食品安全国家标准 食品添加剂 羟丙基甲基纤维素(HPMC)
中文名称: 羟丙基二淀粉磷酸酯 中文商品名称:羟丙基磷酸双淀粉 英文名称: Hydroxypropyl distarch phosphate 别名: HPDSP 详情: 理化性质:白色粉末,无臭,无味,易溶于水,不溶于有机溶剂。在醚化的基础上,适当地交联所得到的HPDSP,其膨润力、透明度仍显著高于原淀粉。糊液对温度、酸度和剪切力的稳定性高。 来源与制法: 淀粉与三偏磷酸钠或磷酰氯(≤0.1%)与环氧丙烷(≤10%)伴同酯化而成。 编辑本段毒理学依据 1、ADI:无须规定(FAO/WHO,1994)。 2、可安全用于食品(FDA,§172.892,1994)。 质量要求:质量标准(FAO/WHO,1990;CXAS,1991) 羟丙基含量/% 7.0 氯丙醇/(mg/kg)≤ 1 土豆或小麦类淀粉/% ≤ 0.14 其他类淀粉/5 ≤ 0.04 二氧化硫 谷物类/(mg/kg)≤50 其他类/(mg/kg)≤10 砷(以As计)(mg/kg) ≤ 3 重金属(以Pb计)(mg/kg) ≤ 40 铅/(mg/kg)≤ 2 编辑本段用途与注意事项 我国《食品添加剂使用卫生标准》(GB2760―2007)表A.3(可在各类食品中按生产需要适量使用的添加剂名单)第46为羟丙基二淀粉磷酸酯,功能为增稠剂。未限定最高用量,可按需添加。 FAO/WHO规定:可单独使用或与其他增稠剂合用。用于蛋黄酱,5 FAO/WHO;罐装胡萝卜(产品含有奶油或其他油脂)、发酵后经加热处理的调味酸奶及其制品,10 g/kg;冷饮制品,30 g/kg;罐装沙丁鱼和沙丁鱼类产品,20 g/kg;罐装鲐鱼和竹荚鱼,60 g/kg(仅用于填料);速冻鱼条和鱼块(仅指用面包粉和面包拖料包裹),以GMP为限。羟丙基二淀粉磷酸酯Hydroxypropyl Distarch Phosphate编码 GB 20.016;INS 1442性状 白色粉末,无臭,无味,易溶于水,不溶于有机溶剂。在醚化的基础上,适当地交联所得到的HPDSP,其膨润力、透明度仍显著高于原淀粉。制法 由淀粉在碱性条件下,与环氧丙烷进行醚化,再与磷酸交联剂进行酯化反应制得。质量标准 参见羟丙基淀粉。鉴别方法 本品呈一般食变性淀粉反应和磷酸盐反应。1.一般食用变性淀粉反应 同羟丙基淀粉鉴别方法1、2、3。2.磷酸盐反应 参见磷酸三钙。毒理学依据1.GRAS FDA-21CF
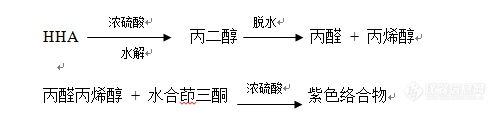
[align=center][b]羟丙基透明质酸质量标准的建立[/b][/align][align=center]杨桂兰,臧恒昌[b][/b][/align][b]摘要:[/b]透明质酸(HA)具有保湿、润滑、营养、修复和预防损伤等生理功能,在维持组织完整性方面和促进感染、损伤、胚胎发育过程中组织形成和重塑方面发挥重要作用。在化妆品、食品及医药领域的应用越来越广泛。但HA容易被体内透明质酸酶降解,体内留存时间短。研究者们期望通过对其进行修饰,得到抗酶解的HA衍生物,延长体内保留时间。修饰HA的衍生物近年来主要致力于将其修饰为两亲性衍生物,对抗酶解活性也有研究;这种亲油亲水性使其不仅能够降低降解速率,而且能够降低表面张力。其次,两亲性HA可以解决美容填充时HA分子量过大,黏度过高,注射困难的问题,修饰后的两亲性HA具有黏度降低(相同分子量相同浓度)的优点。HA两亲性衍生物也可作为生物可降解性的药物载体。 本文参考羟丙基淀粉取代度测定方法,建立了采用分光光度法测定羟丙基透明质酸(HHA)取代度的方法。同时摸索了HHA的抗酶解活性检测法、干燥失重、pH、蛋白含量及微生物等关键指标的测定方法。[b]关键词:[/b]透明质酸;羟丙基透明质酸[align=left] 本研究为确保自制羟丙基透明质酸的质量,特制定一系列产品的质量检验标准。[b]1分子量测定1.1材料[/b] NaCl(AR),NaN[sub]3[/sub] (CP) ,BSA(Roch);高效液相色谱仪,(美国Agilent);多角度激光光散射仪,DAWNEOS,美国Wyatt。[b]1.2方法[/b] 测定条件:流动相:0.2mol/L NaCl (包含0.02% NaN[sub]3[/sub]);流速:0.6ml/min,样品浓度:0.05 mg/ml;柱温:35 ℃,进样体积:500 μl。按照仪器操作规程进行操作。[b]2取代度测定2.1原理[/b][/align][align=center][b][img=,497,113]http://ng1.17img.cn/bbsfiles/images/2018/07/201807251603456954_6718_3389662_3.png!w497x113.jpg[/img][/b][/align][align=left][b]2.2材料[/b] HHA;水合茚三酮、1,2-丙二醇、浓硫酸、亚硫酸氢钠、可见分光光度计、具塞比色管(25 ml),容量瓶(100 ml、1000 ml)[b]2.3方法 [/b] 丙二醇标准溶液的配制:准确称量1.0g丙二醇溶液于1000 ml容量瓶中,加纯化水稀释至刻度,然后分别取2、4、6、8、10 ml于100 ml容量瓶中,定容至刻度,得到丙二醇含量分别为20、40、60、80、100 mg/ml的溶液。[b]2.3.1丙二醇标准曲线的制备[/b] 分别吸取上述丙二醇溶液0.5 ml于25 ml具塞比色试管中,置于冰浴中,逐滴加入4 ml浓硫酸(不宜加入过快,并不时震荡)混合均匀后置100 ℃的水中加热3 min(秒表控制),取出后立即放入冰浴中,冷却至15℃,沿管壁加入水合茚三酮试剂0.3 ml,边加边摇匀;在25 ℃的水浴中放置80 min,再用浓硫酸稀释至12.5ml(约7.7 ml浓硫酸)。缓慢倾倒混匀后(不要用混合器震荡),静置5 min,用1 cm比色皿于590 nm波长处测定溶液的吸光度,绘制吸光度—浓度曲线,拟合丙二醇标准曲线方程。 空白:以相同条件下不加丙二醇溶液作空白。[b]2.3.2试样的测定[/b] 分别称取0.05 g~0.1 gHHA及制备该批HHA所用HA粉末于100ml的量瓶中,量取25 ml的0.5 mol/L的硫酸,缓缓加入量瓶中。置于100℃水浴中加热,缓缓摇动,至试样完全溶解,冷却,用纯水定容,量取0.5ml此溶液置25ml比色管中,其余如上述丙二醇的配制方法。羟丙基含量和取代度算法分别如公式1、2所示。[/align][align=center][img=,411,64]http://ng1.17img.cn/bbsfiles/images/2018/07/201807251608249134_5590_3389662_3.png!w411x64.jpg[/img][/align][align=center]注: C:试样中丙二醇含量,由吸光度计算得出; m:取样量;0.7763:转换系数;[/align][align=center][img=,387,58]http://ng1.17img.cn/bbsfiles/images/2018/07/201807251610349286_9676_3389662_3.png!w387x58.jpg[/img][/align][align=center]注:6.9190:HA分子量/环氧丙烷分子量[/align][align=left][b]3抗HAase降解特性3.1材料[/b] 注射用透明质酸酶(HAase)(上海第一生化药业有限公司、1500单位/瓶);缓冲液(磷酸二氢钠:0.0057 g、磷酸氢二钠:0.0230 g、氯化钠:9.0 g、纯化水:1.0 kg);平氏黏度计,Φ1.0 mm、Φ2.0 mm;恒温水槽,上海仪表仪器厂; DK-8D数显恒温水浴锅,金坛市医疗器械厂。[b]3.2方法[/b] 称取HHA和对照HA各 0.5 g份于150 ml肖特瓶中,加入50 ml缓冲液,震荡至完全溶解。用氢氧化钠溶液或HCl溶液调节pH值6.0~7.2,取溶解液10.0 g,纯水稀释5倍;作为起始样品测黏度。取1500单位的酶用缓冲液稀释10倍,分别吸取40单位加入上述HA和HHA溶液中,摇匀,放入37℃的水浴中降解,24 h取样:称取10.0gHA溶液于50 ml容量瓶中,加入纯化水稀释至刻度线,加热煮沸2min,冷却至室温,测其在25℃下的运动黏度,算法如公式3所示。[/align][align=center] [img=,449,41]http://ng1.17img.cn/bbsfiles/images/2018/07/201807251629104708_3045_3389662_3.png!w449x41.jpg[/img][/align] 24小时黏度下降率Δη低于75%。[align=left][b]4透光率的测定4.1材料[/b] 紫外-可见分光光度计、电子天平(精度0.01g)[b]4.2方法[/b][/align][align=left] 取本品0.50g至盛有100 ml水的锥形瓶中,在冰箱中放置过夜,溶解后,纯水作为空白,参考紫外-可见分光光度计操作规程,550 nm波长处测定溶液的透光率。[/align][align=left][b]5pH的测定5.1材料[/b][/align][align=left] 电子天平(精度0.01g)、pH计、磁力搅拌器、磁子、100 ml锥形瓶、100 ml量筒、新沸放冷的纯化水。[/align][align=left][b]5.2方法[/b][/align][align=left][b]5.2.1 溶解[/b][/align][align=left] 称取供试品0.10 g,置锥形瓶中。加新沸放冷的水100 ml和磁子,将锥形瓶用封口膜封口,将锥形瓶置磁力搅拌器上搅拌约4小时,完全溶解,目测为均一透明溶液。[b]5.2.2 测定[/b][/align][align=left] 按照所用pH计的操作规程,先对pH计进行校准,之后将电极和温度探头深入被测溶液中,缓慢搅拌,读取pH值。[b]6运动黏度的测定6.1材料[/b][/align][align=left] 电子天平(精度0.1 mg);平氏黏度计(毛细管内径为1.0 mm ± 0.05 mm);恒温水浴:控温精度±0.01 ℃;秒表:分度0.01秒;振荡器。[b]6.2方法[/b] 称量样品0.1 g(折干),置100 ml容量瓶中,加水振荡至溶解后作为供试液。取毛细管内径为1.0mm ± 0.05 mm的平氏黏度计,加入5 ml供试液,置水浴中,25 ℃下放置15分钟后,秒表测定供试液流过黏度计两条线之间的时间,取两次测定的平均值按下式计算,即为供试品的运动黏度,计算方法如公式4所示。[/align][align=left] 运动黏度ν(mm[sup]2[/sup]/s)=[i]Kt [/i]公式(4)[/align][align=center]式中 [i]K[/i]为用已知黏度的标准液测得的黏度计常数,mm[sup]2[/sup]/s[sup]2[/sup];[/align][align=center][i]t[/i]为测得的平均流出时间,s;[/align][b]7干燥失重7.1材料[/b] 卤素水份测定仪,HHA样品;[b]7.2方法[/b][align=left] 取本品约1.0g,置HG53 型卤素水分测定仪托盘内。110 ℃测定15分钟,记录测定结果。[b]8细菌、霉菌及酵母菌测定8.1供试液制备 [/b][/align][align=left] 取34ml无菌磷酸盐缓冲液1瓶,将1500U HAase加入其中,用吸量管各吸取1ml分别加入至4个平皿中,作为阴性对照。再取样品1.5 g,加入到做完阴性对照的含有HAase的30 ml磷酸盐缓冲液中,42℃下振荡溶解,制得 1﹕20的供试品溶液。[b]8.2 细菌总数测定[/b](1)阴性对照试验将温度低于45℃溶化的营养培养基分别注入上述2个含有1 ml的磷酸盐缓冲液的平皿中,每个平皿约15~20 ml左右,凝固,倒置培养。均不得有菌生长。(2)样品测定用吸量管准确吸取上述1∶20的供试液2ml加入至8 ml的磷酸盐缓冲溶液中,混匀,作为1∶100的稀释级。向平皿中分别加入1∶20、1∶100的供试液各1 ml,向每个平皿注入温度低于45℃的事先溶化的营养琼脂约15~20 ml,待凝固后倒置放入培养箱中。每个稀释级均制备2个平板。[b]8.3 霉菌及酵母菌数测定[/b](1)阴性对照试验 分别注入向2个含有1ml的上述磷酸盐缓冲液的平皿中将温度低于45℃溶化的玫瑰红钠琼脂培养基,每个平皿约15~20 ml左右,凝固,倒置培养,均不得有菌生长。(2)样品测定 用吸量管准确吸取上述1∶20供试液2ml加入至8 ml的磷酸盐缓冲溶液中,混匀,作为1∶100的稀释级。各吸取1∶20、1∶100的稀释级的供试液1 ml加入至平皿中,注入温度不超过45 ℃的溶化玫瑰红钠琼脂培养基,每个平皿约15~20 ml,待凝固后,倒置培养。每个稀释级均制备2个平板。[b]8.4 结果[/b] 将营养琼脂培养基和玫瑰红钠琼脂培养基平板分别倒置于30~35℃、23~28℃生化培养箱中,营养琼脂平板培养3天,用于细菌计数;玫瑰红钠琼脂培养平板培养5天,用于霉菌、酵母菌计数,按照稀释比例,计算出每克样品中的微生物数。[b]9 结论[/b] 采用制定的质量标准对产品检验,结果表明,HHA能保持HA的润滑性和流动性,也具有明显的抗HAase降解的特性;克服了HA衍生物抗酶解但缺少润滑性的缺点,预期用途是开发成骨关节注射液或皮下注射填充剂用于美容,期望能够延长体内保留时间起到长效治疗的作用,减少患者注射次数,减轻患者痛苦。[/align][align=center]参考文献[/align] 赵凯, 刘丽艳, 刘婧婷. 分光光度法测定羟丙基淀粉取代度. 食品科学,2011, 32(22) : 201-203.[align=center][b][/b][/align][align=center][b][/b][/align]
羟丙基淀粉取代度的测定有什么注意点?与茚三酮络合后为何要快速测定?丙二醇的标准曲线是否每次测量都需绘制?标准曲线是大概是多少?100度的油浴能否用水浴代替?
请教各位一下 有谁做过羟丙基淀粉取代度试验 那其中用到的浓硫酸是不是对其测定有很大的影响?那么所用硫酸的标准是什么?
EN71-3 2013附录G有机锡分析方法中10种有机锡化合物标准品均为固体,但是采购的时候二正丙基二氯化锡这个供应商报过来的全都是10ml 10ug/ml的液体,请问大家也遇到这样的问题吗?是如何解决的呢?跪求答案,谢谢!
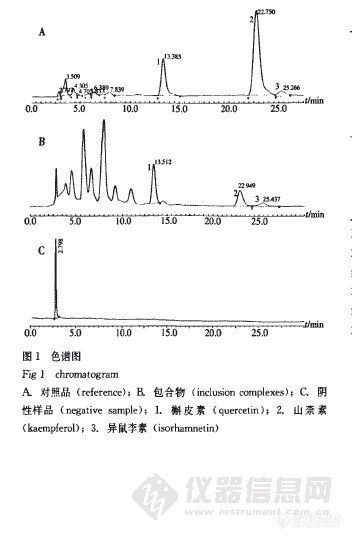
作 者:赵建彬 陈建海(南方医科大学南方医院药学部,广州,510515)摘要: 目的 建立银杏酮酯-羟丙基-β-环糊精包合物的质量标准.方法 采用高效液相色谱法测定制剂中的总黄酮苷含量,色谱柱:Diamonsil C18柱(250 mm×4.6 mm,5 μm),流动相:甲醇-0.4%磷酸(45∶55),检测波长368nm,流速:1 mL·min-1;采用红外光谱分析法鉴别包合物.结果 槲皮素在2~132 μg·mL-1与峰面积具有良好的线性关系,r=0.9995;山奈素在0.9~60 μg·mL-1与峰面积具有良好的线性关系,r=0.9998;异鼠李素在0.1~9 μg·mL-1与峰面积具有良好线性,r=0.9998.槲皮素、山奈素、异鼠李素的平均回收率分别为101.6%、100.4%、99.3%,RSD分别为1.3%,1.4%,0.63%.红外光谱法能有效鉴别包合物.结论 本法可作为该制剂的质量控制参考标准.谱图:http://ng1.17img.cn/bbsfiles/images/2012/08/201208061012_381701_1606903_3.jpg

[align=center][img=,600,457]https://ng1.17img.cn/bbsfiles/images/2019/09/201909251626050517_8200_932_3.jpg!w500x381.jpg[/img][/align]我们今天给大家带来的是羟丙基-β-环糊精有关物质的测定。羟丙基-β-环糊精是应用最广泛的环糊精衍生物之一。主要应用于食品、医药、化妆品行业。1、在食品、香料领域,可提高营养分子的稳定性和长效性,可掩盖或矫正食品营养分子的不良气味和口味,可改进生产工艺和产品品质。2、在化妆品原料中用作稳定剂、乳化剂、去味剂等,可降低化妆品中有机分子对皮肤粘膜组织的刺激,增强有效成分的稳定性,防止营养成分的挥发、氧化。它有一定的相对吸湿性。3、在医药工业中,由于相对表面活性和溶血活性比较低且对肌肉没有刺激性,所以它是一种理想的注射剂增溶剂和药物赋形剂。我们今天就一起来看下月旭Xtimate SEC-700(7.8×300mm,5μm)色谱柱对羟丙基-β-环糊精的测定效果如何。[b]色谱条件[/b]色谱柱:月旭Xtimate SEC-700(7.8×300mm,5μm);流动相:0.1mol/L硝酸钾/乙腈=35/65;检测波长:示差检测器;温度:40℃;柱温:40℃;流速:0.8mL/min;进样量:20μL。[b]谱图和数据[/b]1、混合溶液图[align=center][img=,600,310]https://ng1.17img.cn/bbsfiles/images/2019/09/201909251626100982_7478_932_3.jpg!w690x357.jpg[/img][/align][align=center][img=,600,94]https://ng1.17img.cn/bbsfiles/images/2019/09/201909251626155127_2008_932_3.png!w592x93.jpg[/img][/align]2、羟丙基-β-环糊精[align=center][img=,600,313]https://ng1.17img.cn/bbsfiles/images/2019/09/201909251626196371_979_932_3.jpg!w690x360.jpg[/img][/align][align=center][img=,600,80]https://ng1.17img.cn/bbsfiles/images/2019/09/201909251626247666_6386_932_3.png!w690x92.jpg[/img][/align]3、β-环糊精[align=center][img=,600,313]https://ng1.17img.cn/bbsfiles/images/2019/09/201909251626295011_5994_932_3.jpg!w690x361.jpg[/img][/align][align=center][img=,600,76]https://ng1.17img.cn/bbsfiles/images/2019/09/201909251626337665_1162_932_3.png!w690x88.jpg[/img][/align]4、空白溶液[align=center][img=,600,315]https://ng1.17img.cn/bbsfiles/images/2019/09/201909251626374979_8817_932_3.jpg!w690x363.jpg[/img][/align][align=center][b]结 论[/b][/align]使用月旭Xtimate SEC-700(7.8×300mm,5μm),在此色谱条件下检测,能满足检测需求。
请教,羟丙基取代度测定时硫酸的加入量以及样品在处理过程中如何选择加热的时间?
查找了中国药典,没有找到H-HPC的标准,如果用HPC的方法去测定其含量,对结果会产生什么影响?谁家可进行此项检测,比如什么样的机构或实验室,谢谢。[color=#00FFFF][size=4]H-HPC高取代羟丙基纤维素[/size][/color]
需要买一根R,S-羟丙基-β环糊精为填充剂的色谱柱,以前没用过,大侠给推荐一下哪家的好
毕业在即,毕业论文中涉及2-丙基苯并咪唑的红外标准图谱图,望各位朋友帮帮忙了!本人将万分感谢!如果哪位朋友能找到,请发到我的邮箱,piao23luo◎163.com,谢谢拉!
有关单位: 经国家食品药品监督管理局化妆品审评专家委员会审核,拟批准“二甲氧基甲苯基-4-丙基间苯二酚”和“聚甲基丙烯酰基赖氨酸”作为化妆品原料使用。现公开征求意见,请于2011年6月27日前将反馈意见电子版发送至chenzh@sfda.gov.cn。 附件:1.“二甲氧基甲苯基-4-丙基间苯二酚”技术要求 2.“聚甲基丙烯酰基赖氨酸”技术要求 国家食品药品监督管理局食品许可司 二〇一一年六月十五日
各位兄弟姐妹,请问羟丙基-β-环糊精在乙酸乙酯的溶解度是多少?我查了很多文献都找不到答案,打算自己去测定了!但是具体该怎么做?希望各位能指点一下小弟!
HPLC法1 应用范围本法适用于唇膏、雪花膏、化妆品及痱子粉等化妆品中的异丙基甲酚的测定。2 原理样品中的异丙基甲酚在H2SO4介质条件下,用二氯甲烷萃取,然后蒸干萃取液,用乙醇定容。将乙醇溶液注入高效液相色谱仪,以荧光检测器检测,与同样处理的已知标准溶液比较进行定性和定量测定。3 试剂3.1 异丙基甲酚的标准溶液:精确称取异丙基甲酚50.0mg溶于乙醇中定容至50.0ml。取此溶液5.0ml用乙醇稀释至50ml,此液1.0ml含100.0μg异丙基甲酚。3.2 内标准溶液:精确称取百里酚25.0mg溶于水500.0ml乙醇中。3.3 柱填充剂:氨丙基硅烷键合硅胶,十八烷基硅烷键合硅胶。3.4 流动相:已烷 乙醇(9 1) 水 乙腈(1 1)。4 仪器4.1 高效液相色谱仪:具荧光检测器。5 分析步骤5.1 样品的预处理精确称取样品约0.5~2.5g(2),加饱和NaCl溶液50ml,移入分液漏斗中,加10%H2SO4 1ml,30ml二氯甲烷,振摇5min,静置分层.水层再各用20ml二氯甲烷提取二次,合并全部二氯甲烷,用50ml水洗涤,用无水硫酸钠脱水后(3),在旋转蒸发器上(水浴约40℃)蒸去二氯甲烷(4),于残留物中加入内标液5.0ml(5),用乙醇定容至50ml(6)作为待测溶液。5.2 测定5.2.1高效液相色谱条件5.2.1.1色谱柱(7):氨丙基硅烷键合硅胶(内径4.6mm、长250mm)。流动相:己烷一乙醇(9+1)。流速:1.2m1/min。5.2.1.2色谱柱(8):十八烷基硅烷键合硅胶(内径4.6mm、长15Omm)。流动相:水 乙腈(1 1)。流速:1.0ml/min。5.2.2荧光检测器波长:激发波长280nm,荧光波长305nm。5.2.3定性:取5.0ml标准溶液与5.0ml内标溶液于50.0ml容量瓶中,混匀,然后加乙醇至刻度。取此液2.5进行高效液相色谱分析,从得到的色谱图求出内标物对异丙基甲酚的相对保留时间。同样取2.5μl待测溶液,如上述方法操作。从得到的峰求出对内标物的相对保留时间与标准溶液时行比较而定性。5.2.4定量:取2.5μl待测溶液进行高效液相色谱分析。通过异丙基甲酚的峰高(或者峰面积)与内标物质的峰高(或者峰面积)之比,从预先做成的标准曲线中求出待测溶液中异丙基甲酚的浓度A(μg/m1)。5.2.5标准曲线的制备:分别取异丙基甲酚标准溶液0.5、1.0、2.0、3.0、4.0、5.0及6.0ml于50.0ml容量瓶中,分别于每个容量瓶中加入内标准溶液5.0ml,立即加乙醇至50.0m1。分别取2.5μl此溶液进行高效液相色谱分析,求出异丙基甲酚与内标物的峰高(或峰面积)之比值,做标准曲线。6 计算c=A×V/(m×1000×1000)×100式中;c-一样品中异丙基甲酚的含量,%;A――从标准曲线上查得待测溶液中异丙基甲酚浓度,μg/m1。V一-测定用待测溶液的体积,m1;M-一样品质量,g。
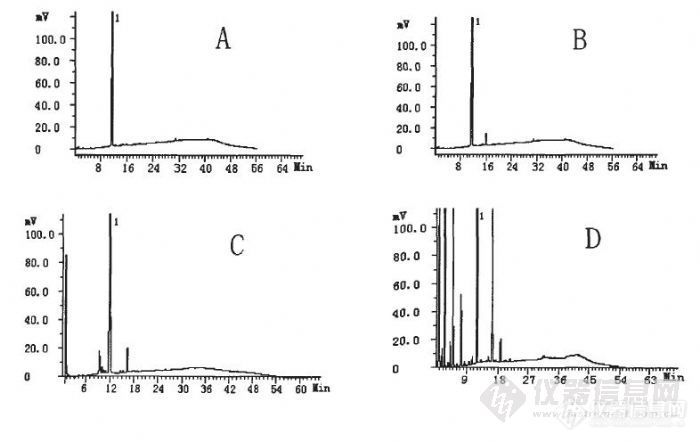
【作者】 葛文娜(东南大学)【摘要】 多烯紫杉醇(Docetaxel,商品名为Taxotere)是新一代紫杉烷类抗肿瘤药物,对乳腺癌、非小细胞肺癌、胰腺癌、头颈癌、卵巢癌以及前列腺癌等均具有良好疗效。目前,多烯紫杉醇上市剂型为注射用浓溶液,该剂型稳定性较差,临床用药不方便并时常引发严重的过敏反应。利用羟丙基-磺丁基-β-环糊精作为药物赋形剂水溶性好、溶血性小、毒性低的特点,实验室开发了多烯紫杉醇羟丙基-磺丁基-β-环糊精包合物冻干粉制剂,以期达到避免或者缓解现有制剂缺点的目的。 本文建立了多烯紫杉醇冻干粉质量的高效液相(HPLC)分析方法,以及生物样本(血浆和组织)中多烯紫杉醇的检测方法,进行了该制剂质量的评价和动物体内的药代动力学研究,为制剂的有效性和安全性评价提供方法学基础;首次提出基于电纺尼龙6纳米纤维的固相膜萃取技术,并以多烯紫杉醇为目标分子,进行了这一样品前处理的新技术在生物样本分析中的应用研究。论文主要工作如下: 一、反相高效液相色谱-紫外法测定多烯紫杉醇冻干粉含量及有关物质 采用Dikma Platisil C18柱(150mm×4.6mm,5μm),检测波长227nm,柱温:30℃。有关物质检查时以乙腈-水(40:60,v:v)为流动相A,乙腈为流动相B,进行梯度洗脱,流速1.5mL/min;含量测定时紫杉醇为内标,以乙腈-水(60:40,v/v)为流动相,进行等度洗脱,流速为1.0mL/min。结果表明:多烯紫杉醇与各有关物质分离良好。在0.05~100μg/mL的浓度范围内具有良好的线性关系(r=0.9993),检出限(LOD)为0.01μg/mL(以S/N=3计)。三个批次样品的多烯紫杉醇标示含量分别为98.79%、99.56%、100.9%,有关物质含量分别为2.8%、2.2%、2.5%。本法准确可靠,专属性强,能满足多烯紫杉醇包合物冻干粉质量控制的要求。 二、多烯紫杉醇包合物冻干粉家兔体内药代动力学的研究 本文建立了灵敏、准确的家兔血浆中多烯紫杉醇的HPLC-UV测定方法,为多烯紫杉醇包合物冻干粉家兔体内药代动力学的研究提供方法学基础。血浆中杂质不干扰样品的测定,多烯紫杉醇在0.04~10μg/mL浓度范围内线性关系良好(r=0.9996),检出限为0.02μg/mL(以S/N=3计),平均提取回收率为90.1%~93.7%,相对标准偏差(RSD)为3.2%~12%。以上方法验证结果表明,此方法满足药代动力学研究的要求。 采用血药浓度法研究了多烯紫杉醇包合物冻干粉的家兔体内药代动力学,通过测定血药浓度经时变化曲线,得出了多烯紫杉醇冻干粉(受试制剂)药代动力学参数;以上市的多烯紫杉醇注射液为参比制剂,研究了受试制剂的相对生物利用度。参比制剂的t1/2β为5.32±1.72h,AUC和CL分别为1.29±0.459μg/mL·h和5.82±1.27mL/h;受试制剂t1/2β为7.00±1.90min,AUC和CL分别为1.59±0.798μg/mL·h和4.72±2.12 mL/h。其平均相对生物利用度为123%。结果表明家兔给予受试制剂t1/2β和AUC均大于参比制剂,清除率CL小于参比制剂(P0.05),提示在相同剂量的条件下,多烯紫杉醇羟丙基-磺丁基-β-环糊精包合物冻干粉在体内可保持相对较高的血药浓度,提高了生物利用度,在一定程度上增强药物对肿瘤细胞的杀伤作用,从而产生较好的临床效果。 三、基于电纺尼龙6纳米纤维的固相膜萃取技术及其在生物样本中的应用研究 研究了基于电纺尼龙6纳米纤维的同相膜萃取技术,研制了相应的装置,在此基础上,对生物样本(家兔血浆和小鼠组织)进行前处理,全面考察了影响萃取效率的因素:蛋白酶的种类、酶解时间和温度、介质pH和离子强度、洗脱溶剂、洗脱溶剂体积,建立了基于电纺尼龙6纳米纤维的固相膜萃取富集生物样本中多烯紫杉醇的分析方法。在优化的条件下,多烯紫杉醇平均提取回收率为72.1%~78.5%(RSD8%),检出限0.015μg/mL(以S/N=3计)。与传统液-液萃取法、C18柱固相萃取法相比,此萃取方法解决了常规提取方法有机溶剂用最大、提取时间长、净化效果差的缺点,不仅使得检测灵敏度增高、干扰减少,而且提取过程快速、环保,符合“绿色化学”发展趋势。【谱图】 http://ng1.17img.cn/bbsfiles/images/2012/08/201208211806_385130_1609970_3.jpghttp://ng1.17img.cn/bbsfiles/images/2012/08/201208211803_385128_1609970_3.jpg
最近做二异丙基萘含量测定,由于购买的只是一种混合物(含7种异构体),但是只有一个cas登记号,按照标准方法(内标法),确实发现并确定了7种组分流出顺序,根据标准方法只是计算二异丙基萘的总量。但是突然想计算不同异构体的具体含量,这又该如何计算呢?由于异构体中有几种是很难找到(买到)单标的,此时可用面积归一化法计算可以吗?是否需要通过面积归一化法来单独建立不同异构体的标准曲线?这样算出来的结果具有说服力吗?如何不行,该如何计算呢?望老师赐教,谢谢!
买了二异丙基萘混合标准(7种),跑出来,检索发现都是2,6-DINP出现在前2位,如何鉴定是其他的异构体呢?先上传全扫描附件,请老师帮忙确定,谢谢!
论坛里面已经有人提供了水中红外到远红外的了,但是近红外的却没有,求哪位提供一下,此外谁有羟乙级纤维素和羟丙基甲基纤维素的近红外图谱,谢谢
今天看到一标准中用到 (50%氰丙基)-甲基聚硅氧烷 为填充物的柱子,不知道这是哪根柱子?是否是DB225、HP225或者RTX225?
【序号】:8【作者】: 程珍华【题名】:壳聚糖的羟丙基化、季铵盐化修饰及其衍生物的结构与性能研究【期刊】:浙江工业大学【年、卷、期、起止页码】:2018【全文链接】:[url]https://kns.cnki.net/kcms/detail/detail.aspx?dbcode=CMFD&dbname=CMFD201801&filename=1017246430.nh&uniplatform=NZKPT&v=Sj53myVTRIHhcZwzkOJsh3h3El6tssU6oZpVOnZImBUkxEECiiP2f-I53b5DxJ2G[/url]
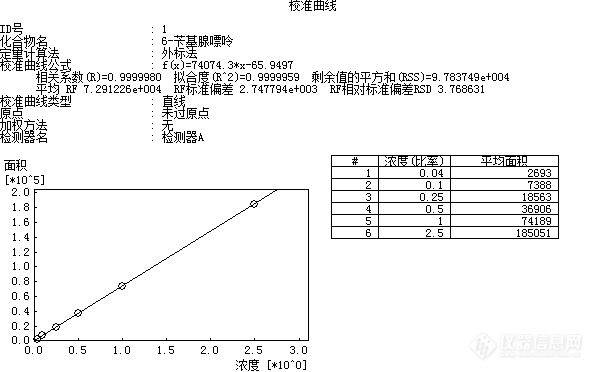
[align=center][b]食品中6-苄基腺嘌呤的测定方法验证报告[/b][/align][align=center][b]GB/T 23381-2009( 高效液相色谱法)[/b][/align][align=center][b]张霞[/b][/align]一、方法概述1.范围 本标准规定了用高效液相色谱法测定食品中6-苄基腺嘌呤(6-BA)含量的方法。 本标准适用于果蔬菜(豆芽、黄瓜、番茄、香菇、草莓、橙类)等植物性食品及其制品中6-苄基腺嘌呤的测定。2.规范性引用文件下列文件中的条款通过本标准的引用而成为本标准的条款。凡是注日期的引用文件,其随后所有的修改单(不包括勘误的内容)或修订版均不适用于本标准,然而,鼓励根据本标准达成协议的各方面研究是否可使用这些文件的最新版本。凡是不注日期的引用文件,其最新版本适用于本标准。GB/T 6682 分析实验室用水规格和试验方法(GB/T 6682-2008,ISO 3696:1987,MOD)3.方法提要 试样经甲醇提取、浓缩并净化后,用高效液相色谱检测,外标法定量。二、仪器与试剂1. 仪器1.1高效液相色谱仪:配有紫外检测器或二极管阵列检测器。1.2 组织捣碎机。1.3离心机:转速不低于4000r/min。1.4超声波清洗仪。1.5旋转蒸发仪。1.6固相萃取装置。1.7电子天平:感量0.1mg。1.8微孔滤膜:0.45μm,有机相。以上仪器符合国标要求。2. 试剂及其配制 除另有规定外,所有试剂均为分析纯,水为GB/T 6682规定的一级水。2.1甲醇:色谱纯。2.2冰乙酸。2.3 C[sub]18[/sub]固相萃取柱:6mL,500mg,或相当者,使用前依次用5mL甲醇、10mL水活化。2.4乙酸铵溶液(0.02mol/L):称取1.54g乙酸铵,用适量水溶解,加入1.0mL冰乙酸,加水定容至1000mL。2.5 6-苄基腺嘌呤标准溶液(100.0μg/mL) [color=#ff0000] [/color][color=#ff0000]来源[/color][color=#ff0000]:[/color][color=#ff0000]农业部环境保护科研监测所[/color][color=#ff0000] [/color][color=#ff0000]货[/color][color=#ff0000]号[/color][color=#ff0000]:[/color][color=#ff0000]SB05-368-2016[/color][color=#ff0000] [/color]三、分析步骤1、标准曲线绘制1.1 标准工作液的配制: 分别吸取适量6-苄基腺嘌呤标准溶液,用甲醇定容至10mL容量瓶中,配制成浓度为0.04μg/mL、0.1μg/mL、0.25μg/mL、0.5μg/mL、1.0μg/mL、2.5μg/mL系列工作液。2、样品的处理2.1提取:称取经组织捣碎机捣碎的样品约10g(精确到0.01g)于50mL离心管中,加入20mL甲醇,超声提取15min,以转速不低于4000r/min离心10min,上清液转入50mL梨形瓶中,样品再次用20mL甲醇超声提取15min,离心合并上清液,用旋转蒸发仪(不超过60℃)浓缩至近干,去除甲醇,残液待净化。2.2纯化:将上述2.1残液以2mL/min流速通过预先活化的固相萃取柱,用少量水(约2mL)洗涤梨形瓶,洗液过固相萃取柱,再用5mL水洗涤固相萃取柱,去除杂质后用甲醇洗脱并定容至5.0mL,混匀后经0.45μm滤膜过滤,作为待测液供HPLC分析。3.仪器测定条件3.1色谱柱:C18柱,柱长250mm,内径4.6mm,粒径5μm或相当型号色谱柱。3.2流速:1.0mL/min。3.3柱温:30℃。3.4检测波长:267nm。3.5进样量:10μL。3.6流动相:甲醇 :0.02mol/L乙酸铵溶液=1:1四、结果处理试样6-苄基腺嘌呤含量按下式进行计算:[table][tr][td=1,2][align=center]X(mg/kg)=[/align][/td][td]C×[i]V[/i]×1000[/td][/tr][tr][td]m×1000[/td][/tr][/table]式中:X-试样中6-苄基腺嘌呤含量,单位为毫克每千克(mg/kg) C-由标准曲线计算出样液中6-苄基腺嘌呤的浓度,单位为微克每毫升(μg/mL) m-试样质量,单位为克(g) V-试样的最终定容体积,单位为毫升(mL)。1000—换算系数。计算结果保留两位有效数字。五、验证结果1.线性结果将标准系列工作溶液分别注入液相色谱仪中,测定相应的峰面积,以标准系列工作溶液的质量浓度为横坐标,以峰面积为纵坐标,绘制标准曲线。同时做空白实验。6-苄基腺嘌呤[u]Y=74074.3*X-65.9497 R^2=0.9999959[/u][align=center][img=,595,372]http://ng1.17img.cn/bbsfiles/images/2018/07/201807242033263735_9846_2904018_3.png!w595x372.jpg[/img][/align]以上结果表明6-苄基腺嘌呤在0.04μg/mL~2.5μg/mL范围内,R[sup]^2[/sup]=0.9999959,6-苄基腺嘌呤浓度和峰面积呈线性关系,线性良好,符合要求。2.检出限结果将0.25μg/mL标准溶液逐级稀释至S/N=3±1,得出6-苄基腺嘌呤的方法检出限为0.0125mg/kg[color=#ff0000],[/color]此检出限结果小于国标GB/T 23381-2009的方法检出限0.02mg/kg,故此方法满足条件。六、方法精密度(重复性)对LBF180700282样品分别进行6次加标重复性的测定,测定结果如下:[table][tr][td][align=center]测定编号[/align][/td][td][align=center]1[/align][/td][td][align=center]2[/align][/td][td][align=center]3[/align][/td][td][align=center]4[/align][/td][td][align=center]5[/align][/td][td][align=center]6[/align][/td][/tr][tr][td][align=center]质量(g)[/align][/td][td][align=center]10.0031[/align][/td][td][align=center]10.0016[/align][/td][td][align=center]10.0025[/align][/td][td][align=center]10.0044[/align][/td][td][align=center]10.0027[/align][/td][td][align=center]10.0048[/align][/td][/tr][tr][td][align=center]浓度(μg/mL)[/align][/td][td][align=center]0.652[/align][/td][td][align=center]0.650[/align][/td][td][align=center]0.652[/align][/td][td][align=center]0.652[/align][/td][td][align=center]0.652[/align][/td][td][align=center]0.651[/align][/td][/tr][tr][td][align=center]含量(mg/kg) [/align][/td][td][align=center]0.33[/align][/td][td][align=center]0.32[/align][/td][td][align=center]0.33[/align][/td][td][align=center]0.33[/align][/td][td][align=center]0.33[/align][/td][td][align=center]0.33[/align][/td][/tr][tr][td][align=center]平均值(mg/kg)[/align][/td][td=6,1][align=center]0.33[/align][/td][/tr][tr][td][align=center]RSD%[/align][/td][td=6,1][align=center]1.24[/align][/td][/tr][/table]本方法的精密度为1.24%,符合GB/T 23381-2009中给出试样测试结果的精密度要求。因此,本次测定均符合要求。七、准确度验证(加标回收)对LBF180700282样品加标,取2.5μg/mL的标液0.09mL、0.35mL、0.64mL同样品同步处理后,结果见下表:[table][tr][td=2,1][align=center]测定编号[/align][/td][td=6,1][align=center]6-苄基腺嘌呤[/align][/td][/tr][tr][td][align=center]序号[/align][/td][td][align=center]m(g)[/align][/td][td][align=center]V(mL)[/align][/td][td][align=center]C(μg/mL)[/align][/td][td][align=center]6-苄基腺嘌呤含量(mg/kg)[/align][/td][td][align=center]平均值(mg/kg)[/align][/td][td][align=center]加标量(mg/kg)[/align][/td][td][align=center]回收率%[/align][/td][/tr][tr][td][align=center]1#[/align][/td][td][align=center]10.0236[/align][/td][td][align=center]5.0[/align][/td][td][align=center]N.D[/align][/td][td][align=center]N.D[/align][/td][td=1,2][align=center]N.D[/align][/td][td][align=center]/[/align][/td][td][align=center]/[/align][/td][/tr][tr][td][align=center]2#[/align][/td][td][align=center]10.0157[/align][/td][td][align=center]5.0[/align][/td][td][align=center]N.D[/align][/td][td][align=center]N.D[/align][/td][td][align=center]/[/align][/td][td][align=center]/[/align][/td][/tr][tr][td][align=center]加标1#[/align][/td][td][align=center]10.0087[/align][/td][td][align=center]5.0[/align][/td][td][align=center]0.042[/align][/td][td][align=center]0.021[/align][/td][td][align=center]0.021[/align][/td][td][align=center]0.022[/align][/td][td][align=center]95.5[/align][/td][/tr][tr][td][align=center]加标2#[/align][/td][td][align=center]10.0103[/align][/td][td][align=center]5.0[/align][/td][td][align=center]0.163[/align][/td][td][align=center]0.081[/align][/td][td][align=center]0.081[/align][/td][td][align=center]0.087[/align][/td][td][align=center]93.1[/align][/td][/tr][tr][td][align=center]加标3#[/align][/td][td][align=center]10.0189[/align][/td][td][align=center]5.0[/align][/td][td][align=center]0.307[/align][/td][td][align=center]0.15[/align][/td][td][align=center]0.15[/align][/td][td][align=center]0.16[/align][/td][td][align=center]93.8[/align][/td][/tr][/table] 由上表可以看出6-苄基腺嘌呤测定的加标回收范围在 60%-120% ,RSD值为1.31%符合规定要求。八、总结从检出限、线性范围、重复性、回收率测试结果可知,均符合方法要求,本实验方法符合GB/T 23381-2009的要求。[img=,595,372]http://ng1.17img.cn/bbsfiles/images/2018/07/201807242033072325_3650_2904018_3.png!w595x372.jpg[/img][img=,595,372]http://ng1.17img.cn/bbsfiles/images/2018/07/201807242033072325_3650_2904018_3.png!w595x372.jpg[/img][img=,595,372]http://ng1.17img.cn/bbsfiles/images/2018/07/201807242033072325_3650_2904018_3.png!w595x372.jpg[/img]
【序号】:5【作者】: 宓英其【题名】:不同阴离子化2-羟丙基三甲基铵类壳聚糖的制备、活性及性能研究【期刊】:中国科学院大学(中国科学院烟台海岸带研究所)【年、卷、期、起止页码】:2019【全文链接】:https://kns.cnki.net/kcms/detail/detail.aspx?dbcode=CMFD&dbname=CMFD201902&filename=1019909665.nh&uniplatform=NZKPT&v=Tb5dxN6PyXtwEP263qxVa4fkRlfspmQqPGAe_cjbNcQGjz0XyrMMcWgIhyqvAxg1
我现在用液质做tepa三-(1-氮杂环丙基)氧化膦,可是买不到标准品,大家咋买到的啊,谁有用液质做的色谱图啊!http://images.basechem.org/struct/2011-03/ff30a0213c0d51d45505574710dd5862.gif
[color=#444444]用安捷伦[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质联用[/color][/url]7890A5977B测纸张中的二异丙基萘,根据标准建立了方法之后,运行时仪器状态显示错误,研究了好久没找出相关原因,请教大家一下,有没有懂这个的,希望能指点迷津,感激不尽![/color][color=#444444][img]http://muchongimg.xmcimg.com/data/bcs/2017/0809/bw133h3202411_1502285021_281.jpg[/img][/color]
不知哪位大侠有做过甲基丙基纤维素的测定。前处理过程需要注意些什么?刚接触,总是含量做不准。
羟丙纤维素-中国药典2010版本品为低取代2-羟丙基醚纤维素。按干燥品计算,含羟丙氧基(-OCH2CHOHCH3)应为7.O%~l6.0%.羟丙甲纤维素-中国药典2010版本品为2-羟丙基醚甲基纤维素。按干燥品计算,含甲氧基(-OCH3)应为l9.0%~30.0%,含羟丙氧基(-OCH2CHOHCH3)应为4.0%~l2.0%。
维权声明:本文为huomeng520原创作品,本作者与仪器信息网是该作品合法使用者,该作品暂不对外授权转载。其他任何网站、组织、单位或个人等将该作品在本站以外的任何媒体任何形式出现均属侵权违法行为,我们将追究法律责任。本实验建立了一种以牛肉中肌肽为代表,反相分离测定亲水性物质的方法。该方法选用丙基酰胺键合硅胶亲水作用色谱柱,反相分析测定牛肉中亲水性成分—肌肽的含量。该方法操作简单,样品无需衍生处理。通过该法结合 H P L C—M S联用技术确定了保留时间为10.276~10.609min的色谱峰就是肌肽峰。将该色谱柱与常规C18色谱柱进行对比后发现,该色谱柱对L-肌肽的保留能力和分离能力均优于C18柱。此法精密度实验显示其相对标准偏差(RSD%)为1.06%,最低检测限为4.59×10-2mg/L。最后实验结果表明:亲水性色谱柱反相使用时,完全适用强极性物质含量的测定,通过实际样品分析检测,每克牛肉的肌肽含量为0.011克。引言L-肌肽(L-carnosine)是一种水溶性二肽,在1900年由Gulewitsch和Amiradzhibi在牛肉提取物中发现。L-肌肽天然存在于多种脊椎动物的骨骼肌以及新陈代谢旺盛的脑中。它具有广泛的生物活性,如抗氧化、保护膜的完整性、抗糖基化、质子缓冲、调节巨噬细胞活性等,是维持机体正常状态的一种含量很低的物质。L-肌肽的结构为β-丙氨酰—L-组氨酸。L-肌肽的结构如图1所示。从化学结构上看,肌肽由于含有较多的极性基团(-OH、-NH2、-COOH),水溶性特别强。肌肽的正辛醇—水分散系数为-2,远远小于0,理论上说明了L-肌肽的强极性,不溶于任何有机溶剂,属于亲水性成分。近年来,L-肌肽的研究一直受到人们关注。其含量测定方法一直在探索中。目前已报道的L-肌肽的分析方法主要以高效液相色谱法为主,且多采用柱前衍生化法,这种方法试剂成本高,样品预处理繁琐,且分析时间长,不利于对样品的快速检测。也曾有报道将离子色谱和毛细管电泳色谱应用于L-肌肽的测定,但两种方法较为复杂,且仪器操作较为繁琐。将氨基柱应用于反相高效液相色谱,能实现对样品中L-肌肽快速、准确地检测,但氨基柱不耐水解,长时间在反相条件下使用,会缩短氨基柱的使用寿命。所以应选择一款既耐水解,柱效又高的色谱柱对牛肉中L-肌肽进行分析。色谱柱填料通常是以硅胶为载体,在硅胶表面进行修饰。C18色谱填料是在硅胶表面键合非极性的十八烷基碳,属于非极性色谱填料。根据“相似相亲原则”,应选用极性较强的色谱柱分析极性物质,普通的C18反相色谱柱属于非极性色谱柱,对亲水性成分没有保留能力,因此不能满足对此类物质的分析要求。实验中选用丙基酰胺键合硅胶柱,该色谱柱填料以硅胶为载体,表面键合丙基酰胺基团,极性强,耐水解,适用于对极性物质的分离。马婧玮采用此柱,实现了对亲水性井冈霉素A快速准确的定量分析。本次实验从L-肌肽的性质出发,结合色谱柱的性质,将丙烯酰胺键合硅胶色谱柱与常规C18柱进行对比,并借鉴田颖刚等人已发表的L-肌肽质谱分析条件,选择分离效果最好的色谱柱与电喷雾质谱串联使用,对牛肉中肌肽进行了分析鉴定。
正在做萘的异丙基化反应,因为2,6-二异丙基萘和2,7-二异丙基萘标样太贵了,请教各位大牛,这两种物质在色谱中出峰顺序哪个在前?哪个在后? 非常感谢!!
各位老师:我在做N,N-二异丙基乙二胺(DIPEA)时,使用Agilent G1888顶空进样器,色谱柱是DB624,进样DIPEA标准溶液时其峰型是好的,但是在将该标液加到样品溶液中以后DIPEA的峰型裂分,裂分后的2个峰的峰面积相加与原峰面积相同。但是同样的溶液在CTC上进样时,加标后的DIPEA的峰型也依然是好的,请问有老师曾遇到过这样的问题么?谢谢!







 我要推广仪器
我要推广仪器
 下载APP
下载APP