和溶出度,但其属于热力学不稳定状态,因此物理稳定性差,容易引起纳米混悬剂粒径分布发生变化,同时溶出速率和溶出度下降;而结晶型药物具有较好的热力学稳定性,随着其粒径的减小,其饱和溶出度会明显提高[30]。根据本实验对LNS中木犀草素物理状态的研究结果可知,本实验制备的LNS中木犀草素以结晶形式存在,这表明LNS可能存在稳定的粒径和溶出度。 在过饱和溶出实验中发现,相比于木犀草素原料药,LNS具有显著的长期高过饱和溶出水平,这可归因于LNS中药物以粒径远小于原料药的状态存在,正如开尔文定律所描述的小粒径药物具有高溶解度一样[31]。药物的长期高过饱和溶出水平将有助于避免或减少口服给药后因胃肠道pH变化而引起的析晶沉淀现象,从而增加药物的吸收速度和时间,提高药物的口服生物利用度。 综上所述,本实验制备的LNS,分散性和储存稳定性良好,方法也简单易行,本实验建立的木犀草素体外分析方法,经方法学验证可知,该方法快速、可靠、准确度高,适合LNS的体外溶出和外翻肠囊吸收实验研究。 同时,外翻肠实验表面,LNS能促进药物在肠道的吸收,可作为木犀草素口服给药的潜在剂型,也为其进一步加工成其他剂型研究提供坚实基础。同时,在木犀草素肠道吸收的具体机制方面还有很大的研究空间。

作者:http://vpn.library.shmtu.edu.cn:2308/Images/head_pic.gif谢一凡 http://vpn.library.shmtu.edu.cn:2308/Images/head_pic.gif顾雅芳 http://vpn.library.shmtu.edu.cn:2308/Images/head_pic.gif周金娥 http://vpn.library.shmtu.edu.cn:2308/Images/head_pic.gif陈泽乃 http://vpn.library.shmtu.edu.cn:2308/Images/head_pic.gif陆阳 Author:XIE Yi-fan GU Ya-fang ZHOU Jin-e CHEN Ze-nai LU Yang 作者单位:上海交通大学医学院药学系,上海,摘要: 目的 建立染料木素-4'-葡萄糖苷的含量测定方法.方法 采用反相高效液相色谱法.Diamonsil C18柱(4.6 mm×250 mm,5 μm),流动相为乙腈-水(用磷酸调节pH至2.73)(25:75),流速1.0 mL·min-1,检测波长261 nm,柱温35℃.结果 染料木素-4'-葡萄糖苷在0.505 5~101.1 mg·L-1内峰面积与浓度呈良好线性关系(r=1.000 0,n=6),平均回收率99.2%(RSD=1.1%,n=9),日内精密度RSD<0.5%,日间精密度RSD<0.7%,最低检测限0.2 ng.同时对样品中所含染料木素、染料木素-7-葡萄糖苷和染料木素-7,4'-二葡萄糖苷等微量有关物质的分离和检测也获得满意的结果.结论 该方法简便、可靠,可用于染料木素及其苷类的质量控制http://ng1.17img.cn/bbsfiles/images/2012/08/201208271807_386613_2379123_3.jpg
http://ng1.17img.cn/bbsfiles/images/2012/09/201209272130_393425_2255248_3.gifHPLC测定木犀草苷对照品为什么峰前面有个小峰????

问题:麻仁润肠丸中橙皮苷的检测药典要求理论板数按橙皮苷峰计算应?答案:药典要求理论板数按橙皮苷峰计算应不低于3000【活动奖励】幸运奖(2钻石币):抽奖软件,当天随机抽取3个回答正确的版友ID号(最后一个ID号,截止至下午3:00),每人奖励2个钻石币梧桐(ID:mengzhou)吕梁山(注册ID:shih20j07)ZHAOGUANGXI(ID:ZHAOGUANGXI)http://ng1.17img.cn/bbsfiles/images/2016/01/201601261650_583560_1610895_3.pnghttp://ng1.17img.cn/bbsfiles/images/2016/01/201601261650_583561_1610895_3.png积分奖励:所有回答正确的版友奖励10个积分(幸运奖获得者除外)。【注意事项】同样的答案,每人只能发一次PS:该贴浏览权限为“回贴仅作者和自己可见”,回复的版友仅能看到版主的题目及自己的回答内容,无法看到其他版友的回复内容。下午3点之后解除,即可看到正确答案、获奖情况及所有版友的回复内容。======================================================================= 麻仁润肠丸中橙皮苷的检测样品制备制备方法1. 对照品:取橙皮苷对照品适量,精密称定,加甲醇制成每1 mL含100 μg的溶液,即得。2. 供试品:取本品大蜜丸,剪碎,取约1 g,精密称定,置乳钵中,加入硅藻土约1.5 g,研匀,置具塞锥形瓶中,另取少量硅藻土置上述乳钵中,研磨,并入上述锥形瓶中精密加入甲醇50 mL,密塞,称定重量,加热回流1小时,放冷,再称定重量,用甲醇补足减失的重量,摇匀,滤过,取续滤液,即得分析条件色谱柱Diamonsil C18 250 x 4.6 mm,5 μm (Cat#:99903)流动相A:甲醇 B:0.1%磷酸 梯度流速1.0 mL/min柱温30 ℃检测器UV 283 nm进样量10 μL色谱图对照品http://ng1.17img.cn/bbsfiles/images/2016/01/201601260935_583525_1610895_3.jpg 峰号 保留时间 min 峰面积 μV*s 峰高 μV 理论塔板数* N USP拖尾因子 分离度 1 20.792 2052991 38439 3527.486 0.941 -- *药典要求理论板数按橙皮苷峰计算应不低于3000供试品 http://ng1.17img.cn/bbsfiles/images/2016/01/201601260935_583526_1610895_3.jpg 峰号 保留时间 min 峰面积 μV*s 峰高 μV 理论塔板数* N USP拖尾因子 分离度 1 20.702 1988492 40554 4159.306 0.944 -- *药典要求理论板数按橙皮苷峰计算应不低于3000本品种同时使用了Platisil ODS色谱柱,在药典规定条件下进行橙皮苷的检测,满足药典要求。
按2010版药典中测金银花的木犀草苷的提取方法测木犀草苷,其图谱中木犀草苷的保留时间总是不一致,然而杂质峰太多,真不知道哪个是所需的峰。图如附件
怎样通过前处理对柠檬苦素糖苷和配基进行分离啊,看到一种用strata-x的方法,但是成本太高,不知道可以重复利用不
[color=#444444]有谁知道新橙皮甙二氢查尔酮,柚皮甙二氢查尔酮的液相检测方法啊???麻烦发一下,谢谢!!!!!![/color][img=,30,30]file:///C:\Users\25163\AppData\Local\Temp\ksohtml10044\wps3.png[/img]
【作者】:曾莉萍 ,刘 敏 ,万凯化,付辉政【摘要】:目的: 建立益肾补骨液中橙皮苷的含量测定方法。方法: 用 Diamonsil C18 色谱柱( 250 mm × 4. 6 mm,5 μm) ,以甲醇 - 2%冰醋酸溶液( 4258) ,流速为 1. 0 mL /min,检测波长为 283 nm。结果: 橙皮苷在 0. 115 2 - 0. 345 6 μg 范围内呈良好的线性关系( r = 0. 999 7) ,平均回收率和 RSD 分别为 101. 8% 和 1. 0% 。结论: 本方法简便、准确、重现性好,可作为该制剂的含量测定方法。【作者单位】: 江西护理职业技术学院; 江西省食品药品检验所; 中国医学科学院【关键词】:反相高效液相色谱法; 益肾补骨液; 橙皮苷; 含量测定http://ng1.17img.cn/bbsfiles/images/2012/07/201207311321_380841_1838299_3.jpg
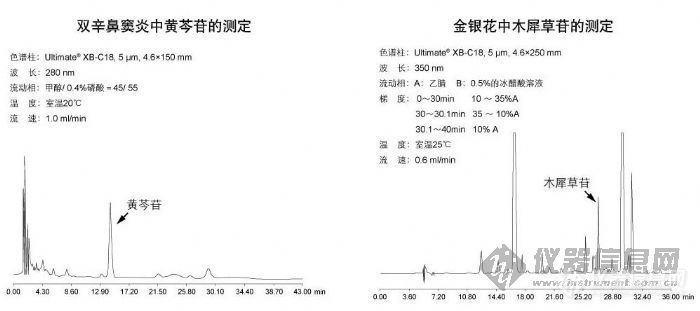
双辛鼻窦炎口服液中黄芩苷的测定和金银花中木犀草苷的测定http://ng1.17img.cn/bbsfiles/images/2009/10/200910161356_175983_1896702_3.jpg
说说大家的金银花的木犀草苷是怎么分开的







 我要推广仪器
我要推广仪器
 下载APP
下载APP