拉米夫定由英国葛兰素史克公司(GlaxoSmithKline)开发研制生产,1995年首次在加拿大、美国上市销售,1998年在中国上市,商品名贺普丁。目前,在我国市场上销售的拉米夫定及其复方制剂是由史克公司独家销售。拉米夫定及其片剂和口服液于1999年4月10日授权在中国获得药品行政保护,保护期将于2006年10月终止。拉米夫定全球专利将于2006年9月到期。拉米夫定是近年新上市的一种抗乙肝病毒新药,其英文名称为Lamivudine,又称3—TC。是一种核苷类抗病毒药,对人类免疫缺陷病毒和乙型肝炎病毒均具有显著的抑制活性,是治疗艾滋病和乙型肝炎的具有显著疗效的药物。拉米夫定是第一个经美国FDA以及中国食品药品监督管理局批准的口服抗乙肝病毒药物。由于拉米夫定问世时间早,很长一段时间都是临床药物研究的“宠儿”,又由于其临床相对比较安全,应用较为广泛。现阶段,拉米夫定是乙肝病毒感染的一线治疗药物。拉米夫定的缺点:拉米夫定易出现耐药现象。但并未影响该药的销售额。据中国医药商业协会最新报道,2003年全国 28家主要医药批发公司畅销药品中,拉米夫定排名第13位,销售额为3.06亿元,在46种抗感染药品中占4.33%。2002年贺普丁世界性销售额为2.95亿英镑,2003 年为2.93亿英镑,折合4.8亿美元。2003年,贺普丁在我国16城市样本医院销售额排名41位,用药金额8011万元,其中进口药品占60.62%,是英国葛兰素和WELLCOME(UK)两家公司,该药在全国市场的销售额约为8亿多元。2004年为7311.43万元,仍占全身用抗病毒药物的38%,而在全国医院销售额已达到6亿~8亿元的市场规模。2005年上半年样本医院购入药物是苏州葛兰素史克、英国葛兰素史克和WELLCOME(UK)公司的药物,各厂商分别占据了51.03%、47.92%和1.05%的份额。迄今为止,全球每年约有80万人使用贺普丁,拉米夫定目前在中国的终端销售额达到10亿元人民币左右,在中国药品市场单品销售排行榜上位列第二。 拉米夫定原料药及其制剂目前均为进口药品。目前原料药及其制剂,国内尚未有生产厂家上市销售,也未有公司进行国内新药申报。另外英国GlaxoSmithKline公司在中国分公司(葛兰素史克制药(苏州)有限公司)也在中国已经获得生产批件,批准文号:国药准字H20030581。目前国内有,拉米夫定原料药以及拉米夫定片(两种规格)、拉米夫定口服液(两种规格)两种进口剂型上市销售。
阿德福韦酯是由美国Gilead Science公司开发的新型核苷类抗乙型病毒性肝炎药物,已在国外进行了Ⅱ、Ⅲ期临床试验。国外的临床研究资料表明,阿德福韦能有效地抑制HBV DNA的复制,使HBV DNA滴度迅速降低,而且在出现拉米夫定耐药的患者中阿德福韦能继续有效地抑制变异株。我国药品监督管理局于2000年12月批准该药在中国进行临床试验,目前,Ⅰ期临床试验已结束,Ⅱ期临床试验也已在2002年12月正式启动。 一、作用机制 阿德福韦酯是腺嘌呤磷酸酯化合物阿德福韦的前药,其分子式为C20H32N5O8P,分子量为501.48。口服后,在体内转化为阿德福韦发挥抗病毒作用。阿德福韦是单磷酸腺苷的核苷酸类似物,在体内通过细胞激酶作用被磷酸化为具有活性作用的二磷酸阿德福韦,二磷酸阿德福韦抑制HBV DNA多聚酶或逆转录酶作用机制包括:(1)竞争脱氧腺苷三磷酸底物;(2)终止病毒DNA链延长。二磷酸阿德福韦对HBV DNA多聚酶的抑制常数为0.1mmol/L;对人类DNA多聚酶α和γ的抑制作用较弱,其抑制常数分别为1.18mmol/L和0.97mmol/L,因此,治疗剂量对正常细胞没有毒性。 二、药效和毒理 在体外实验中,阿德福韦抑制HBV转染人肝细胞瘤细胞株HepG2和HB611细胞病毒复制的半数抑制浓度(IC50)分别为0.2~2.5mmol/L和0.2~1.2mmol/L。二磷酸阿德福韦在细胞内的T1/2为30h,故作用较持久,可以每天给药一次。 拉米夫定耐药株涉及HBV DNA聚合酶M552V、M552I、L528M、L552M/M552V位点的突变。在体外实验中发现这些突变体对阿德福韦仍敏感,与野生株比较,它们的抑制常数增加了不到2.2倍,而拉米夫定对变异株的抑制常数则增加了8~25倍。这些资料表明,阿德福韦可以治疗对拉米夫定耐药的HBV,而且与拉米夫定联合治疗可以有效控制对拉米夫定的耐药。同时,体外实验也发现阿德福韦对泛昔洛韦耐药株也较敏感。 在体内实验中,发现阿德福韦能有效地抑制鸭乙型肝炎病毒和土拨鼠肝炎病毒(WHV)的复制。给予慢性感染WHV的成年土拨鼠每日口服5mg/kg、15mg/kg的阿德福韦或安慰剂治疗12周,治疗后5mg/kg剂量组WHV DNA水平减少了260倍,而15mg/kg剂量组则下降了1000倍以上。 四、国外临床研究进展 阿德福韦已在美国、欧洲、澳大利亚及东南亚进行了Ⅱ、Ⅲ期临床试验。临床试验涉及HBeAg阳性和考虑有前C区变异的HBeAg阴性的慢性乙型肝炎患者、对拉米夫定耐药的代偿性肝病患者、合并人类免疫缺陷病毒感染的拉米夫定耐药患者、肝移植前或移植后对拉米夫定耐药的失代偿性肝病患者。 早期进行的针对HBeAg阳性、ALT异常或正常的二项双盲、安慰剂对照的Ⅱ期临床试验,疗程为12周,并随访24周。ALT异常临床研究的患者,接受剂量为5mg/d、30mg/d和60mg/d。治疗12周后,5mg剂量组血清HBV DNA较基线下降1 Log10,30mg和60mg剂量组血清HBV DNA下降3~4 Log10,而安慰剂组无显著变化;36周后,30mg和60mg剂量组HBeAg转阴率为27%,HBeAg血清转化率为20%,血清转化率增高与基线时ALT水平呈正相关。ALT正常的临床研究患者,接受剂量为30mg/d。治疗12周后,30mg剂量组血清HBV DNA较基线下降3Log10,而安慰剂组无显著变化。所有接受治疗的患者在治疗12周后进行基因检测,没有发现与阿德福韦耐药有关的变异产生。在这二项研究中,30mg和60mg剂量组均出现部分患者的肾功能损害,表现为尿素氮和肌酐的升高,出现肾功能损害的比例与剂量呈正相关,故在以后的延续试验中以10mg剂量组而代替60mg剂量组。 在一项随机、双盲、安慰剂对照的临床试验中,共有515例HBeAg阳性的患者进入研究。在前48周,患者被随机分入阿德福韦30mg组(173例)、阿德福韦10mg组(172例)或安慰剂组(170例)。48周后,30mg组接受安慰剂治疗至96周,安慰剂组接受阿德福韦10mg治疗至96周,10mg组则再次随机按1:1接受安慰剂或继续阿德福韦10mg治疗至96周。所有患者在第一次随机前6月内接受第一次肝活检,在治疗48周、96周后接受第二、三次肝活检。所有患者治疗96周后随访24周。治疗48周后,10mg组和30mg组组织学改善率(组织学改善定义为Knodell坏死炎症计分下降32分,且Knodell肝纤维化计分无恶化)分别为53%和59%,显著高于安慰剂组25%;10mg组和30mg组治疗后血清HBV DNA较基线时下降3.52 Log10和4.76 Log10,安慰剂组为0.55 Log10;10mg组HBeAg阴转率为24%,HBeAg血清转化率为12%,显著高于安慰剂组的6%和11%;10mg组ALT复常率为48%,安慰剂组则为16%。研究中,发现基线ALT水平与肝组织学改善和HBeAg血清转化呈正相关。另一项随机、双盲、安慰剂对照的临床试验,共有185例考虑有前C区变异的HBeAg阴性的患者按2:1比例进入阿德福韦10mg组或安慰剂组。治疗48周后,10mg组组织学改善率为64%,显著高于安慰剂组33%;10mg组治疗后血清HBV DNA较基线时下降3.91 Log10,51%患者HBV DNA转阴,安慰剂组HBV DNA较基线时下降1.35 Log10,没有患者HBV DNA转阴;10mg组ALT复常率为72%,著高于安慰剂组29%。目前本项研究仍在进行中。 五、耐药和病毒变异 阿德福韦较少产生耐药的分子学基础包括:(1)阿德福韦与自然底物dATP在结构上非常相像;(2)阿德福韦具有灵活的开链连接;(3)具有磷酸键。 629例患者在治疗48周后接受了病毒变异的检测,结果未发现产生阿德福韦耐药的病毒变异。2003年美国肝病年会上,Gilead公司报道,238例患者在治疗96周时有4例发现N236T位点的变异,发生率为1.7%,并证实N236T变异与阿德福韦耐药有关。另一可能与阿德福韦耐药有关的A181V位点突变,96周时的发生率为0.8%。另外一项研究是早期进行的针对HBeAg阳性、ALT异常或正常的二项双盲、安慰剂对照研究的延续。患者在治疗中没有出现血清转化,也没有出现与治疗相关的毒性反应,患者自愿继续接受治疗。剂量开始为30mg/d,后改为10mg/d。在长达136周的观察中,阿德福韦对野生株和前C区变异的慢性乙型肝炎具有持续的抗病毒作用,而且没有发现与阿德福韦耐药相关的病毒变异。 七、国内的研究状况 我国食物药品监督管理局于2000年12月批准该药在中国进行Ⅰ期临床试验。2001年6月~9月进行Ⅰ期临床试验,Ⅰ期临床试验包括3个研究方案:(1)在健康中国男性志愿者中,对单次口服阿德福韦片剂的安全性和耐受性进行评估的一项Ⅰ期、单中心、随机、双盲、安慰剂对照的研究;(2)在健康中国男性志愿者中,对阿德福韦片剂的药代动力学进行评估的一项Ⅰ期、单中心、开放、拉丁方设计的研究;(3)在健康中国志愿者中,就连续6d,1次/d,口服阿德福韦片剂的安全性、耐受性和药代动力学进行评估的一项Ⅰ期、单中心、随机、双盲、安慰剂对照的研究。I期研究结果显示在健康中国志愿者中口服阿德福韦片剂的安全性、耐受性良好;10mg剂量下,未观察到肾功能损害;药代动力学参数与国外研究结果相似。2002年10月国家药品监督管理局批准该药在中国进行Ⅱ期临床试验。Ⅱ期临床试验在中国的总病例数为480例,均为HBeAg阳性、HBV DNA阳性、ALT增高的患者。全国有12个中心参与。目前,Ⅱ期临床试验已在2002年12月正式启动,2003年2月底已完成最后一例患者入组。
我需要拉米夫定的标准红外谱图!自己做了一张,但是和客户的那一张对不起来,所以恳请好心人帮忙了.如果有此标准谱图的请发到我的邮箱:wuai-111@163.com先谢谢了!
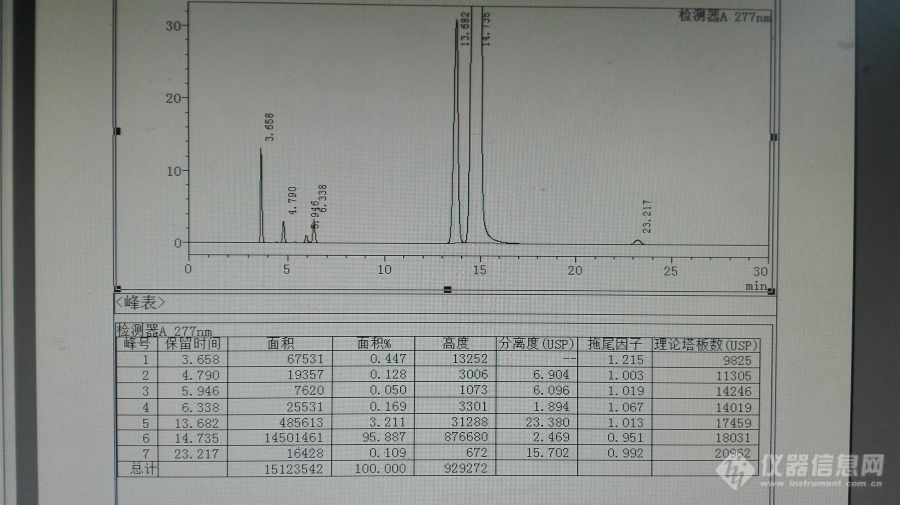
来自中国的自信,月旭Welchrom C18测定拉米夫定有关物质[b]除非经大量试验比较市面主流厂家的色谱柱均不能达到要求,最好不要把色谱柱的品牌定下来。[/b]拉米夫定(Lamivudine)对乙型肝炎病毒和HIV有明显的抑制作用。口服吸收后,拉米夫定可在HIV感染细胞和正常细胞内代谢生成拉米夫定三磷酸盐,它是拉米夫定的活性形式。后者通过竞争抑制作用,终止DNA链的延长,从而抑制HIV和HBV的反转录酶和HBV聚合酶,阻止HIV和HBV的DNA合成和病毒复制。体外实验中与齐多夫定联合,对HIV病毒有协同作用。下面为中国药典2015年版收载的质量标准,其中提到的色谱柱,我们咨询了价格比较贵,我们就用手上的月旭色谱柱进行了探索研究。参照了EP8.0质量标准,进行了系统适用性试验,其中EP8.0质量标准有关物质项下为:Related substances. Liquid chromatography (2.2.29).Test solution. Dissolve 50.0 mg of the substance to beexamined in the mobile phase and dilute to 100.0 mL withthe mobile phase.Reference solution (a). Dilute 1.0 mL of the test solution to100.0 mL with the mobile phase. Dilute 1.0 mL of this solutionto 10.0 mL with the mobile phase.Reference solution (b). Dissolve 5 mg of salicylic acid R in themobile phase and dilute to 100.0 mL with the mobile phase.Dilute 1.0 mL of the solution to 100.0 mL with the mobilephase.Reference solution (c). Dissolve 50.0 mg of lamivudine CRSin the mobile phase and dilute to 100.0 mL with the mobilephase.Reference solution (d). Dissolve 5 mg of cytosine R and 5 mg ofuracil R in the mobile phase and dilute to 100.0 mL with themobile phase. Dilute 2.0 mL of the solution to 10.0 mL withthe mobile phase.Reference solution (e). Dissolve 5 mg of lamivudine for systemsuitability 1 CRS (containing impurities A and B) in 2 mL ofthe mobile phase. Add 1.0 mL of reference solution (d) anddilute to 10.0 mL with the mobile phase.Column:- size: l = 0.25 m, Ø = 4.6 mm - stationary phase: base-deactivated octadecylsilyl silica gel forchromatography R (5 μm) - temperature: 35 °C.Mobile phase: mix 5 volumes of methanol R and 95 volumes ofa 1.9 g/L solution of ammonium acetate R, previously adjustedto pH 3.8 with glacial acetic acid R.Flow rate: 1.0 mL/min.Detection: spectrophotometer at 277 nm.Injection: 10 μL.Run time: 3 times the retention time of lamivudine.Identification of impurities: use the chromatograms obtainedwith reference solutions (b) and (e) to identify the peaks dueto impurities A, B, E, F and C.[b]Relative retention with reference to lamivudine (retentiontime = about 9 min): impurity E = about 0.28 impurity F = about 0.32 impurity A = about 0.36 impurity B = about 0.91 impurity J = about 1.45 impurity C = about 2.32.[/b]System suitability: reference solution (e):- resolution: minimum 1.5 between the peaks due toimpurities F and A minimum 1.5 between the peaks dueto impurity B and lamivudine.Limits:- correction factors: for the calculation of content,multiply the peak areas of the following impurities bythe corresponding correction factor: impurity E = 0.6 impurity F = 2.2 impurity J = 2.2 - impurity A: not more than 3 times the area of the principalpeak in the chromatogram obtained with referencesolution (a) (0.3 per cent) - impurity B: not more than twice the area of the principalpeak in the chromatogram obtained with referencesolution (a) (0.2 per cent) - impurity C: not more than the area of the principal peakin the chromatogram obtained with reference solution (b)(0.1 per cent) any other impurity: for each impurity, not more than thearea of the principal peak in the chromatogram obtainedwith reference solution (a) (0.1 per cent) - total: not more than 6 times the area of the principal peakin the chromatogram obtained with reference solution (a)(0.6 per cent) - disregard limit: 0.5 times the area of the principal peak inthe chromatogram obtained with reference solution (a)(0.05 per cent).中文名拉米夫定外文名lamivudineCAS号134678-17-4分子式C8H11N3O3S[img=,690,400]http://ng1.17img.cn/bbsfiles/images/2017/11/201711302020_01_1621890_3.png!w690x400.jpg[/img][img=,690,388]http://ng1.17img.cn/bbsfiles/images/2017/11/201711302026_01_1621890_3.png!w690x388.jpg[/img][img=,690,388]http://ng1.17img.cn/bbsfiles/images/2017/11/201711302027_01_1621890_3.png!w690x388.jpg[/img][img=,690,388]http://ng1.17img.cn/bbsfiles/images/2017/11/201711302028_01_1621890_3.png!w690x388.jpg[/img][img=,690,388]http://ng1.17img.cn/bbsfiles/images/2017/11/201711302029_01_1621890_3.png!w690x388.jpg[/img]以上均为参照中国药典2015年收载的拉米夫定片质量的截图,然后进行系统适用性试验,进行流动相微调,其典型色谱图见下图:[img=,690,353]http://ng1.17img.cn/bbsfiles/images/2017/11/201711302137_01_1621890_3.png!w690x353.jpg[/img]初步统计色谱峰保留时间,其他色谱图参数很好,理论板数都超级好,分离度和拖尾因子也超级好! [table=167][tr][td]相对保留时间(RRT)[/td][td]保留时间(min)[/td][/tr][tr][td]0.25 [/td][td]3.658[/td][/tr][tr][td]0.33 [/td][td]4.79[/td][/tr][tr][td]0.40 [/td][td]5.946[/td][/tr][tr][td]0.43 [/td][td]6.338[/td][/tr][tr][td]0.93 [/td][td]13.682[/td][/tr][tr][td]1.00 [/td][td]14.735[/td][/tr][tr][td]1.58 [/td][td]23.217[/td][/tr][/table]其中水杨酸的色谱峰如下:[img=,690,338]http://ng1.17img.cn/bbsfiles/images/2017/11/201711302144_01_1621890_3.png!w690x338.jpg[/img]另外最好控制柱温,最好是液相自动控温装置,不要外接质量差的柱温箱,因为在升温的过程有±5℃的差异,会引起水杨酸色谱峰保留时间和峰型变化较大,使其用相对保留时间定位出现偏差,建议用水杨酸对照品同时定位,因为其价格便宜易得,其典型色谱图如下:[img=,690,320]http://ng1.17img.cn/bbsfiles/images/2017/11/201711302159_01_1621890_3.png!w690x320.jpg[/img]拉米夫定对照品定位溶液主峰典型色谱图如下:[img=,690,405]http://ng1.17img.cn/bbsfiles/images/2017/11/201711302150_01_1621890_3.png!w690x405.jpg[/img][img=,690,409]http://ng1.17img.cn/bbsfiles/images/2017/11/201711302211_01_1621890_3.png!w690x409.jpg[/img]以上拉米夫定对照品出峰保留时间基本上变化不大,但是浓度较大是有较大差异,15分钟在14.4分钟变化。其变化情况见下面系统色谱图。但是其相对于水杨酸的变化幅度较小。系统进针另外图谱:[img=,690,375]http://ng1.17img.cn/bbsfiles/images/2017/11/201711302207_01_1621890_3.png!w690x375.jpg[/img][img=,690,415]http://ng1.17img.cn/bbsfiles/images/2017/11/201711302209_01_1621890_3.png!w690x415.jpg[/img]总结:我们没有用到中国药典的色谱柱,但是从色谱图及其系统适用性试验结果,其系统各杂质均能有效分离,和EP8.0提到的主峰保留时间差距很大,约6分钟差异,估计是色谱柱填料性能差异引起,从研究角度来讲,[b]中国产的色谱柱完全适用于本品的有关物质检测和含量测定,为我们后续的研究增强了自信心。作为权威的中国药典,不知道什么原因把色谱柱的品牌给定下来了,个人愚见,除非经大量试验比较市面主流厂家的色谱柱均不能达到要求,最好不要把色谱柱的品牌定下来。作为国产优良色谱柱的生产商月旭公司不得不由衷给个赞![/b]

LC-MSMS做拉米夫定标准品,不知为啥一直有个明显肩峰。图谱在附件里http://ng1.17img.cn/bbsfiles/images/2013/09/201309051141_462516_2167114_3.bmp
[center]盐酸芬氟拉明全国叫停 北京未招标采购该药[/center]由于会引发心脏损害等严重不良反应,昨天(8日),国家食品药品监管局紧急叫停对减肥药盐酸芬氟拉明原料药和制剂的生产、销售和使用,并表示已购买盐酸芬氟拉明的患者可将剩余的药品退回原购买医院或药店。 药监局新闻发言人颜江瑛昨日(8日)在例行新闻发布会上介绍,国家药品不良反应监测中心的监测数据表明,使用盐酸芬氟拉明可引起心脏瓣膜损害、肺动脉高压、心力衰竭、心动过速、心慌、胸闷、血尿、皮疹、恶心、头晕等严重不良反应。 颜江瑛说,国家药监局组织专家对该品种进行了综合评价,认为国内外监测和研究资料表明,该药品用于减肥风险大于利益。 为保障公众用药安全,根据《药品管理法》和《药品管理法实施条例》,国家药监局决定自1月7日起,停止盐酸芬氟拉明原料药和制剂在我国的生产、销售和使用,撤销其批准证明文件。已上市销售的药品由生产企业负责召回,在所在地药品监督管理部门监督下销毁或者处理。 资料显示,早在上世纪末、本世纪初,欧美国家已经先后停止了盐酸芬氟拉明在本国家(地区)销售和使用。 - 名词解释 盐酸芬氟拉明 盐酸芬氟拉明作为一种化学减肥药,主要用于单纯性肥胖及患有糖尿病、高血压、心血管疾患、焦虑症的肥胖患者,是一种食欲抑制剂。盐酸芬氟拉明在中国目前只有一个品种,即盐酸芬氟拉明片。中国于上世纪80年代批准盐酸芬氟拉明片及其原料药在中国制售。目前共有30多家企业持有该药品的批准文号。 - 北京情况 北京未招标采购盐酸芬氟拉明 市药监局表示,医保药品目录中无该药品 昨天(8日),北京市药品监督管理局相关负责人表示,盐酸芬氟拉明片是一种作为二类精神药品管理的处方药,但并非北京地区医疗机构定点招标采购药品,更不在医保药品目录中。 记者昨天(8日)走访金象大药房、嘉事堂连锁药店和老百姓大药房,店方均由于不良反应较多,盐酸芬氟拉明片早已在药店消失。 - 相关新闻 药监局表示“山寨药”根本不是药,无药品功能 “不要用山寨药说法误导公众” 昨天(8日),国家药监局新闻发言人颜江瑛在回答本报提问时表示,号称“山寨药”的非药品本身就不是药品,建议不要用“山寨药”这种说法来误导公众。 在新闻发布会上,本报记者提问:现在有媒体报道中用的比较火的一个词是“山寨药”,“山寨药”到底是不是药品? 对此,颜江瑛表示,“我不太赞同“山寨药”的说法,我建议不要用“山寨药”这种说法来误导公众。” 颜江瑛说,现在有一些山寨手机,某种程度手机还是一个手机,但是所说的“山寨药”根本不是药,绝对没有药品的功能,甚至会误导公众、影响公众的健康,延误病情的治疗,“我不同意大家用“山寨药”的说法。” 信息来源:新京报
旋光性物质的核磁与相应外消旋物质有什么不同?
一個分析方法要選擇使用內標或外標法來定量,並沒有一定的規定,所以這裡就嘗試來分析一下, 如何找到一個合適的定量方式, 怎麼去選擇一個內標準品 .........內標準品的選擇 傳統上在教科書中都會很模糊的告訴學生內標準的選擇方式,例如說要選安定性好 與分析物性質要相近 在分析的基質中不能出現等,現在我對這些個" 選擇方式"沒有太大的興趣,因為這個大家都知道,那現在就從另一個角度的來看看內標準品的選擇還有哪些需要注意的.1. 只選一個內標準品? 當然, 如果你的分析目標物就只有一個, 在正常的狀況下,內標準"應該"也只會有一個才對! 但是如果你的分析是多成份的,那就必須十分小心地看待在一個分析方法內標準品的選擇, 如果分析物的在層析圖中是平均分布在各處, 那你就必須看看你的檢測方法中是否有規定內標物與分析物之間的滯留時間差範圍是多少,依此規定來選擇內標,但是如果並沒有規定,最好也是選擇一個以上的內標來使用,因為即使化學性質不會差太多,但在沸點方面卻會有滿大的差異, 內標與分析物的沸點差異過大,在GC的注射口中就無法把因為discrimibation所造成的誤差校正回來. 如果你的分析物是性質相差頗大的 (例如說同時含有醇,酸...),那別懷疑一定是要使用一個以上的內標準品,如果多個物種再加上多成份,那就很複雜了. 最低的限度也要依照分析物的沸點高低來使用多個不同的內標準品. 在美國環保署的檢驗方法USEPA 8270C,是一個檢測半揮發性的污染物的規範,前後列了不下一百種的分析物,就使用了六個不同的內標準品作為校正的依據,來照顧到各個不同沸點的分析物!! 濫用藥物分析大概是最嚴謹的了,即使是結構性質極相近的分析物,例如morphine和codeine,amphetamine和methamphetamine,在分析時為求準確,都是以各自的D同位素取代的標準品作為內標.2.基質中一定不能存在? 這個問題當然是肯定的, 不然定量結果會很不穩定或者是很淒慘. 但是有些時候你根本不知道哪些東西在分析樣品的基質中不會存在! 這時候怎麼辦? 找以往的文獻看看別人是用甚麼,這是一個方法, 但要注意的是文獻不一定就是對的! 使用分析物的氫同位素(D)取代物,是最妥當的,但問題是價格昂貴,而且不是每一種分析物的氫同位素(D)取代物都有販售,當然,如果本錢夠,你可以去訂購專門替你合成氫同位素(D)取代物. 如果要自己選, 那就必須了解一下分析物的成分了. 以前曾經替人定性和定量過一陣子海水魚中所含不飽和脂肪酸,這魚中不飽和脂肪酸的碳數都是奇數(或是偶數, 我已經忘了), 所以內標就使用了幾個偶數(或奇數)的不飽和脂肪酸, 像這種內標物絕不可能出現在分析物中,且性質十分相近,所以定量的結果一般誤差都不會太大! 如果完全無法確定怎辦? 有時候就是賭一賭囉.........舉例來說:如果你的分析物結構中含有氯的話,就可以尋找一個化合物,而這個化合物是把其中的氯換成氟或溴,例如你可以使用2-氟聯苯或2-溴聯苯來當作分析2-氯聯苯的內標準品. 以環境分析為例,通常在自然界中的氟及溴化物並不多, 在EPA的方法中,就經常把結構相似的氟及溴化物添加入樣品中,來做為分析含氯化合物的QC樣品.所以找氟及溴來取代氯原子的化合物來作為內標,基本上還算是很安全的. 如果實在連上述轉換一個基團的內標都找不到,那至少在選擇時一定要找同一類的,分析酸就找酸當內標,分析醇就找醇當內標,直鏈結構分析物就不要找一個環狀的化合物來當內標,分子量不要相差太大,這算是最基本的要求!!3.內標和分析物的滯留時間必須接近? 這個說法原則上沒錯,因為如果滯留時間接近,它們的物理或者是化學的性質在某種程度上是接近的,所以有些分析方法會有這樣的規定. 但在某些特殊的狀況下卻出了問題, 事實上還真的碰過, 結果在更換到第三個內標準品後, 它的RT遠離分析物, 反而解決了定量誤差的問題!! 這個案例以後有空在來聊聊. 4.內標的濃度應該是多少? 除了某些分析方法會規定必須加入多少濃度的內標外,其他的就只能靠分析員自己來決定了. 以我為例, 通常會先決定一個分析方法的檢量線範圍,然後開始配製不同濃度的內標準品來注射入儀器中,分析完後選出一個大概是檢量線最高濃度的波峰高度三分之二左右的濃度,作為該項分析的內標濃度. 這樣的選擇方式,可以兼顧到高低濃度的需要,一般來說,內標濃度過高除了會增加成本之外,對於低濃度的校正是會產某些程度的誤差. 在某些分析方法中會強制妳使用內標法而不是外標法或線性迴歸的方式來定量,但是如果你的基質很複雜,一針打進儀器中,結果大大小小的出來一堆波峰, 這個時候如果你又不是使用質譜作為偵測器的話,就必須考慮加大內標準品的使用濃度, 來降低內標準品的可能發生的積分誤差!!5.內標準怎麼配製? 一但決定了添加內標準品的濃度,就可以開始配製分析時所用的內標準品標準溶液,相對於測試時的小量, 分析員必須先估計妳在這一批的分析樣品有多少, 然後計算整個分析需要添加入多少的量,例如說妳需要大概100mL的內標準品標準溶液,這時就必須再加多30%以上,一次就配好整個分析要用的量! 分裝或者是整瓶放到-20度的冰箱中存放,免得做到一半發覺內標準品標準溶液用完了,再重新配製一次,如果你是分析的新手, 搞不好前後兩次配得不一樣濃度,那就會很悽慘了…..有些領域的分析,很重視這種內標波峰的積分值是否是有一定的再現性!!講完了內標準品, 下次就可以來看看內標法和外標法了………..
1.药物鉴别:具有旋光性的药物,在“性状”项下,一般都收载有“比旋度”的检验项目。测定比旋度值可用来鉴别药物或判断药物的纯杂程度。《中国药典》要求测定比旋度的药物很多,如肾上腺素、硫酸奎宁、葡萄糖、丁溴东莨菪碱、头孢噻吩钠等。 例如,肾上腺素比旋度的测定方法为:取本品,精密称定,加盐酸溶液(9→200)溶解并定量稀释制成每1ml中含20mg的溶液,照“旋光度测定法”测定,比旋度应为。 2.杂质检查:具有光学异构体的药物,一般具有相同的理化性质,但其旋光性能不同,一般有左旋体、右旋体和消旋体之分,通过测定药物中杂质的旋光度,可以对药物的纯度进行检查。 例如,硫酸阿托品中杂质莨菪碱的检查,硫酸阿托品为莨菪碱的外消旋体,无旋光性,而莨菪碱为左旋体,莨菪碱虽然作用较强,但毒性也大,常将其作为杂质加以控制。 检查方法为:取本品,按干燥品计算,加水制成每1ml中含50mg的溶液,依法测定,旋光度不得超过。已知莨菪碱的比旋度为,控制莨菪碱的限量为24.6%. 考试大-全国最大教育类网站(www.Examda。com) 3.含量测定:具有旋光性的药物,特别是在无其他更好的方法测定其含量时,可采用旋光度法测定。《中国药典》采用旋光度法测定含量的药物有葡萄糖注射液、葡萄糖氯化钠注射液、右旋糖酐氯化钠注射液、右旋糖酐葡萄糖注射液等。
如果中间产物有一个手性碳,并不分离,是外消旋体,环合之后,又出现一个手性碳, 上面两个不同的氢,是会出现两个小峰吗?对其他的影响呢?谢谢!!
下記農薬について、食品中の残留基準を設定・イソキサフルトール(Isoxaflutole,异恶唑草酮,用途:除草剤)・イマザピック(Imazamethapyr,甲基咪草烟; 甲咪唑烟酸,用途:除草剤)※・エタルフルラリン(Ethalfluraline,丁氟消草,用途:除草剤)・フェンブコナゾール(Fenbuconazole,腈苯唑,用途:殺菌剤)・フロニカミド(FLONICAMID,氟啶虫酰胺,用途:殺虫剤)・ぺノキススラム(Penoxsulam,五氟磺草胺,用途:除草剤)・マンジプロパミド(Mandipropamid,双炔酰菌胺,用途:殺菌剤)※今回基準値を設定するイマザピックはイマザピックアンモニウム塩として暫定基準が設定されていたため、イマザピックアンモニウム塩として経過措置を設定しているが、各種試験はイマザピックを用いて実施されていること、海外における基準値はイマザピックの残留量を考慮して設定されていることから、今後は告示においては、イマザピックアンモニウム塩は「イマザピック」とする。・フェンブコナゾール:かき等6食品・フロニカミド:小豆等27食品・ぺノキススラム:ぶどう等5食品・マンジプロパミド:だいこん類(ラディッシュを含む。)の葉等7食品・イソキサフルトール:米(玄米をいう。)等7食品・イマザピック:豚の筋肉等17食品・エタルフルラリン:きゅうり(ガーキンを含む。)等9食品・フロニカミド:羊の筋肉等15食品・イソキサフルトール:とうもろこし等19食品・イマザピック:牛の脂肪等9食品・フェンブコナゾール:みかん等10食品・フロニカミド:クレソン等32食品・マンジプロパミド:はくさい等20食品≪施行・適用期日≫ 平成24年6月14日 ※ただし、下記の農薬等ごとに掲げる食品に係る残留基準値については、 平成24年12月14日から適用。 ◆イソキサフルトール 米、小麦、大麦、ライ麦、とうもろこし、そば、その他の穀類、 その他のスパイス、豚の肝臓、その他の陸棲哺乳類に属する動物の肝臓、 乳、鶏の卵及びその他の家きんの卵 ◆イマザピック 豚の筋肉、豚の脂肪、豚の肝臓、豚の腎臓、豚の食用部分及び乳 ◆エタルフルラリン きゅうり、かぼちゃ、しろうり、すいか、メロン類果実、まくわうり、 その他のうり科野菜、えだまめ及びべにばなの種子
木犀草素(luteolin),别称草木犀、黄示灵等,大多以糖苷的形式广泛存在于多种中药材、天然药用植物[1]及蔬菜[2]中的一种黄酮类化合物,是一种天然色素成分,可以作为食用色素添加于食品中。木犀草素的化学名为3′,4′,5,7-四羟基黄酮(3′,4′,5,7-tetrahydroxyflavone),物理状态为淡黄色结晶状粉末,熔点为330 ℃,包含4个酚羟基,具有弱酸性,可溶于碱性溶液中,因脂溶性高而难溶于水,从而阻碍了其在体内的吸收与利用[3]。木犀草素具有抗炎和抗菌[4-5]、抗氧化[6]、抗肿瘤[7]、神经保护[8]、抑制肺纤维化[9]及肺癌[10-11]和心血管疾病[12]等多种药理作用。由于水溶性差(仅为6.0 mg/L)、生物利用度率低等原因限制了其成药性和临床应用。针对这一问题,近年来许多学者开展了增加木犀草素溶解度的研究,如微球[13]、纳米胶束[14]、金属配合物[15]、自微乳[16]、脂质体[17]等,并明显提高了其生物利用度,这表明木犀草素的肠道渗透性不是限制其生物利用度的关键因素,其属于生物药剂学系统II类药物。因此,采用制剂技术提高木犀草素的溶解性是可以改善其成药性和生物利用度的,将有利于推广其临床应用。然而上述开发的剂型仍存在诸多的缺点,如工艺复杂、载药量低、生物安全性差、成本高等,难以大范围推广应用。近年来,逐步发展成熟的纳米混悬剂[18]作为一种新剂型,与传统纳米制剂相比,它具有载药量高、溶出度高、添加剂用量少、易于放大生产等优点。因此,本实验尝试将难溶性木犀草素制备成纳米混悬剂以提高其水溶性和生物利用度,改善其成药性和临床优势。 为此,本实验首先采用微沉淀-高压匀质法制备口服木犀草素纳米混悬剂(luteolin nano-suspension,LNS),并以纳米粒的粒径、稳定性、多分散性指数(polydispersity index,PDI)、ζ电位等为考察指标,采用单因素考察法筛选LNS的稳定剂和最优药物-稳定剂比;接着,对LNS的理化性质进行考察,并分析其物理状态和体外溶出行为;最后通过大鼠外翻肠模型考察药物在肠道不同部位的吸收转运情况,探索药物在肠道内的吸收速率和最佳部位,预测纳米混悬剂可能存在的体内吸收行为,既可以用于木犀草素口服给药的潜在剂型,也为其进一步加工成其他剂型研究提供基础。 1 仪器与材料 1.1 仪器 ZNCL-BS180型恒温磁力搅拌器,北京市永光明医疗仪器有限公司;AL104-1C型精密分析天平,上海鼎科科学仪器有限公司;NS1001L型高压匀质机,意大利GEA [url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]NIR[/color][/url]o Soavi公司;Nanotrac wave II型激光粒度仪型激光粒度仪,美国麦奇克有限公司;LC3100型高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱仪[/color][/url],安徽皖仪科技股份有限公司;ZWY-103D型恒温振荡仪,上海智诚分析仪器制造有限公司;H1650-W型医用离心机,湖南湘仪实验室仪器开发公司;DZF-6030型真空干燥箱,上海精宏实验设备有限公司。JEOL 2010型透射电子显微镜(TEM),日本JEOL公司。 1.2 试剂 木犀草素原料药,批号JZ19021403,质量分数97.0%,南京狄格尔医药科技有限公司;木犀草素对照品,批号ps1032-0025,HPLC质量分数≥98%,成都普思生物科技有限公司;十二烷基磺酸钠(sodium dodecyl sulfonate,SDS),医药级,河南圣拓实业有限公司;泊洛沙姆188(Poloxamer 188,Pluronic,F68),医药级,西安天正药用辅料有限公司;维生素E聚乙二醇琥珀酸酯(D-α-tocopherol polyethylene glycol 1000 succinate,TPGS),医药级,上海惠诚生物科技有限公司;二甲基亚砜(dimethyl sulfoxide,DMSO),分析纯,天津市德恩试剂有限公司。 1.3 动物 SD大鼠购买于河南省实验动物中心,体质量(200±20)g,合格证号:SCXK(豫)2017-0001。所有动物实验均经过河南大学动物伦理委员会审核批准(HUSOM2019-216)。 2 方法与结果 2.1 LNS的制备 2.1.1 LNS中稳定剂的选择 将40 mg木犀草素原料药超声溶解于1 mL的DMSO中作为有机相,再取等量的稳定剂(SDS、F68、TPGS)溶解于纯水中(作为水相,或称反溶剂相);在室温下,将有机相通过注射器快速注入转速为1 800 r/min的反溶剂相中,继续搅拌10 min,得到预混悬剂;将预混悬剂转移至高压匀质机中,分别以20.0、50.0、80.0 MPa的压力循环匀质5、5、25次,得到LNS。 利用动态光散射仪分别考察LNS的粒径、多分散系数(polydispersity index,PDI)、表面电荷(ζ电位)和稳定性。本实验以不同稳定剂(SDS、F68、TPGS)制备的LNS粒径大小、PDI、ζ电位结果如表1所示。3种稳定剂所制备的粒径均在100~500 nm。以SDS为稳定剂制备的纳米混悬剂粒径最大,以F68为稳定剂制备的纳米混悬剂PDI最大,以TPGS为稳定剂制备的纳米混悬剂ζ电位最大,但是3者没有较大的差异,因此对于预测稳定性来说,上述结果难以判断哪个稳定剂制备的LNS会有良好的贮存稳定性。 因此,本实验又对各种条件的贮存稳定性进行了研究,结果见图1。以SDS、F68为稳定剂制备的纳米混悬剂在1周内粒径呈现持续增长的趋势,而以TPGS为稳定剂制备的LNS粒径未出现明显变动,由此可知,本实验中以TPGS为稳定剂制备的LNS具有较好的物理稳定性。 2.1.2 LNS中药物-稳定剂质量比的筛选 将40 mg的木犀草素原料药超声溶解于1 mL的DMSO中作为有机相,再分别按照木犀草素与TPGS的质量比为1∶2、1∶1、2∶1称取TPGS,溶解于水中,得到反溶剂相;再按上述工艺制备LNS,得到不同药物-稳定剂质量比的LNS。利用动态光散射仪分别考察纳米混悬剂的粒径、分布、ζ电位和稳定性。不同药物-稳定剂比制备的LNS的理化性质研究结果见表2和图2。如表2所示,3种不同药物-稳定剂比制备的LNS的粒径分别为(289.3±6.6)、(210.7±2.0)、(34.6±3.7)nm,3种LNS的PDI接近,1∶2时ζ电位最大,2∶1时ζ电位没测到。虽然药物与稳定剂的质量比为2∶1时,其粒径与1∶2、1∶1时相差较大,但是粒径难以反映稳定性情况。因此,接下来考察了1∶2、1∶1、2∶1 3种不同比例下制备的LNS的稳定性,结果如图2所示。当药物-稳定剂比为2∶1和1∶2时,在2周内粒径变化幅度都较为明显,说明其稳定性表现均极差;而当药物-稳定剂比为1∶1时,制备的纳米混悬剂的粒径基本保持稳定,表明其稳定性较好。因此,本实验最终选用药物-稳定剂比为1∶1。 2.1.3 最优制备处方和方法的确定 依照LNS的稳定剂及药物-稳定剂比的筛选结果,初步确定LNS的最优制备处方与方法如下:将精密称取40 mg的木犀草素原料药超声溶解于1 mL的DMSO中作为有机相;将40 mg TPGS搅动溶解于40 mL纯水中作为水相,将有机相快速注入转速为1 800 r/min的水相中,搅动10 min,得到预混悬剂;将制备的预混悬剂倒入高压匀质机的导入槽中,分别以20.0、50.0、80.0 MPa的压力,分别循环匀质5、5、25次,得到LNS。重复制备3批,以粒径、PDI和ζ电位考察制剂处方和制备工艺的稳定性。 2.2 LNS的表征 2.2.1 粒径、ζ电位及形态分析 将最优处方制备的3批LNS分别通过激光粒度分析仪测定其粒径、PDI、ζ电位,结果LNS的粒径为(209.00±3.24)nm(n=3),PDI都低于0.228±0.013(n=3),粒径分布图见图3;ζ电位值为(?16.80±0.27)mV (n=3),较小的PDI和绝对值较大的ζ电位,意味着LNS可能具有较好的长期稳定性[19]。 再取适量的LNS加蒸馏水稀释到适当倍数后,滴在覆有支持膜的铜网上,自然环境下干燥后,通过TEM观察其形态特征及大小,并成像,结果见图4。LNS呈现均匀分散的球形或椭圆形颗粒,粒径约为180 nm,比动态光散射测定结果较小,这可能是由于TEM样品为干燥品,导致粒子外层亲水部分失水而收缩[20]。 2.2.2 储存稳定性 将制备的LNS分别放在4 ℃和室温环境中,在预定的时间点取样,通过激光粒度分析仪测定其粒径和PDI,连续考察14 d,每个样品平行操作3份,结果见表3。LNS在4 ℃和室温下储存2周后,粒径和PDI稍有增加,但变化范围都较小,说明该LNS的储存稳定性较好。 2.2.3 体外胃肠环境中的稳定性 以pH 1.2和pH 6.8的缓冲溶液模拟胃液和肠液,将制备的LNS分别以1∶1与上述2种缓冲溶液混合,并于37 ℃水浴中放置,在预定的时间点0、2、4、6、8、12、24 h时取样,通过激光粒度分析仪测定其粒径,连续考察24 h,每个样品平行操作3份,结果见表4。在2种37 ℃的缓冲溶液中孵育24 h内,LNS的粒径和PDI几乎无变化,表明LNS在2种环境中能保持稳定,这表示LNS口服给药后,在经胃肠道给药时能保持良好的稳定性,这有利于木犀草素到达肠道后仍以纳米晶存在,从而有利于木犀草素的快速释放而获得较高的生物利用度。 2.2.4 纳米混悬剂的物理状态研究 本实验选用DSC来确定LNS中的木犀草素晶型是否发生了改变,测试样品有木犀草素、TPGS、木犀草素与TPGS的物理混合物和LNS。以空铝盘作为空白对照,分别精密称取3~5 mg的木犀草素、TPGS、物理混合物(木犀草素+TPGS)、LNS干粉放于差式扫描量热分析(differential scanning calorimetry,DSC)仪中,N2流(40 mL/min)保护下,以10 ℃/min升温速度持续升温,升温范围设置为40~600 ℃,记录差式扫描量热分析图谱,所有测试样品重复分析3批,结果见图5。木犀草素和LNS、物理混合物均是结晶,其熔融温度为339.38 ℃,稳定剂对木犀草素的熔融温度基本无影响。这表明LNS中的木犀草素仍处于结晶状态,稳定剂的存在不会改变木犀草素的晶型。在木犀草素和LNS中,在50~150 ℃出现了1个宽峰,这可能是由于药物吸收了水分造成的。 再分别称取适量的木犀草素、TPGS、物理混合物(木犀草素+TPGS)、LNS置于X射线粉末衍射(X-ray powder diffraction,XRPD)仪中,以步进测定方式,散射角扫描范围设为5°~60°,电压设为40 kV,电流为30 mA,结果见图6。由图6可知,木犀草素在19.12、23.20、26.32 ℃有3个衍射峰,衍射峰的峰形较为尖锐,峰值较高,表明木犀草素的晶型为结晶型。稳定剂TPGS在15.72、17.48、22.86、25.60、29.26 ℃有衍射特征峰。制备成纳米混悬剂后,虽然LNS图谱中木犀草素的特征峰有所减弱,但与木犀草素相比,在相应位置特征峰均存在,进一步证实制备成LNS后木犀草素并未显著改变晶型,说明稳定剂的加入不会影响木犀草素的晶型,这与DSC分析的结果一致。 2.3 平衡溶解度与过饱和溶出度测试 为了测定木犀草素的平衡溶解度与木犀草素纳米混悬剂的过饱和溶出度,本实验参考文献方法[21]建立了HPLC法。 2.3.1色谱条件 色谱柱为Sino Chrom ODS-BP色谱柱(250 mm×4.6 mm,5 μm);流动相为甲醇-0.3%磷酸水溶液(60∶40);柱温30 ℃;检测波长350 nm;体积流量1 mL/min;进样量10 μL。 2.3.2对照品溶液的配制 精密称取木犀草素对照品2.50 mg,放入100 mL棕色量瓶中,以适量色谱甲醇使之完全溶解,并定容至刻度线,摇匀得到质量浓度为25 μg/mL的木犀草素对照品储备液。 2.3.3 线性关系考察 采用色谱甲醇稀释成质量浓度分别为0.5、1.0、2.0、5.0、7.0、10.0 μg/mL系列的木犀草素对照品溶液,按“2.3.1”项下色谱条件进行分析,以对照品质量浓度为横坐标(X)、峰面积为纵坐标(Y)进行线性回归,得线性回归方程为Y=44 670 X-2 498.3,R2=0.999 8,结果表明木犀草素在0.5~10.0 μg/mL线性关系良好。 2.3.4 专属性、精密度和准确度考察 在建立的HPLC色谱条件下,木犀草素色谱峰不会受pH 1.2和pH 6.8的溶出介质、稳定剂TPGS、Tyrode液以及肠吸收液中所有成分的干扰(图7),表明本实验所建立的含量测定方法具有较好的专属性,能够满足体外溶出和肠吸收试验中木犀草素的含量测定要求。另外,其精密度实验的RSD为1.2%,高、中、低3个质量浓度的样品加样回收率在99.67%~101.47%,RSD均小2%,符合《中国药典》2020年版的规定。 2.3.5 平衡溶解度的测定 为了测定木犀草素在pH值为1.2、6.8缓冲溶液中的平衡溶解度,取5 mL 2种缓冲溶液各3份于西林瓶中,加入过量的木犀草素,将西林瓶置于恒温振荡箱中,在温度为37℃,转速为75 r/min条件下振荡24 h。取出各样品,3 000 r/min下离心10 min后取上清液,然后用0.2 μm滤膜滤过,取续滤液于进样瓶中,按照“2.3.1”项下色谱条件进样测定,并计算木犀草素的平衡溶解度,结果可知,木犀草素在pH值为1.2、6.8的缓冲溶液中的平衡溶解度分别为(3.83±0.23)、(7.81±0.13)μg/mL。 2.3.6 过饱和溶出度的测定 为了考察LNS体外溶出行为,参照《中国药典》2020年版中桨法进行。具体操作如下:在智能溶出仪中,以500 mL模拟胃液为溶出介质,温度为37℃,桨旋转速度为75 r/min,将30 mL LNS加入溶出介质中,以相同质量浓度的木犀草素乙醇溶液作为对照,二者均平行操作3份。以药物刚接触溶出介质开始计时,分别于5、15、30、60、120、130、150、180、240、360、480 min时取样4 mL,取完样后立即补充4 mL相应的新鲜溶出介质。另外,于120 min取样后,每个溶出杯中分别加入适量的Na3PO4溶液,调节pH值为6.8,以模拟肠液。将所取样品溶液经0.2 μm微孔滤膜滤过,取续滤液置于进样瓶中,照“2.3.1”项下色谱条件测定,计算累积溶出度,结果见图8。为了测定过饱和溶出水平,在整个实验过程中,介质中药物的质量浓度都应保持远远大于药物的饱和溶解度[22]。结果如图8所示,在pH 1.2和pH 6.8时,木犀草素-原料药的过饱和溶出始终低于对应的平衡溶解度,LNS的过饱和溶出始终高于对应的平衡溶解度,说明制剂的过饱和度高;在溶出介质的pH值调为6.8后,过饱和溶出水平明显下降,在150 min后过饱和溶出水平逐渐稳定,说明LNS能维持较高的过饱和溶出水平。 结果表明,LNS较木犀草素原料药具有明显优势,其饱和溶出度约是木犀草素原料药的15倍,过饱和度高并能维持较长时间,可以延缓药物在体内因析出晶体而沉淀的过程,从而使稳定剂在较小用量下也能保证药物分子成溶解态,提高了原料药的溶解度,有利于增加其生物利用度[23]。 2.4 小肠吸收实验 为了探索LNS对木犀草素在胃肠道的吸收部位和吸收速率的影响,采用外翻肠囊法[24]研究LNS在肠道不同肠段的吸收特征,以探究药物在肠道内的最佳吸收部位。 2.4.1 对照品溶液的制备 精密量取“2.3.1”项下相应体积的储备液,置于50 mL棕色量瓶中,用Tyrode液定容至刻度,摇匀,配制出质量浓度为1、2、4、8、16、32、40 μg/mL木犀草素对照品溶液。 2.4.2 线性关系考察 按照“2.3.1”项下色谱条件测定,以木犀草素对照品质量浓度为横坐标(X),峰面积为纵坐标(Y)进行线性回归,得到回归方程为Y=45 475 X-19 575,R2=0.999 6,结果表明木犀草素在1~40 μg/mL线性关系良好。 2.4.3 供试品溶液的制备 大鼠按实验质量浓度随机分为3组,每组4只,实验前12 h禁食,自由饮水。颈椎脱臼处死,打开腹腔,小心分离出小肠,分别截取十二指肠、空肠、回肠、结肠相应肠段各10 cm,用生理盐水冲洗至无内容物流出。将肠段放入37 ℃ Tyrode液中,冲洗,在不损伤肠管的情况下,小心剥离肠表面的脂肪及血管,取出,用滤纸吸干表面水分。 将肠管一端结扎,用光滑的玻璃棒外翻,用Tyrode液冲洗过后,向不同肠段中注入3 mL的空白Tyrode液后将另一端也进行结扎形成囊状的肠管。将肠管放入盛有Tyrode液的烧杯中,实验中始终保持37 ℃的恒温,并不断通入95% O2/5% CO2的混合气体。平衡5 min后,将烧杯中的液体倒出,分别加入不同质量浓度(0.15、0.30、0.60 mg/mL)的木犀草素及LNS药液。以肠囊和药液接触时开始计时,取样时间点分别为15、30、45、60、75、90、105、120 min,每个时间点从肠囊内取样500 μL,同时补充同温同体积的空白Tyrode液。待试验结束后,将各段肠囊置于空白Tyrode液中孵育1 h,以清除掉肠囊及肠组织中残留的药物;随后将上述用于木犀草素和LNS吸收实验的各肠段互换,再按上述操作同法重复试验,以进行自身对照交叉试验的后段实验。取上述肠吸收液,加入甲醇500 μL,超声混匀,15 898×g离心(离心半径6.32 cm)2次,每次15 min,取上清液用0.2 μm滤膜滤过,取续滤液适量即得。 按照“2.3.1”项下色谱条件测定,并计算药物在各时间点的累积吸收量(Q,μg)和药物吸收速率常数[Ka,μg/(mincm2)],结果见图9。 由公式计算不同质量浓度下木犀草素在各个时间点的累积吸收量(Q)。 Q是每个时间点木犀草素的累积吸收量,Ci是每个时间点的实际检测质量浓度,V1是加入肠囊内的空白Tyrode液,V2是每次取样的体积 由图9可知,通过对比2种制剂在各肠段中不同质量浓度的药物吸收情况,可以发现药物的同一时间点的吸收量表现出质量浓度相关性。相同质量浓度下,在各肠段中2制剂组吸收量相比,LNS组的药物累积吸收量显著大于木犀草素溶液组,表明LNS相比于木犀草素溶液能够促进药物在肠道的吸收。 根据小肠内(4个肠段)的Q值,通过线性拟合,由公式Ka=L(斜率)/A(肠管平铺面积)求得吸收速率常数(Ka)和相关系数(R2),结果见表5。2种制剂中木犀草素在肠道的不同部位中的吸收速率大小顺序均为十二指肠>空肠>回肠>结肠,这可能归因于十二指肠和空肠肠段的吸收面积较大;这一结果还表明LNS并没有改变木犀草素在肠道内的主要吸收部位和机制。对比相同质量浓度、相同肠段中2种制剂的吸收情况可以发现,LNS中木犀草素的吸收速率显著高于木犀草素溶液的情况,尤其是十二指肠和空肠中LNS和木犀草素溶液的木犀草素吸收速率差异更加明显,这表明LNS可以增加木犀草素的肠吸收,且十二指肠和空肠是主要吸收部位。 另外,还可以发现2种制剂在每一肠段中的吸收速率都存在显著的质量浓度相关性(P<0.01),但是2种制剂在同一肠段中的吸收速率随质量浓度增加而提高的程度有明显差异,即木犀草素溶液随质量浓度的增加,各肠段中吸收速率增幅增大,而LNS随质量浓度的增加,各肠段中吸收速率增幅减小,这些结果表明2种制剂在各肠段中的吸收均有质量浓度相关性,但其吸收速率与质量浓度之间均存在非线性关系,且仅在Ka<0.052时,木犀草素的肠吸收过程可能只受木犀草素溶解度限制,而不受吸收速度限制。然而,木犀草素的实际口服吸收情况是否符合上述规律以及其具体吸收机制如何,将有待于后期开展体内外吸收途径探索和体内药动学研究来进一步证实。 3 讨论 3.1 稳定剂的选择及药物-稳定剂比的确定 由于不同的稳定剂中化学基团的差异,导致稳定剂与药物微粒之间的分子间作用力以及胶粒间的作用力都有明显差异,所以稳定剂种类会影响到纳米混悬剂的稳定性[25]。因此,本实验首先以粒径和稳定性为考察指标,通过单因素筛选法优化了LNS的稳定剂种类,并确定了以TPGS作为稳定剂能达到较好的预期效果;考虑到稳定剂用量对稳定效果的影响[26],随后本实验又考察了药物-稳定剂比对纳米混悬剂的粒径、稳定性、PDI、ζ电位的影响,最终确定最佳药物-稳定剂比为1∶1。 3.2 LNS体外分析方法的建立及研究 3.2.1 波长的选择 木犀草素对照品与稳定剂TPGS在紫外波长200~800 nm扫描,结果显示木犀草素在207、254、350 nm 3处波长处有强吸收;而TPGS在219、286 nm显示出强吸收,350 nm处没有显示出强吸收。为了排除稳定剂TPGS对木犀草素测定的干扰,选用350 nm作为木犀草素的测定波长。 3.2.2 Tyrode溶液的配制 在木犀草素的肠吸收情况研究中,虽有文献报道了外翻肠囊模型和在体单向肠灌流模型[27-29],但关于木犀草素及其制剂在大鼠不同肠段中的吸收情况鲜有报道,且大多数文献对其吸收情况所提甚少。 本实验采用离体外翻肠囊法,可操作性强、重复性好;能够保留较为完整的肠道组织和黏膜特性,其实验结果与机体药物吸收水平比较接近,具有说服力;但肠外翻肠囊法也存在缺点,如长时间暴露在体外,肠管没有血管和神经的控制,肠黏膜功能和形态会失去作用。因此,本研究为解决这一问题,利用Tyrode培养液改善肠管的存在环境,具体配制方法如下:将NaCl(8.0 g/L)、KCl(0.2 g/L)、CaCl2(0.2 g/L)、NaHCO3(1 g/L)、NaH2PO4(0.05 g/L)、MgCl2(0.1 g/L)、葡萄糖(1.0 g/L),用蒸馏水定容至1 000 mL,稀盐酸调pH值为7.2~7.4,由于CaCl2不好溶解,应在其他无机盐溶解完全后再加入,葡萄糖于临用前再加入。并且在实验过程中连续通入95% O2/5% CO2,保证了在实验期间肠管上肠黏膜的活性。实验证明用该模型了解药物的离体吸收,其结果可靠。 3.3 LNS的过饱和溶出 药物在纳米混悬剂中所处的物理状态关系着其粒径和溶出稳定性,通常无定形药物微粒具有较高的饱和溶出度,但其属于热力学不稳定状态,因此物理稳定性差,容易引起纳米混悬剂粒径分布发生变化,同时溶出速率和溶出度下降;而结晶型药物具有较好的热力学稳定性,随着其粒径的减小,其饱和溶出度会明显提高[30]。根据本实验对LNS中木犀草素物理状态的研究结果可知,本实验制备的LNS中木犀草素以结晶形式存在,这表明LNS可能存在稳定的粒径和溶出度。 在过饱和溶出实验中发现,相比于木犀草素原料药,LNS具有显著的长期高过饱和溶出水平,这可归因于LNS中药物以粒径远小于原料药的状态存在,正如开尔文定律所描述的小粒径药物具有高溶解度一样[31]。药物的长期高过饱和溶出水平将有助于避免或减少口服给药后因胃肠道pH变化而引起的析晶沉淀现象,从而增加药物的吸收速度和时间,提高药物的口服生物利用度。 综上所述,本实验制备的LNS,分散性和储存稳定性良好,方法也简单易行,本实验建立的木犀草素体外分析方法,经方法学验证可知,该方法快速、可靠、准确度高,适合LNS的体外溶出和外翻肠囊吸收实验研究。 同时,外翻肠实验表面,LNS能促进药物在肠道的吸收,可作为木犀草素口服给药的潜在剂型,也为其进一步加工成其他剂型研究提供坚实基础。同时,在木犀草素肠道吸收的具体机制方面还有很大的研究空间。
1.测果树中的有机酸,标准品该怎么选择,应该选DL型吗?2.测DL苹果酸会出现两个峰,应该选择哪一个?最高的峰吗?
各位专家前辈好!我选择了几款柱子用美国UPS方法做拉米夫定,但不是保留时间太久就是拖尾,各位专家前辈做过拉米夫定吗?你们是用什么柱子做的?恳求各位专家前辈不吝赐教,谢谢!
[align=center][font='times new roman'][size=16px]超速离心法[/size][/font][font='times new roman'][size=16px]富集[/size][/font][font='times new roman'][size=16px]细胞分泌的外[/size][/font][font='times new roman'][size=16px]泌[/size][/font][font='times new roman'][size=16px]体[/size][/font][/align][align=left][font='times new roman'][size=16px]引言[/size][/font][/align]本章利用超速离心法富集细胞分泌的外泌体。采用透射电子显微镜,Western Blot实验,纳米颗粒示踪分析等表征了外泌体的形态结构及分布特征。最后,用增殖实验和划痕实验测试了富集得到的外泌体的生物完整性[font='宋体']。[/font][align=left][font='times new roman'][size=16px]细胞分泌的[/size][/font][font='times new roman'][size=16px]外[/size][/font][font='times new roman'][size=16px]泌[/size][/font][font='times new roman'][size=16px]体表征[/size][/font][/align][img]" style="max-width: 100% max-height: 100% [/img]利用透射电子显微镜对超速离心法得到的外泌体进行了表征。如图(a)所示,洗脱后的外泌体具有典型的杯状结构。纳米颗粒示踪分析技术显示富集的外泌体粒径分布较窄,平均粒径为115.3 nm(图 b)。Western blot方法验证了富集方法的有效性。如图(c)所示,经超速离心法富集后,典型的低丰度外泌体标记物HSC70、TSG101、CD63和CD9可得到有效检测。以上结果证明,基于超速离心法对外泌体的富集可行性。[align=center][font='黑体'][size=14px]图[/size][/font][font='黑体'][size=14px] (a)重悬液中外[/size][/font][font='黑体'][size=14px]泌[/size][/font][font='黑体'][size=14px]体的透射电子显微镜表征,(b)[/size][/font][font='黑体'][size=14px]纳米颗粒示踪分析技术[/size][/font][font='黑体'][size=14px]表征外[/size][/font][font='黑体'][size=14px]泌[/size][/font][font='黑体'][size=14px]体的尺寸分布,(c)[/size][/font][font='黑体'][size=14px]Western blot方法[/size][/font][font='黑体'][size=14px]表征[/size][/font][font='黑体'][size=14px]外[/size][/font][font='黑体'][size=14px]泌[/size][/font][font='黑体'][size=14px]体标记物HSC70、TSG101、CD63和CD9[/size][/font][font='黑体'][size=14px]蛋白条带[/size][/font][/align][align=left][font='times new roman'][size=16px][color=#000000] [/color][/size][/font][font='times new roman'][size=16px][color=#000000] [/color][/size][/font][font='times new roman'][size=16px]完整性验证[/size][/font][/align][img]" style="max-width: 100% max-height: 100% [/img]首先用细胞增殖用于评估超速离心法分离的外泌体的生物完整性。不同浓度(10[font='times new roman'][sup][size=16px]6[/size][/sup][/font]、10[font='times new roman'][sup][size=16px]8[/size][/sup][/font]、10[font='times new roman'][sup][size=16px]10[/size][/sup][/font]颗粒/孔)的H1299细胞外泌体分别与H1299细胞共培养24、48和72小时。如图(a)所示,H1299细胞来源的外泌体以剂量依赖性方式显著促进细胞增殖。此外,伤口愈合实验显示,H1299细胞来源的外泌体加速了伤口愈合速度,并以剂量和时间依赖的方式提高了伤口愈合质量(图 b)。上述结果表明,通过超速离心法富集的外泌体具有完整性,可以促进细胞增殖、分化和迁移。[align=center][font='黑体'][size=14px]图[/size][/font][font='黑体'][size=14px](a)H[/size][/font][font='黑体'][size=14px]1299[/size][/font][font='黑体'][size=14px]细胞来源的外[/size][/font][font='黑体'][size=14px]泌[/size][/font][font='黑体'][size=14px]体对细胞增殖的影响结果,(b)[/size][/font][font='黑体'][size=14px]H1299细胞来源的外[/size][/font][font='黑体'][size=14px]泌[/size][/font][font='黑体'][size=14px]体对细胞[/size][/font][font='黑体'][size=14px]划后伤口愈合[/size][/font][font='黑体'][size=14px]的影响结果[/size][/font][/align][align=left][font='times new roman'][size=16px]小结[/size][/font][/align]本章利用超速离心法富集了细胞来源的外泌体,并对富集得到的外泌体进行了一系列表征表征。通过透射电子显微镜和纳米颗粒示踪分析技术分析表明得到的外泌体具有典型的杯状结构和小的粒径分布,证明了富集方法的可行性。Western Blot结果表明,富集后得到的外泌体溶液经裂解后,特征蛋白HSC70、CD63、TSG101和CD81,其含量比富集前显著提高。利用细胞增殖实验和划痕实验证明超速离心法富集得到的外泌体具有良好的生物学活性,可应用于下游的实验研究。
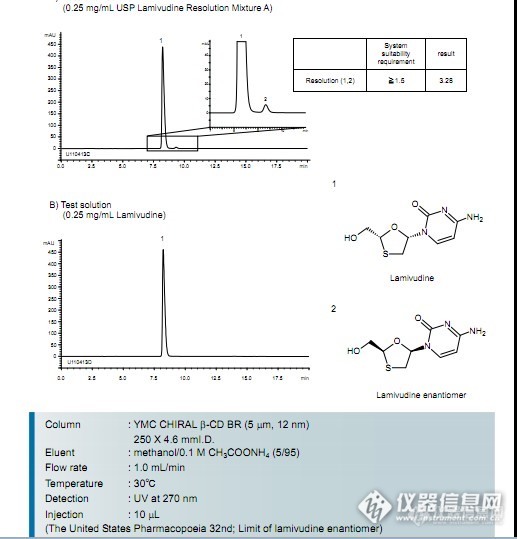
按USP规定色谱条件:YMC CHIRAL β-CD BR柱分离拉米呋啶手性异构体。http://ng1.17img.cn/bbsfiles/images/2013/07/201307091659_450387_1638724_3.jpg
氟乐灵又叫特福力、氟特力、茄科宁,是一种低毒除草剂。我国已有国家标准GB/T 5009.172用于大豆油和花生油中氟乐灵的检测,但辣椒油与大豆油和花生油有较大的差别,除了脂肪酸外,还有精油、色素等,这给辣椒油中氟乐灵的检测造成了一定的麻烦。本人参照GB/T 5009.172,开发了辣椒油中氟乐灵的检测方法,结果比较满意。具体方法如下:提取:称取辣椒油样1.0g(标准称取油样5.0g,本人认为没必要称这么多)于20mL规格的螺口试管中,加入10mL丙酮溶解,加2mL水,快速涡旋1min,离心,上清液转移入50mL试管中,下层再提取一次,合并提取液,加入10mL硫酸钠溶液(50g/L),5mL正己烷,振荡3min,离心,取出上清液,下层再加正己烷提取一次,合并提取液,浓缩至约1mL,待净化。此为提取过程,方法同国标,仅称样量与试剂量不同而已。净化:本人首先考虑可不可以用磺化法净化,因为磺化法最简单,最经济,最快。虽然文献没有磺化氟乐灵的报道,但本人还是试了一试。氟乐灵标样用浓硫酸磺化后,氯乐灵的峰不见了,在氟乐灵峰的前面出了一个峰,如果能定量地转化成另一化合物也有可能对其检测,但两个平行样的色谱峰响应相差较大,因而放弃了磺化法。下图中蓝色为未磺化的标样谱图,红色和绿色为磺化处理的标样的色谱图。

色素+辅料+碳酸钙=螺旋藻氨基酸螯合钙片我常接触药品,了解氨基酸螯合钙补钙效果比第一第二代的钙片好,去药店找过含氨基酸螯合钙的商品不多,店员推荐了金参堂牌的氨基酸螯合钙片,正因为是氨基酸螯合钙,后来我还推荐买给我的父母、外母。该厂家金参堂牌的氨基酸螯合钙片分为儿童型成人型妇女型好几种(如图一)。本人是质量检验员,一次偶然的机会对长期食用的该钙片作了检验,不检验还好,一检验吓我一跳,这钙片竟然名不附实,居然是色素+辅料+碳酸钙=螺旋藻氨基酸螯合钙片!。。。。。。http://ng1.17img.cn/bbsfiles/images/2011/01/201101192115_275111_1621232_3.jpg惊吓一:氨基酸螯合钙=碳酸钙首先我要了解一下氨基酸螯合钙的化学性质,氨基酸螯合钙是水溶性物质,比食盐的溶解度还要大,所以把氨基酸螯合钙放入水中是马上溶解的,且氨基酸螯合钙遇到酸(比如盐酸)是没有气泡产生的。碳酸钙放入盐酸液中产生气泡(CO2)大家都应该知道的,我无意中放了一粒金参堂牌的氨基酸螯合钙片入盐酸液中,想不到,吱。。。吱。。。冒泡,导入氢氧化钙水溶液中产生白色沉淀,我觉得奇怪了,不应该有的现象哦。我意识到钙片中可能含有碳酸盐物质,出于谨慎,我开始对这种钙片作了一系列的鉴别实验。首先我将本品研成细粉,加水搅拌溶解,发现上清液变为黄绿色溶液,下面有一些白色的不溶物,我再用离心机离心出不溶物,这样反复水洗离心几次,除去水溶性物质,得到一白色不溶物(如图二)。离心后的上清液经焰色鉴别没有钙的砖红色焰色反应,即这钙片中水溶液不含有钙离子。跟着向白色不溶物滴加稀盐酸,吱。。。吱。。。马上产生气泡,白色不溶物渐渐溶解,对溶解液作焰色鉴别,产生明显钙的砖红色焰色反应,证明白色不溶物中含有钙。碳酸钙,我脑海中闪过这个词。天啊,我每天给我儿子吃的螺旋藻氨基酸螯合钙片竟然是碳酸钙片,一种被欺骗的感觉蛹到心头。后用白色不溶物测试了含钙量,经用直接混合酸消化液(硝酸+高氯酸=4:1)测定钙片中全钙量和离心水洗法测定本品的钙含量,结果消化法测得钙含量为217mg/粒,离心水洗法测得钙含量为203mg/粒。考虑离心水洗时可能有损失,离心法测得的钙含量与消化法结果一致。http://ng1.17img.cn/bbsfiles/images/2011/01/201101192114_275110_1621232_3.jpg由此推断,本品标示以氨基酸螯合钙作为原料投料很有可疑了,水洗离心后,氨基酸螯合钙应随水洗去,白色不溶物不应含有钙的了,而且,消化法测得的是钙片中全部钙元素(包括水溶性和水不溶性的钙)的含量,水洗离心法测得的是水不溶性的钙量,但两种方法测得结果一样,这样算来,金参堂牌氨基酸螯合钙片中氨基酸螯合钙的含量为零。可以得出其投料很可能就是碳酸钙的结论。http://ng1.17img.cn/bbsfiles/images/2011/01/201101192117_275116_1621232_3.jpg惊吓二:色素+辅料=螺旋藻 我见到上述离心时的上清液为黄绿色溶液,沉淀不溶物为白色,没有发现有黑绿色或蓝绿色螺旋藻。我们知道螺旋藻是一种蓝藻,同我们见到的绿色植物一样含有叶绿素,其粉未颜色为黑绿色或蓝绿色。我想到这钙片标示含有螺旋藻是否有假呢?我做了以后的试验作了一鉴别。用已知的纯螺旋藻粉和该钙片用水和石油醚分别提取色素,纯螺旋藻粉水溶液呈蓝色,该钙片水溶液呈黄绿色(如图三),两种溶液在阳光下放置数天后,纯螺旋藻粉水溶液完全褪色,而该钙片水溶液仍呈黄绿色。纯螺旋藻粉石油醚溶液呈绿色植物一样的绿色(叶绿素),该钙片石油醚溶液呈纯蓝色(如图四)。将纯螺旋藻粉和该钙片放到显微镜下比较,纯螺旋藻粉具有细胞结构和绿色色素物质,而该钙片只有一些矿质物质,没发现有细胞结构和绿色色素物质(如图五)。由此,我们可以得出结论,该钙片标示含有螺旋藻也是假的。随后我又用薄层色谱对两者成分作了比较,同时用石油醚提取叶绿色进行层析(如图六),结果显示,螺旋藻提取液中具有正常叶绿素a等斑点,而钙片提取液什么也没,从这更印证了显微镜的鉴别,钙片中的螺旋藻只不过是色素罢了。http://ng1.17img.cn/bbsfiles/images/2011/01/201101192115_275113_1621232_3.jpg后来我又拿了家里的儿童型和成人型的该厂家钙片进行了化验,情况一模一样。真气人,自我儿子出生开始到现在三岁多,我一直信任这个厂家的氨基酸螯合钙片,点知吃的还是普通钙,还是假的!http://ng1.17img.cn/bbsfiles/images/2011/01/201101192116_275114_1621232_3.jpg该厂家的金参堂牌一系列的螺旋藻氨基酸螯合钙片无论外包装,还是说明书,均称该钙片是第三代钙源,其说明书是这样的:金参堂牌螺旋藻氨基酸螯合钙精选深海无污染的螺旋藻,运用高科技螯合超微细技术,科学配制需成。主要原料为:螺旋藻、[/
20X106 copy/ml,并持续至少一年,其中四例HbeAg阳性。加用替诺福韦酯后,经24到30周治疗,患者HBV载量降至均数为5460 copy/ml(4000-7800),前后对比差异显著。10例经拉米夫定治疗出现耐药,换用阿德福韦再次出现耐药的慢性乙肝患者,经替诺福韦酯单一治疗,至12个月时HBV DNA下降4.4log10 copy/ml,治疗17个月时,5例患者HBV DN
我按照《药品检验仪器检定规程》(第44页)检定旋光仪,其中准确度和精密度的没有公式,不知道应该怎么算啊。求教!!下附《药品检验仪器检定规程》。
紫外分光光度计的波长校正和检定由于温度变化对机械部分的影响,仪器的波长经常会略有变动,因此除应定期对所用的仪器进行全面校正检定外,还应于测定前校正测定波长。常用汞灯中的较强谱线237.83、 253.65、 275.28、296.73、313.16、334.15、365.02、404.66、435.83、546.07与576.96nm,或用仪器中氘灯的486.02与656.10nm谱线进行校正,钬玻璃在279.4、287.5、333.7、360.9、418.5、460.0、484.5、536.2与637.5nm波长处有尖锐吸收峰,也可作波长校正用,但因来源不同会有微小的差别,使用时应注意。 吸收度的准确度可用重铬酸钾的硫酸溶液检定。取在120摄氏度干燥至恒重的基准重铬酸钾约60mg,精密称定,用0.005mol/L硫酸溶液溶解并稀释至1000ml,按下表规定的波长处测定并计算其吸收系数。 规定的吸收系数如下表,相对偏差可在±百分之1以内。波长(nm):235(最小) 257(最大) 313(最小) 350(最大)吸收系数E百分之1 1cm:124.5 144.0 48.62 106.6 (与上面数值一一对应)杂散光的检查可按下表的试剂和浓度,配制成水溶液,置1cm石英吸收池中,在下表规定的波长处测定透光率,应符合表中的规定。试剂 浓度(g/ml) 测定用波长(nm) 透光率碘化银 百分之1.00 220 百分之0.8亚硝酸钠 百分之5.00 380 百分之0.8
想测一地区地表径流的总氮,采用过硫酸钾消解紫外分光光度法,最后用分光光度计读数时数据老跳动。开始不能自动调零,换了石英比色皿后,自动调零有点小跳,-0.001到0.001,参比数据也还稳定,但是样品和标准曲线全都非常不稳定,相差可以达0.1。有朋友说是反应没完全,就把消煮时间增加到一小时,结果还是一样不稳定,有人知道是什么原因吗?
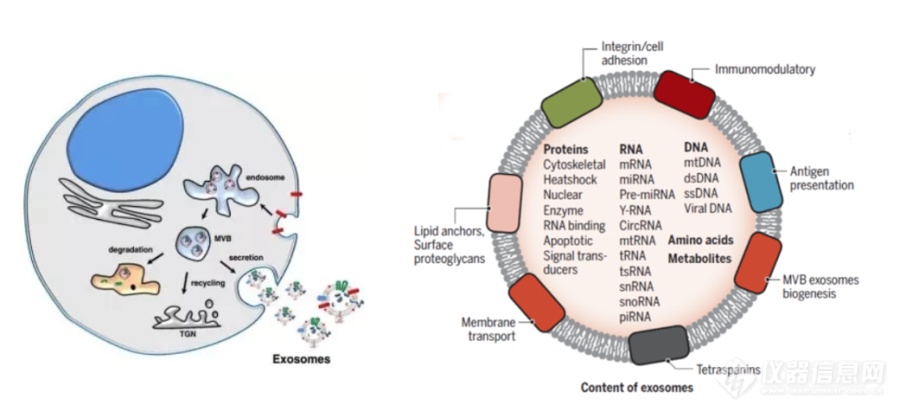
[align=center][font='times new roman'][size=16px]外[/size][/font][font='times new roman'][size=16px]泌[/size][/font][font='times new roman'][size=16px]体[/size][/font][font='times new roman'][size=16px]富集及在肿瘤诊断中的作用[/size][/font][/align]外泌体是直径30-200 nm,由细胞内陷产生的具有磷脂双分子层结构的细胞外小囊泡,是细胞间通讯的重要介质。此外,由于其易于获取、高度稳定且在体液中广泛分布,外泌体已成为液体活检中最有前途的生物标志物[font='宋体']。[/font][align=left][font='times new roman'][size=16px]外[/size][/font][font='times new roman'][size=16px]泌[/size][/font][font='times new roman'][size=16px]体的生发与分布[/size][/font][/align]外泌体是具有磷脂双分子层结构的细胞外小囊泡,它携带原始母细胞重要的物质信息,能够反应原始细胞的一些重要信息。随着研究的深入,已有研究证实外泌体是通过细胞内陷产生的胞外囊泡,如图1-1所示,它是起源于细胞内吞系统中的多囊泡内体,通过出芽、内陷、多泡体形成和分泌等形成的一种具有磷脂双分子层结构的小泡,形态呈球形、扁形或杯状小体,直径为30-200 nm。此外,外泌体的分布非常广泛,几乎分布在所有的体液中,如血液,尿液,乳汁,汗液等。携带活性生物分子(蛋白质、核酸和脂质等)和他们的母细胞有相似的特征,这可能适用于正常细胞和病理细胞的鉴别。特别是,外泌体膜蛋白的水平与癌症的动态密切相关,从而为癌症诊断和治疗提供了新的机会预后。[img]https://ng1.17img.cn/bbsfiles/images/2023/10/202310171529195086_7813_6197418_3.png[/img][align=center][font='黑体'][size=14px]图1.[/size][/font][font='黑体'][size=14px]1 外[/size][/font][font='黑体'][size=14px]泌[/size][/font][font='黑体'][size=14px]体的生发与组成[/size][/font][/align][align=left][font='times new roman'][size=16px]外[/size][/font][font='times new roman'][size=16px]泌[/size][/font][font='times new roman'][size=16px]体的通讯作用[/size][/font][/align]外泌体含有多种生物活性物质,这些被包裹的生物大分子可以在靶细胞中发挥一系列作用,是细胞间接通讯的重要媒介。外泌体与靶细胞间的信息传递主要通过三条途径实现:受体-配体相互作用;质膜直接融合;吞噬作用内吞。外泌体的生物学功能包括物质传递、信息交流、细胞增殖分化、血管生成、免疫调节等,在肿瘤发生发展、侵袭、转移、预后等过程中起重要的调控作用。肿瘤细胞较正常细胞分泌更多的外泌体,来源母细胞不同,所分泌的外泌体量与成分也不同。其次,由于其具有磷脂双分子层结构,生物学相对稳定,难于降解。外泌体的一个关键功能是将其内含物从供体细胞转运到受体细胞,使受体细胞的基因和表型修饰[font='宋体']。[/font][align=left][font='times new roman'][size=16px]外[/size][/font][font='times new roman'][size=16px]泌[/size][/font][font='times new roman'][size=16px]体的富集方法[/size][/font][/align]由于外泌体丰度相对较低且大分子干扰,高质量外泌体的分离仍然具有挑战性。基于不同的分离机制,已经提了各种分离方法。作为金标准的超速离心法在外泌体的分离中使用最广泛。其他策略,如免疫亲和的富集方法,微流控芯片等,这些方法大多依赖于抗体和适配体。分离的外泌体的质量和纯度受限于外泌体表面抗原等活性分子的缺失,失活和降解,并且不适合大规模捕获应用。[align=left][font='times new roman'][size=16px]外[/size][/font][font='times new roman'][size=16px]泌[/size][/font][font='times new roman'][size=16px]体与肿瘤[/size][/font][/align]肿瘤发病机制复杂,早期诊断困难,病程进展快,是严重威胁人类生命和社会发展的重大公共卫生问题。外泌体是由细胞分泌产生的纳米级囊泡,在肿瘤的发生发展、诊断和治疗方面发挥着重要作用。外泌体携带多种肿瘤相关因子,可以作为治疗靶点。外泌体以其天然的物质转运特性、相对较小的分子结构、优良的生物相容性及免疫原性低可避免被单核吞噬系统吞噬,可作为优良的药物载体,允许它们穿过生理病理屏障将其携带的物质递送到下面的组织。基于这些特性,越来越多的人关注外泌体在疾病特别是靶向治疗领域的研究[font='宋体']。[/font]
想请问一下各位同行,用紫外法测定水中丁基黄原酸,需要用酸化前后的吸光度值来做纵坐标,在实际操作时,是先做一下酸化前的吸光度值打印出来,再做一下酸化后的吸光度值打印出来,然后算好两者的差值,输到分光光度计里面做曲线吗?做实际样品的时候呢?
今年6月底前后,跨国药企美国默克公司降胆固醇药舒降之(辛伐他汀)和全球药业老大辉瑞公司的抗抑郁药左洛复(舍曲林)先后失去专利保护权,去年这两种药品全球销量分别为44亿美元、34亿美元。同时,包括诺华的兰美抒、赛诺菲安万特的思诺思、百时美施贵宝的普拉固和葛兰素史克(GSK)的枢复宁等在内的全球多个重磅药品,均将在两年内结束其专利保护期。有数据显示,今年年内将有全球年销量约230亿美元的专利药专利到期,而到2008年之前,全球专利到期药品的总价值将超过800亿美元。 全球专利药的集中到期,将给以仿制药为主的中国药企提供巨大商机,国内药企无论在仿制药开发、原料供应还是制剂生产方面都面临很多机会。 研发机构、药企提前“盯”上到期专利药 美国一项名为《全球非专利处方药市场》的最新报告称,未来4年超过30种药物面临专利到期的命运,全球非专利处方药销售将快速上扬,从2005年的282亿美元增至2010年的480亿美元。2004年非专利处方药市场即达到253亿美元,2005年增长了11.2%.预计未来数年内该市场仍将以近似的增速稳定增长。在未来数月内,畅销药物如络活喜、帕罗西汀专利均将陆续到期。对于这一市场空间,业内分析,最先受益的将是靠开发仿制药生存的中国药品研发机构,他们的生意将显得日益兴隆。 “到期专利药的仿制开发在国内十分普遍,包括药厂自己的研究所在内,全国至少有6000个药品研发机构都提前在做类似的事情。”研发型企业广东固志医药科技有限公司总经理吴智南告诉记者,目前委托他们做这种开发的有10多家药企,他们正在对20多个专利即将到期的药品进行开发,主要是根据其已公开的专利技术知识,研究药物的合成路径。吴智南称,一般开发这样的药品要花9个月左右的时间,每个品种收取研发费用约50万元。 其实,与收益相对稳定的研发型企业相比,药厂面对的专利药到期市场无疑是一场苦战。“我现在主要还是生产抗菌类普药,暂且不会考虑去做到期专利药仿制。”广州诺贝华乐制药公司总经理许守良称,专利到期药的研发成本并不高,但报批手续特别烦琐,没有一定渠道很难拿到批文。“往往有上百家药厂去开发一个品种,国内厂商利润已非常低,竞争近乎惨烈。”做药品研发近20年的吴智南认为,这种过度竞争的模式他并不看好。 另外,不仅是仿制药企之间存在竞争,现在原研药厂家也开始介入这一场仿制药的争夺战。据悉,正是瞅准这一市场前景,制药巨头诺华去年已将其位居全球第二位的非专利药业务“山德士”引进中国,并在华积极寻求合作伙伴;而日前,就在美国一家仿制药宣布其生产的辛伐他汀将比默克“舒降之”价格低30%~50%之后,默克即与美国最大健康保险公司UnitedHealth达成协议,专利品牌药“舒降之”今后价格将比任何一家上市的辛伐他汀产品都低。 乙肝药仿制市场集中上演“血拼”大战 这么多“重磅炸弹”可以仿制,对于药企和研发机构而言,哪些品种对他们更有吸引力呢? 据了解,刚在美国专利到期的默克“舒降之”进中国11年并无专利保护,而辉瑞“左洛复”在华行政保护也在几年前到期,这两种药品的仿制品早就“遍地开花”了。业内人士分析,目前到期专利药的仿制大战中,最为激烈的恐怕是乙肝药市场的争夺了。据GSK相关人士介绍,现在国内已有几十家药厂把目光聚焦在专利即将到期的一线乙肝抗病毒药“贺普丁”(拉米夫定)。若以零售价计算,2005年贺普丁在中国销售收入约有10亿元。而今年9-10月,贺普丁在华专利保护就将到期。 最新资料显示,全球目前有4亿乙肝病毒感染者,而中国就占到1/3,慢性乙肝治疗等相关费用每人年均2万元左右,中国整个乙肝病市场至少有100亿元的容量。去年以来,GSK、施贵宝又先后向中国引进了贺维力、博路定两款乙肝新药,诺华公司乙肝新药也即将在华上市。与这些价格不菲的专利药相比,专利行政保护即将到期的贺普丁成了众多患者和药厂的“抢手货”。“目前,公司正在研发一系列抗病毒品种,包括拉米夫定在内总共有几十个品种。”上海新先锋药业副总经理李建文向记者表示,一般他们会提前两年就对将到期的目标药品合成路径及生产工艺进行研发,等到临过期时再开发,市场早就被别人抢走了。“研发费用本身并不贵,像开发拉米夫定一个品种约200万元,最头疼的事情是仿制厂家太多,市场竞争将十分激烈。”李建文称。 不过,对于国内正酝酿的拉米夫定仿制热潮,贺普丁生产商GSK有关人士则表示了不同意见。“贺普丁专利的行政保护权,确实将在2006年第四季度到期,但目前贺普丁在华仍有其他专利可保护。”负责贺普丁市场的GSK抗感染药高级经理黄怡峥向记者表示,贺普丁在华专利包括生产制备方法专利以及拉米夫定活性成分与药用载体相合成的方法专利,这几项专利将在2010年至2015年间到期。问及GSK乙肝新药贺维力与老药贺普丁会不会形成营销冲突,黄怡峥表示,两者可以互补,今后贺普丁也不会撤出中国市场。 专家说法 “首仿药”更应关注 一直以来,国内4500多家药企90%以上在做仿制药,而往往同一个品种集中了上百家甚至几百家药厂在生产,造成了严重的资源浪费和恶性竞争。有关专家据此认为,国内厂商与其一哄而上去拼抢在华专利到期的某一个品种,不如把精力投向已在国外上市却未进入中国市场的“首仿药”市场,虽说投入稍高,但回报将更为客观。 在美国,一旦某个药品专利到期,首家获得仿制权的药企在市场和定价上都有更多优先权。“目前,专利到期的舒降之已被全球最大非专利药公司以色列TEVA首家取得FDA仿制批文,TEVA将获得180天市场独占权”,默沙东(即美国默克)中国公司公共事务高级主任丁燕宁介绍。 今年3月初,国家发改委向相关企业下发了《药品定价方法》征求意见稿,首次提出“首仿药”概念,要求对首仿药(即“首先研究申报国外已上市而在国内未上市的药品”)给予定价优先。该方案称,国内首先仿制生产并上市销售的同种仿制药品的价格经专家论证后,允许在统一定价的基础上适当上浮,上浮范围不超过政府规定的最高上浮范围。 上海市食品药品监督管理局科技情报研究所主任干荣富向媒体表示,国家拟允许“首仿药”价格比同类产品上浮15-20个百分点,且享有一段时间的价格独享权,是国内制药企业不可错过的好机会。干荣富称,他汀类、普利类、沙坦类、抗抑郁药、抗溃疡药等特色原料药已给许多企业创造了利润,这些企业避开了专利,并且在国际上获得了注册、认证以及合同加工订单,特别是在2008-2010年前后,将有一批专利药到期,这会给一大批特色原料药创造巨大的市场需求。
有人用碱性过硫酸钾-紫外分光法做出来土壤总氮的吗?这个方法只有水的国标,我根据文献的方法做出来的结果和土壤标准的标定值差距很大。先讲下我的实验流程:1.纯化国产过硫酸钾,过硫酸钾(GR科密欧)用怡宝55摄氏度以下过饱和溶解后,4摄氏度冷却析出,用冰怡宝冲洗两遍后继续过饱和溶解冷却析出,此过程纯化两次后低温烘干,避光保存2.消解液配制,1.5g氢氧化钠(科密欧AR)溶于怡宝,冷却,和4g纯化过硫酸钾溶于100mL容量瓶,40摄氏度超声溶解。3.空白样制备,旋塞比色管中依次加10mL怡宝和5mL消解液后,颠倒摇匀一次。4.样品制备,分别称取20mg、50mg、80mg、100mg、150mg、200mg土壤标准GSD-31置入旋塞比色管中,用怡宝定容至10mL后加入5mL消解液,手摇使土壤散开。5.消解过程,置入高压灭菌锅中,反应条件为121摄氏度保持45分钟,待气压接近0MPa,此时温度约为75摄氏度,放气开盖,待冷却。6.后续处理,冷却过程中颠倒摇匀一次,冷却至室温后加入1mL盐酸(1+9怡宝),并定容至25mL,颠倒摇匀一次后,将土壤样品溶液转移至离心管中离心。7.测试过程,标曲用干燥硝酸钾(GR)配制,消解过程同上,能达到0.9998。A220-2A275空白值小于0.003。标曲采用吸光度减去空白吸光度的校正吸光度和实际浓度拉方程。样品测试吸光度减去空白吸光度后,采用校正吸光度算出溶液浓度,此时 [溶液浓度 × 25mL / 土壤质量(g)] 为实际氮含量。土壤测试过程中用滴管吸取离心后的上清液进样。测试结果为:GSD-31标定值为318PPm,测试值为150-200PPm。差距很大我找出了几个可能的原因,有了解的请指点一下,谢谢1.GSD-31水系沉积物标准中的N标定值是318±24,我用此方法做出的氮含量和这个N指的是一个参数吗?标曲浓度我算的硝酸根中的N2.关于纯化过硫酸钾,纯化后空白吸光度小于0.003能否说明过硫酸钾具备要求纯度?因为我测试的TN值较标准值偏小,我怀疑消解能力不够,但文献中采用5mL消解液能在100mg土中测出1700PPm,增加至10mL消解液又会使空白值增大。国产的天津和上海品牌也纯化过,空白值都很低,难道空白值和消解能力没关系吗?真的要买进口的吗3.为了测试消解能力,本人采用过配制乙酸铵进行试验,结果符合计算理论值,这一点说明消解液能够充分转化铵氮吗?谢谢,人要疯了
多种不饱和脂肪酸能直接用紫外检测器检测吗?如果能,紫外吸收波长选多少?
请问外消旋体在碳谱和氢谱上有什么特点,如何利用核磁来区分对映异构体和非对映异构体
甜蜜素、环拉酸拉和环拉酸的区别是什么?环拉酸怎么检测?谢谢各位![em24]
[B][center]55种西药药品的合成方法介绍[/center][/B] [B]目录[/B]1.7β-氨基-3-氯甲基-7α-甲氧基-1-氧代-3-头孢烯-4-羧酸二苯甲酯的合成路线图解.pdf2.11α-羟坎利酮的高特异性生物转化合成.pdf3.CAPIC的合成路线图解.pdf4.Ezetimibe合成路线图解Graphical Synthetic Routes of Ezetimibe.pdf5.L-酒石酸托特罗定的合成.pdf6.阿巴卡韦的合成.pdf7.阿巴卡韦合成路线图解.pdf8.阿伐麦布的合成.pdf9.阿戈美拉汀的合成.pdf10.阿立哌唑合成路线图解.pdf11.氨溴索合成路线图解.pdf12.奥卡西平合成路线图解.pdf13.奥利司他合成路线图解.pdf14.比阿培南的合成.pdf15.从GCLE制取头孢噻利的合成路线图解.pdf16.恩曲他滨的合成.pdf17.恩替卡韦合成路线图解.pdf18.法西多曲合成路线图解.pdf19.伏立康唑合成路线图解.pdf20.氟西汀盐酸盐合成路线图解.pdf21.富马酸福莫特罗的合成线路图解.pdf22.坎地沙坦酯合成路线图解.pdf23.抗高血压药奥美沙坦酯合成新路线和相关杂质的研究.pdf24.克拉屈滨的合成.pdf25.拉米夫定合成路线图解.pdf26.赖诺普利合成路线图解.pdf27.兰索拉唑合成路线图解.pdf28.雷尼酸锶的合成.pdf29.磷酸奥司米韦合成路线图解.pdf30.氯法拉滨的合成.pdf31.罗美昔布合成路线图解.pdf32.麦考酚酸高产菌株的选育.pdf33.莽草酸合成路线图解.pdf34.米格列奈的合成.pdf35.帕珠沙星消旋体的合成及HPLC法拆分.pdf36.氢溴酸达非那新的合成.pdf37.群多普利合成路线图解.pdf38.瑞舒伐他汀合成路线图解.pdf39.塞曲司特的合成.pdf40.舒尼替尼合成路线图解.pdf41.通过重组大肠杆菌的高细胞密度培养热诱导生产人生长激素.pdf42.托曲珠利的合成.pdf43.微生物甾体羟化技术及其应用.pdf44.西他沙星合成路线图解.pdf45.西维来司钠的合成.pdf46.消旋卡多曲的合成.pdf47.新药甲磺酸帕珠沙星的手性拆分.pdf48.盐酸阿比朵尔的合成路线图解.pdf49.盐酸阿扎司琼的合成.pdf50.盐酸吡格列酮合成路线图解.pdf51.盐酸多奈哌齐的合成.pdf52.盐酸米那普仑反式异构体的合成及结构确证.pdf53.盐酸舍曲林的合成路线图解.pdf54.盐酸坦索罗辛合成路线图解.pdf55.盐酸头孢替安合成路线图解.pdf资料下载:[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=161039]55种西药药品的合成方法介绍[/url]







 我要推广仪器
我要推广仪器
 下载APP
下载APP