有没有人做过:三甲基氧苄二氨嘧啶?请各位高手指点一下用什么方法进行检测,谢谢!
原料是4,6-二羟基嘧啶、三氯氧磷、N,N-二甲基苯胺、三氯乙烯经反应→水解→分层后,下层出成品4,6-二氯嘧啶,上层酸液通氨或氢氧化钠中和至pH=7,然后再分层,上层二甲基苯胺套用,下层废液是需要处理的,请问该废液中主要含哪些物质?如果用GC-MS检测的话又需要怎样的前处理才能进样?或者直接顶空进样是否能达到检测目的?谢谢
有没有做过N-亚硝基胺的?我用N-二甲基亚硝基胺标准物质进样,但是根本观察不到信号,TIC和SIM都没有信号,这是什么原因呢?
增效联磺片为磺胺类抗菌消炎药的新型复方制剂,每片含磺胺甲基异(口恶)唑200mg、磺胺嘧啶200 mg、甲氧苄氨嘧啶80 mg,各地方标准均有收载,对前两种成分以纸色谱法鉴别,而对甲氧苄氨嘧啶则另行鉴别。本文以薄层色谱法同时鉴别磺胺甲基异(口恶)唑、磺胺嘧啶、甲氧苄氨嘧啶3种成分,专一性强,斑点明显,操作简便,结果较为满意。1 仪器与试药 三用紫外线分析仪(上海顾村电光仪器厂),硅胶GF254薄层板(10cm×20 cm,自制);磺胺甲基异(口恶)唑、磺胺嘧啶和甲氧苄氨嘧啶对照品(中国药品生物制品检定所);增效联磺片(市售品);硅胶GF254(青岛海洋化工厂生产,化学纯);其它试剂均为分析纯。2 溶液的配制2.1 单一对照品溶液 分别精密称取磺胺甲基异(口恶)唑、磺胺嘧啶、甲氧苄氨嘧啶对照品适量,加50%丙酮溶液分别制成0.5 mg/mL磺胺甲基异(口恶)唑、0.5 mg/mL磺胺嘧啶、0.2 mg/mL甲氧苄氨嘧啶的单一对照品溶液。2.2 混合对照品溶液 精密称取磺胺甲基异(口恶)唑、磺胺嘧啶、甲氧苄氨嘧啶对照品适量,加50%丙酮溶液制成? mL含磺胺甲基异(口恶)唑0.5 mg、磺胺嘧啶0.5 mg和甲氧苄氨嘧啶0.2mg的混合对照品溶液。2.3 样品溶液的配制 取供试品细粉适量(约相当于磺胺甲基异(口恶)唑50mg),加50 %丙酮溶液100 mL,振摇使溶解,过滤,滤液作为供试品溶液。
[size=4][font=宋体]求香菇上氟乐灵,甲基毒死蜱,毒死蜱,氯杀螨,乙硫磷,氯苯嘧啶醇[/font][/size][size=4][font=Times New Roman] [/font][/size][size=4][font=宋体]氟虫腈的前处理方法,能够有效去除杂质的,谢谢大家,这个香菇上的杂质太多了,用弗洛里硅土不管用,杂质很多。[/font][/size]
[color=#444444]各位大侠,我用FPD做马拉硫磷和甲基嘧啶磷,柱子是DB-1701的,但是两个峰,分不开,请求大家帮助[/color]
亚硝基二甲胺和二甲基亚硝胺是不是一种东西?
据欧盟食品安全局消息,鉴于农药甲基嘧啶磷(pirimiphos-methyl)的交叉污染风险,11月15日欧盟食品安全局对甲基嘧啶磷的最大残留限量进行了审查,经审查欧盟食品安全局建议对甲基嘧啶磷在多种作物中的最大残留限量进行修订。 欧盟食品安全局在风险评估过后,做出如下决定:商品代码商品现行MRL(mg/kg)建议MRL(mg/kg)401000油料作物种子0.050.5500010大麦55500020荞麦50.5500030玉米50.5500040小米55500050燕麦55500060大米50.5500070黑麦50.5500080高粱55500090小麦(包括黑小麦)55810000调味料(种子)55820000调味料(果实与浆果)0.10.1830000其它的植物商品见附录B0.051000000动物源食品0.050.01 原文链接:http://www.efsa.europa.eu/en/efsajournal/doc/2436.pdf
有朋友做过6-甲基尿嘧啶的LC分析吗?请问流动相和波长分别是什么?C18可以分开吗?
各位大侠,我用FPD做马拉硫磷和甲基嘧啶磷,柱子是DB-1的,但是怎么也分不开两个峰,请求大家帮助
[color=#444444]各位大侠[/color][color=#444444],[/color][color=#444444]我用[/color][color=#444444]FPD[/color][color=#444444]做马拉硫磷和甲基嘧啶磷[/color][color=#444444],[/color][color=#444444]柱子是[/color][color=#444444]DB-1701[/color][color=#444444]的[/color][color=#444444],[/color][color=#444444]但是两个峰[/color][color=#444444],[/color][color=#444444]分不开[/color][color=#444444],[/color][color=#444444]请求大家帮助[/color]
毛细管柱是岛津的Rtx-1,固定液是100%聚二甲基硅氧烷,由于我们这里没有单标,也没有质谱检测器,不知道每个成分的出峰顺序,固定液相同的毛细管柱应该差不多,回复时请提供柱子类型,先谢谢各位同行啦。六六六四种(α-BHC β-BHC γ-BHC δ-BHC),滴滴涕四种( pp'-DDE pp'-DDD op'-DDT pp'-DDT)、五氯硝基苯(PNCB)以下是具有相对填料的商品柱:100%Dimethyl polysiloxane,100%聚二甲基硅氧烷,商品名:AC1,OV-101,OV-1,DB-1,SE-30,HP-1,RTX-1,BP-1
要做某样品中的二氯对甲基苯甲酸的残余。该物质沸点330.51.首选了ffap柱二氯对甲基苯甲酸标品用溶剂溶解,不出峰。衍生化,出很小的峰。2.rtx-1柱这么高的沸点,6min出峰了。
毛细管柱是岛津的Rtx-1,固定液是100%聚二甲基硅氧烷,由于我们这里没有单标,也没有质谱检测器,不知道每个成分的出峰顺序,固定液相同的毛细管柱应该差不多,回复时请提供柱子类型,先谢谢各位同行啦。六六六四种(α-BHC β-BHC γ-BHC δ-BHC),滴滴涕四种( pp'-DDE pp'-DDD op'-DDT pp'-DDT)、五氯硝基苯(PNCB)以下是具有相对填料的商品柱:100%Dimethyl polysiloxane,100%聚二甲基硅氧烷,商品名:AC1,OV-101,OV-1,DB-1,SE-30,HP-1,RTX-1,BP-1
请教专家做对氯邻硝基苯胺 和 2,5-二氯硝基苯的实验方法?越详细越好。
我今天做有机磷的农药残留,发现毒死蜱,倍硫磷、喹硫磷、马拉硫磷、甲基嘧啶磷这几种有机磷的出峰时间均好接近啊,如果是混标了话,那这几个有机磷岂不是不能分开了,我的[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]条件是:[url=https://insevent.instrument.com.cn/t/Mp]gc[/url] 2010plus,色谱柱:Rtx-5 30m*0.25mm, 0.25um 升温程序:80℃保持1min,以20℃/min升至130℃,再以5℃/min升至200℃,最后以15℃/min升至250℃,并保持11min,检测器:FPD,温度250℃,进样口温度220℃。
最近在做有机锡测试,EN71-3中要求测9种有机锡,但美泰要求加测二甲基二氯锡(DMT),测试过程中发现二甲基二氯锡(DMT)很难衍生,不知道各位有没有测试二甲基二氯锡(DMT)?有的话请老师指教指教!二甲基二氯锡(DMT)的特征离子是多少?7890 GC-5975C MS上最低检测浓度?
大家谁有2-硝基-3甲基苯甲酸分析条件?谢谢
[color=#444444]请问各位大神知不知道检测4,4'-二氯-3,3'-二硝基二苯甲酮的高效液相色谱条件,其主要副产物是单硝基化合物,能不能指点一下,万分感谢![/color]
大家好,谁知道2-甲基-3-硝基苯甲酸的分析方法
请问2-氯嘧啶应该如何检测含量??[em09511]
硅烷化处理可以选用DMDCS(二甲基二氯硅胺)这出自A的培训资料,不知道是不是叫硅胺还是叫硅烷,请帮帮忙???谢谢
本人用含CMC的零价铁溶液降解硝基苯,如果直接提取反应溶液经过0.22微米的有机滤头并进入[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相[/color][/url],会因为粘性较大的CMC堵住色谱柱。所以本人用了二氯甲烷进行了萃取,但依然导致了压力升高,有没有哥哥姐姐能帮忙解答一下的呀?
用气相色谱检测奶粉中的肌醇时,用的是肌醇与三甲基氯硅烷 六甲基二硅胺烷 NN二甲基酰胺等硅烷化试剂,我想请教一下大神们肌醇与这些试剂的反应产物是什么,能提供相关的反应原理或者反应式吗??
最近扩项皮革N亚硝基胺 GB/t24153-2009 5ppm 12种混标打下去, N-亚硝基吡咯烷,N-亚硝基N-甲基苯胺,N-亚硝基乙基苯胺 ,N-亚硝基二苯基胺都没有,查找离子发现有N-甲基苯胺,N-乙基苯胺,二苯胺,推测热分解。那么N-亚硝基吡咯烷应当分解为吡咯烷才对,然而附近并没有找到对应峰......手边没有N-亚硝基吡咯烷单标,不好验证。有没有经验丰富的老师讲解一下,这种情况是不是热分解导致的,N-亚硝基吡咯烷是不是也分解了呢?如果分解了,产物是什么,特征离子多少。还是其他原因造成的?进样口温度260 质谱280 DB-35柱子 分段升温38-300
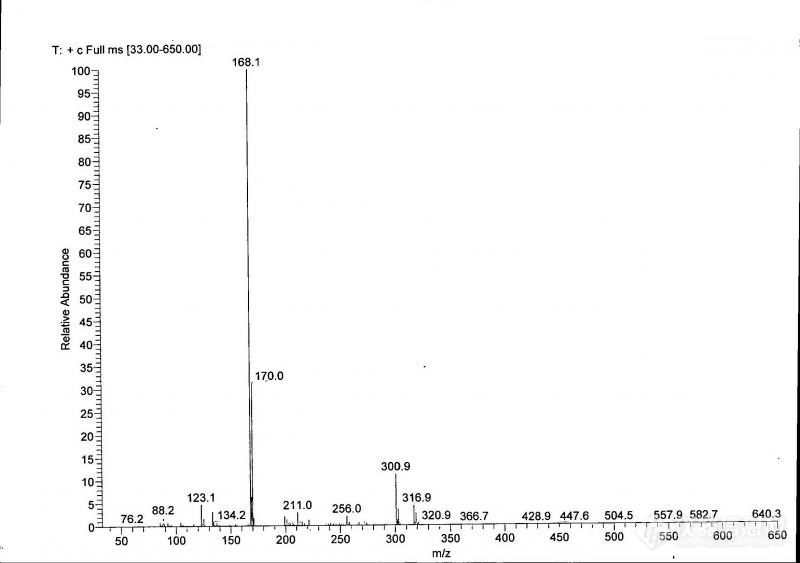
[color=#444444]下图,为2,4,6-三氟-5-氯嘧啶(CAS号:697-83-6)的质谱图,分子离子峰为168.1,请问300.9和316.9是什么碎片离子,我做了这种含氟嘧啶的一系列中间体,都在分子离子峰后有较大的碎片,我计算了一下,正好是分子离子峰的2倍减去一个氯,我怀疑是两分子聚合,脱掉一个氯,但是脱掉一个氯又没法接上,求各位朋友解释[/color][color=#444444][img=,690,485]https://ng1.17img.cn/bbsfiles/images/2019/09/201909090944077709_1029_1752329_3.jpg!w690x485.jpg[/img][/color]
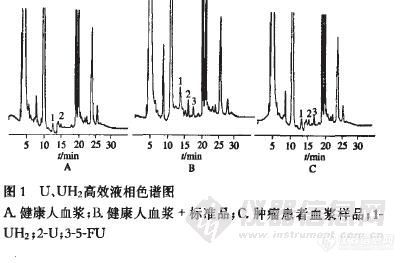
作者:肖力 任斌 陈小陆 李瑞明 刘怡 容颖慈 蓝缨(作者单位:中山大学附属第一医院药学部,广东,广州,510080 )摘要:目的:建立准确测定内源性尿嘧啶和二氢尿嘧啶血药浓度的高效液相色谱法.方法:以氟尿嘧啶(5-FU)为内标,醋酸乙酯-异丙醇混合液(85:15)为提取溶剂;色谱柱Diamonsil C18柱(250 mm×4.6 mm,5 靘);流动相A-0.01 mol·L-1磷酸二氢钾缓冲液(pH 5.5),B-乙腈,梯度洗脱;流速为0.8 mL·min-1;柱温为4 ℃;检测波长为204 nm(0~14.5 min),254 nm(14.5~35 min).结果:尿嘧啶和二氢尿嘧啶线性范围为8~500 靏稬-1,线性回归方程分别为C(UH2)=61.760 8Y+0.506 5,r=0.999 8;C(U)=95.201 1Y-3.064 0,r=0.999 3,(n=7).最低检测质量浓度均为5 靏稬-1.尿嘧啶方法回收率为99.3%~107.0%,二氢尿嘧啶方法回收率为95.0%~98.3%.尿嘧啶日内RSD小于6.5%,日阍RSD小于11.7%,二氢尿嘧啶目内RSD小于9.2%,日间RSD小于12.4%.结论:本方法可用于内源性尿嘧啶和二氢尿嘧啶血药浓度的常规监测.谱图:http://ng1.17img.cn/bbsfiles/images/2012/08/201208142006_383845_1609970_3.jpg
有谁知道叔丁基二甲基氯硅烷含量怎么测定啊?
目前需要建立方法测试饮用水中亚硝基二乙胺和二氯异氰尿酸两个组分的含量,但是好像没有相关标准是针对水中这两个组分的,群里有哪位前辈做过这方面实验的吗?WHO里说明测试二氯异氰尿酸含量是通过测试氰尿酸(三聚氰酸)含量来计算的,那测试水中的氰尿酸有谁做过吗?样品的前处理是怎么样的,微量含量的富集方法?
谁有没有比较好的4-6二羟基嘧啶的液相分析方法,我在主峰附近有杂峰无法分开,有没有哪位大佬有比较好的方法?







 我要推广仪器
我要推广仪器
 下载APP
下载APP