同样的混标液10ug/mL(安赛蜜、苯甲酸、山梨酸、糖精钠、脱氢乙酸),同一根色谱柱C18柱,同样的色谱条件:(甲醇:0.02mol/L乙酸铵=5:95),流速1.0mL/min,在不同仪器下的分离度不同,效果还差挺多的:使用Waters2695e的糖精钠与脱氢乙酸分不开,而且脱氢乙酸还拖尾的厉害,而使用安捷伦1100的液相,这五个混标却分的不错,能实现基现分离,请问各位版友这个是什么原因造成的,
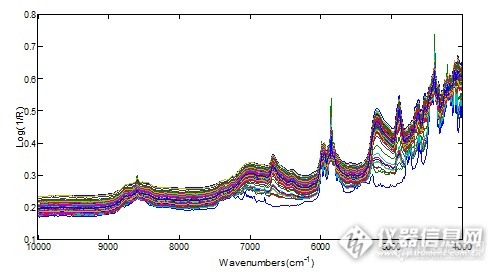
[align=center][b][url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱[/color][/url]分析技术用于美洛西林钠舒巴坦钠药物混合过程在线混合均匀度终点监测[/b][/align][align=left][b]摘要: [/b]利用[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱[/color][/url]技术,对美洛西林钠、舒巴坦钠混合过程进行了在线监测。在研究中,分别建立了基于MBSD法的定性分析模型和基于舒巴坦钠百分含量的定量分析模型,通过3个平行实验的在线混合过程,结果显示MBSD法和舒巴坦钠百分含量测定法均能有效的监测其混合过程,有效的证明了[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]NIR[/color][/url]光谱分析技术用于舒巴坦钠、美洛西林钠混合在线监测的可行性。[/align][b]关键词[/b]:[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱[/color][/url];分析模型;混合均匀度;在线监测自从2004年美国食品与药品监督管理局提出“过程分析技术”以来,全球的药品生产企业正在向着更高技术含量的生产方式和质量控制方式进军。近红外(Near infrared,[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]NIR[/color][/url])光谱分析技术因其快速,无损的特点成为“过程分析技术”的重要组成部分,是制药企业进行产品中间体质量控制的重要方法之一。传统的检测方法为高效液相色谱法,紫外可见分光光度法等需要停止混合操作时才能取样检测,并且等待检测结果所需的时间也比较长,工作效率比较低,而[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]NIR[/color][/url]光谱可以进行在线检测,连续记录不同混合时间内混合物的光谱图,建立数学模型对采集数据进行分析,从而判断各组分之间是否已经达到质量均一,工作效率大幅度的提高。本研究利用 [url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]NIR[/color][/url] 光谱分析技术在线监测美洛西林钠舒巴坦钠的药物混合过程,从而实现混合终点的准确判断。[b]1 材料1.1试剂[/b]美洛西林钠(13102041,山东瑞阳制药有限公司)舒巴坦钠(SS201310-26,江西东风制药有限公司)[b]1.2仪器和软件[/b]AntarisII型傅里叶变换[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱仪[/color][/url](美国ThermoFisher公司),附有积分球采样模块;RESULT采样软件;电子分析天平(Sartorius BT224S,德国);TQ数据处理软件;表面皿;药匙;自制搅拌器。[b]2 方法2.1样品的准备[/b]精密称取舒巴坦钠固体原料药10.00g,美洛西林钠固体原料药40.00g,以备进行在线混合光谱的采集。平行制备3批样品,进行混合光谱的采集。[b]2.2模型的建立[/b]目前,[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]NIR[/color][/url]光谱分析技术用于混合过程在线监测的方法可分为活性药物成分(API)定量分析模型监测和基于移动块标准偏差(MBSD)的定性分析模型监测。前者为基于API药物含量的定量监测模型,当达到混合终点时,API的含量趋于一定值,可以依据模型监测的含量是否达到理论值并趋于稳定进行混合终点的监测;后者为基于光谱的标准偏差的定性监测模型。MBSD法的基本原理为:连续采集的若干张光谱间的标准偏差变化率趋于稳定并小于限定的一阈值时可认为达到了混合终点。其具体的计算步骤为:首先确定用于计算光谱标准偏差的光谱的条数n(即移动块的宽度),当[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]NIR[/color][/url]光谱分析仪器采集到n张光谱后计算n张光谱的峰面积(或最大峰高、平均峰高等)的标准差,当采集到n+1张光谱时将第一张光谱移除,计算最近n张光谱的标准差,如此类推,最终得到随时间变化的光谱的标准偏差,根据标准差的变化进行混合终点的监测。本研究中建立了舒巴坦钠含量的定量分析模型和基于MBSD法的定性分析模型同时对用于混合终点的判断。[b]2.3在线混合光谱的采集[/b]将称取的美洛西林钠、舒巴坦钠原料药样品放入表面皿中,然后将表面皿放在Antaris II型傅里叶变换[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱仪[/color][/url]积分球采样模块的上面,采用积分球漫反射采样方式进行光谱的采集。在运行在线混合工作流的同时采用自制的搅拌器进行样品的混合,采集得到混合过程的原始光谱,同时监测混合过程。波长范围10000-4000cm[sup]-1[/sup],每张光谱扫描次数4,混合过程中每间隔5s进行一张光谱的采集,光谱分辨率为8.0cm[sup]-1[/sup],每4个小时进行背景光谱的采集。每张[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]NIR[/color][/url]光谱由1557个变量点组成。[b]2.4定量定性分析模型用于终点判断数据分析[/b]将在线混合过程进行监测,得到在线混合过程数据进行分析,以便了解混合全过程信息以及混合过程的监测。[b]2.5混合终点分析[/b]当得到混合终点时分别采集混合后的样品6处的原始[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]NIR[/color][/url]光谱,利用舒巴坦钠的定量分析模型预测混合终点时不同样品点处的舒巴坦钠的含量,判别是否混合均匀。[b]3 实验结果3.1分析模型的建立[/b]本研究中分别建立了在线混合过程的舒巴坦钠定量监测模型和基于移动块标准偏差的定性监测模型。[b]3.1.1 定性分析模型的建立[/b]目前混合均匀度在线监测常用的方法为MBSD法,本研究中MBSD法定性建模的参数为:选择的3个光谱区间包括全光谱、5275.6-4806.3cm[sup]-1[/sup](称为Region1)及7096.76-6344.66cm[sup]-1[/sup](称为Region2);用于计算光谱偏差的光谱的条数为5(即移动块的宽度为5)。[b]3.1.2 定量分析模型的建立[/b]本研究中所建立的定量分析模型用于监测混合过程中舒巴坦钠的百分含量的变化,因为本实验中舒巴坦钠和美洛西林钠两者间的混合比为4:1,当达到混合终点时,舒巴坦钠的百分含量应该在20%左右。其模型的具体参数见上一章中得到的舒巴坦钠百分含量的定量分析模型。[b]3.2混合在线过程数据分析[/b]本研究中平行进行了3次混合过程的在线监测,分别对3次实验结果进行分析,以充分了解混合监测过程。[b]3.2.1 第一批实验结果分析3.2.1.1 原始光谱图[/b]图1给出了混合过程中采集得到的208张原始光谱,由图中可知,处于下面的光谱较稀疏,可能属于混合刚开始的阶段,光谱会有较大的差异;处于上面的光谱较密集,其原因为随着混合的不断进行,光谱间差异越来越小,所以光谱较集中。[align=center][img=,498,274]http://ng1.17img.cn/bbsfiles/images/2017/09/201709141912_01_1626619_3.png[/img][/align][align=center]图1 第一批混合过程原始光谱[/align][align=center] [/align][b]3.2.1.2 在线混合过程结果分析[/b]图2为定性分析模型中得到的3个光谱区间的峰面图,其中M1为全光谱建模的峰面积变化,M2为Region 1(5275.6-4806.3cm-1)的峰面积变化,M2为Region 2(7096.76-6344.66cm-1)的峰面积变化,由峰面积的变化图可知,混合过程的前100s其变化较为明显,M1不断升高,M2和M3(7096.76-6344.66cm-1)不断下降,之后峰面积值趋于稳定。[align=center][img=,525,234]http://ng1.17img.cn/bbsfiles/images/2017/09/201709141913_01_1626619_3.png[/img][/align][align=center]图2 光谱区间峰面积图[/align]图3为舒巴坦钠含量及标准偏差变化图,由图中显示在混合的初期阶段,尤其是前100s左右,四个表征混合均匀度的参数均有着较大的变化趋势,在200-300s间四个参数有稍微较小的波动,此后随着混合过程的不断进行,表征混合均匀度的四个参数变化范围均变小,模型给出的舒巴坦钠的百分含量在20%左右,舒巴坦钠和美洛西林钠混合较为均匀,达到了混合终点。由图可知前100s是混合的主要阶段,此阶段舒巴坦钠的百分含量和标准偏差均有着明显的变化。[align=center][img=,538,292]http://ng1.17img.cn/bbsfiles/images/2017/09/201709141914_01_1626619_3.png[/img][/align][align=center]图 3 含量和标准偏差变化图[/align][align=center](a舒巴坦钠百分含量变化 b全光谱峰面积标准差 c Region1峰面积标准差 d Region2峰面积标准差)[/align][align=left] 当达到混合终点时分别采集表面皿下6个点的[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]NIR[/color][/url]光谱,根据建立的模型测定其舒巴坦钠的百分含量,看混合是否均匀。表2给出了用所建模型得到的6个点的舒巴坦钠的百分含量值,6个点舒巴坦钠的百分含量值在20%左右,说明混合较为均一,但是最大的值达到了22.41%,可能是由于混合装置过于简陋,加上是人为搅拌进行混合,不能达到很好的混合,部分地方没有进行很好的混合。从实验的可行性方面,初步证实了[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]NIR[/color][/url]技术用于美洛西林钠舒巴坦钠混合的可行性。[/align][align=center]表1混合后不同点舒巴坦钠百分含量值[/align][align=center] [img=,570,70]http://ng1.17img.cn/bbsfiles/images/2017/09/201709141915_01_1626619_3.png[/img][/align][b]3.2.2 第二批实验结果分析3.2.2.1 原始光谱图[/b]图4给出了第二批混合过程中采集得到的203张原始光谱,其混合过程原始光谱的特征和第一批混合过程较为相似,混合初期光谱变化较为明显,随着混合的进行,光谱差异变小,光谱较为密集。[align=center][img=,488,280]http://ng1.17img.cn/bbsfiles/images/2017/09/201709141915_02_1626619_3.png[/img][/align][align=center]图4 第二批混合过程原始光谱[/align][align=left] [b]3.2.2.2 在线混合过程结果分析[/b][/align]图5为各个光谱波段峰面积的变化图,由图中显示开始的100s内峰面积有着较大的变化幅度,随着混合的不断进行,峰面积的变化趋势不断减小并逐渐趋于稳定。[align=center][img=,516,307]http://ng1.17img.cn/bbsfiles/images/2017/09/201709141916_01_1626619_3.png[/img][/align][align=center]图5 光谱区间峰面积图[/align][align=center](a 全光谱峰面积 bRegion 1峰面积 cRegion 2峰面积)[/align]图6为舒巴坦钠含量及标准偏差变化图,由图可知在混合的初期阶段大约0-100 s时,舒巴坦钠百分含量值及峰面积的标准偏差值有着明显的变化,全光谱峰面积的标准偏差(Full Range STD)在200-400 s间有较为明显的波段,此后随着混合过程的不断进行,四个参数变化范围均变小,模型给出的舒巴坦钠的百分含量在20%左右。由此可知前100 s是混合的主要阶段,此阶段舒巴坦钠的百分含量和标准偏差均有着明显的变化。[align=center][img=,551,327]http://ng1.17img.cn/bbsfiles/images/2017/09/201709141917_01_1626619_3.png[/img][/align][align=center]图6 含量和标准偏差变化图[/align][align=center](a 舒巴坦钠百分含量 b 全光谱峰面积标准偏差 c Region 1峰面积标准偏差 d Region 2峰面积标准偏差)[/align]当达到混合终点时,采集表面皿底部6处的[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]NIR[/color][/url]光谱,检测混合过程是否达到均一,表2列出来了6处的舒巴坦钠的百分含量值,由表2可知达到混合结束后得到的6处的舒巴坦钠的百分含量均在20%左右,说明混合较为均匀。同时,由于实验条件的限制加上搅拌时人为因素的影响等,各点之间含量也着较大的差异。[align=center]表2 舒巴坦钠百分含量[/align][align=center] [img=,566,84]http://ng1.17img.cn/bbsfiles/images/2017/09/201709141918_01_1626619_3.png[/img][/align][b]3.2.3 第三批实验结果分析3.2.3.1 原始光谱图[/b]图7给出了混合过程中采集得到的207张原始光谱,由图中可知,得到的原始光谱图与第一批和第二批有着相似的结果,即混合的初期光谱差异大,因此光谱较为稀疏(偏下方的光谱),随着混合的进行,光谱间差异变小,光谱变得密集(偏上方的光谱)。[align=center][img=,505,262]http://ng1.17img.cn/bbsfiles/images/2017/09/201709141919_01_1626619_3.png[/img][/align][align=center]图7 第三批混合过程原始光谱[/align][b]3.2.3.2 在线混合过程结果分析[/b]图8给出了混合过程中3个光谱区间峰面积的变化趋势值,由图中可知0-100s间三个光谱区间的峰面积有着明显的变化,100-200s间峰面积有着明显的变化,但是变化幅度没有前100s大,200s以后峰面积变化趋势变小。说明前200s是混合的主要阶段,峰面积变化较为明显。[align=center][img=,519,343]http://ng1.17img.cn/bbsfiles/images/2017/09/201709141919_02_1626619_3.png[/img][/align][align=center]图 8 光谱区间峰面积图[/align][align=center](a 全光谱峰面积 bRegion 1峰面积 cRegion 2峰面积)[/align]图9为舒巴坦钠百分含量及光谱峰面积的标准偏差随时间变化的趋势图,其变化趋势和峰面积的变化趋势相似,前100s变化幅度较大,100-200s间也有较为明显的变化,但是变化幅度不是很明显,200s后舒巴坦钠的百分含量和峰面积的标准偏差均趋于稳定,说明此时光谱差异变小,混合趋于均匀。[align=center][img=,529,352]http://ng1.17img.cn/bbsfiles/images/2017/09/201709141920_01_1626619_3.png[/img][/align][align=center]图9 含量和标准偏差变化图[/align][align=center](a舒巴坦钠百分含量变化 b全光谱峰面积标准差 c Region1峰面积标准差 d Region2峰面积标准差)[/align]表3为达到混合终点时采集表面皿底部的[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]NIR[/color][/url]光谱得到的不同点的舒巴坦钠的百分含量值,由表中显示6个点的舒巴坦钠的百分含量值在20%左右,但是6个点之间舒巴坦钠百分含量间存在较大的差异,测得的最小值为17.80%,其原因可能是一方面由于实验条件的限制混合不够均匀,一方面用于舒巴坦钠含量测定的[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]NIR[/color][/url]定量分析模型也有一定的偏差,可能引起含量检测的差异存在。[align=center]表3 混合后不同点舒巴坦钠百分含量值[/align][align=center] [img=,564,66]http://ng1.17img.cn/bbsfiles/images/2017/09/201709141921_01_1626619_3.png[/img][/align][b]3.3小结[/b]通过3个混合平行实验的进行可知所建立的基于MBSD法的定性分析模型和基于舒巴坦钠百分含量的定量分析模型能够有效的监测舒巴坦钠、美洛西林钠的混合过程。由舒巴坦钠百分含量和标准偏差变化图可知两者的变化有着相关性,当舒巴坦钠的百分含量变化幅度大时,其标准偏差的变化幅度也较大,因此两者均可以用于混合过程的在线监测,证实了实验的可行性。[b]4 结论和讨论[/b]本研究采用AntarisII傅里叶变换[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱仪[/color][/url]对美洛西林钠、舒巴坦钠混合过程进行了在线监测。在研究中,分别建立了基于MBSD法的定性分析模型和基于舒巴坦钠百分含量的定量分析模型,然后Antaris II傅里叶变换[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱仪[/color][/url]漫反射采样方式采集混合过程中的光谱,实时监测混合过程的进行。通过3个平行实验的在线混合过程,结果显示MBSD法和舒巴坦钠百分含量测定法均能有效的监测其混合过程,有效的证明了[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]NIR[/color][/url]光谱分析技术用于舒巴坦钠、美洛西林钠混合在线监测的可行性。此外,MBSD法因为无需进行一级数据的采集,方法较为简单且容易理解,目前常用于混合过程的在线监测。本研究中有效证实了[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]NIR[/color][/url]光谱分析技术在舒巴坦钠美洛西林钠样品在线混合过程中应用的可行性,在样品的在线混合监测中有着重要的应用价值和应用前景。该技术能够克服传统方法费时、繁琐等缺点,而且可以实现过程的实时在线监测,让生产者充分了解整个生产过程中的参数变化。 [b]参考文献[/b]陆婉珍, 褚小立. [url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱[/color][/url]([url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]NIR[/color][/url])和过程分析技术(PAT). 现代科学仪器, 2007(004):13-17.SieslerH, Ozaki Y, Kawata S, et al. Near-infrared spectroscopy: principles .Instruments, Applications, 2002:35-181.Bhushan,K.R.,et al.Detection of breastcancer microcalcifications using a dual-modality SPECT/[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]NIR[/color][/url] fluorescent probe. J Am Chem Soc, 2008. 130(52):17648-17649.贾燕花. [url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱[/color][/url]分析技术在化学药品生产过程控制应用初探. 北京协和医学院, 2011.Fevotte.G,et al.Applications of [url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]NIR[/color][/url]spectroscopy to monitoring and analyzing the solid state during industrialcrystallization processes . Int J Pharm, 2004, 273(1):159-169.张敏.盐酸林可霉素多晶型分子构象对其红外光谱行为的影响.中国抗生素杂志, 2005, 30(009):529-532.Blanco M,R Goz"01ez Ba,E.Bertran,Monitoring powder blending in pharmaceutical processes by use of nearinfrared spectroscopy . Talanta, 2002, 56(1):203-212,田科雄.不同装载系数和混合时间对添加剂预混料混合均匀度的影响.河北畜牧兽医, 2004, 20(9):52-53.孙栋. 基于[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱[/color][/url]分析技术的几种固体粉末混合均匀度快速检测研究. 山东大学硕士学位论文, 2012年.
(1)细胞核的分离: 将培养的细胞制成细胞混悬液,或以胰蛋白酶消化法于培养盖上收集培养细胞。应用MgSO4染色分离方法分离细胞核(Van den Engh et al 1986, 在Traok和Van Den Engh 34章 )。细胞混悬液浓度为5×106/ml。利用RNA酶消化后,使核从细胞分离,细胞核混悬液浓度为4~5×106细胞核/ml。(2)细胞固定和酸的处理:在5 ml试管内加冷的100%酒精不断旋转以达到满意的固定。在冰上停留10 min。在4 ℃离心(×150 g)10 min。重复加三次冷的100%酒精入试管内,离心,倾去。置于冰上10 min,再离心。然后加入相当核悬液1/2量的0.1 n HCl , 0.5% Triton X-100。室温停留10 min。加入IBM-0.25%Triton X-100(IBM配方:50 mmol/l KCl, 10 mmol/L MgSO4, 5 mmol/L HEPES pH8.0)。再离心,重复IBM漂洗(这时细胞核可在不染色情况下,以荧光显微镜观察)后,以2×SSC-0.1%Tween漂洗1×3min,继之加入等量2%的多聚甲醛在1×PBS-5 mmol/l MgSO4。在室温静置站立10min。倾去上清液,加IBm -Triton X-100漂洗,离心,使细胞核混悬液最终浓度为108/ml,(可用IBM-Triton X-100稀释约50倍,在血球计数器计数),混悬液镜检应含单个,完整的细胞核。(3)细胞核混悬液杂交①配制杂交混合液:甲酰胺5份,20×SSC1份,50%硫酸葡聚糖2份,pH调至7.0。此原液(stock solution)可贮存在4 ℃冰箱内。应用时加1份10 mg/ml鲱鱼精子DNA(herring sperm DNA)。②混合1 μl的细胞核混悬液(108/ml)与18μl的杂交混合液,充分混匀。将此19 μl混合液移入1.5 ml容积的Eppendorf 管中(核含量约为105)。③加入100 ng/每管的AAF标记DNA探针(如为生物素标记DNA探针浓度为20~40 ng/每管)。④置70 ℃10min使DNA探针和核DNA变性。⑤和组织切片与DNA探针杂交方法相较,不同的是在加热变性后切勿置冰上迅速冷却以终止反应,而应迅速转入37 ℃孵育过夜。(4)杂交后漂洗在每管中加入1.25 ml 50%甲酰胺-2×SSC(pH7.0),在42 ℃静置10~15min。偶尔旋转以助混匀。冷却至室温。加100 μl经dimethylsuberimidate(DEMS)处理的血细胞(107/ml)混匀,离心,室温,10min,轻弹试管使沉淀的小块散开,加入1.25 ml 2×SSC(pH7.0),42 ℃,继之,静置于室温10~15min,如前离心,再加1.25 ml IBM-Triton X-100,室温静置5min,离心。注:DEMS处理红细胞方法:经漂洗并离心去除白细胞和血清的红细胞在盐液如PBS中,细胞含量为108/ml,以K2CO3和DEMS溶液处理3次,第1次:K2CO3为20 mmol/L,DEMS为3 mmol/L,以后2次:K2CO3依然为20 mmol/L,而DEMS为10 mmol/L。在应用前将K2CO3和DEMS液混合加入红细胞混悬液中。在最后2次漂洗液中,应用100 mmol/l K2CO3将pH调至9~10。在25 ℃,15min后,加入50 μl,100 mmol/l 的柠檬酸(citric acid)/每ml细胞混悬液的浓度以达固定红细胞的目的。固定的红细胞离心倾去上清液后,用2×SSC稀释到108/ml,加0.1%叠氮钠可在4 ℃保存至少1年。(5)AFF标记的荧光显示:加200 μl的PBS含0.05%Tween和2%正常血清(NGS),轻轻振荡混匀,室温静置10min,加20 μl 1:100的单克隆抗AFF抗体,37 ℃孵育45min,加1.25 ml的PBS-Tween,室温静置10min,加20间歇性振荡,离心,倾去上清液,加200 μlPBS含0.05%Tween –2%NGS,振荡,室温静置10min,加20 μl的羊抗小鼠–FITC荧光标记抗血清,稀释度1:100~1:300。孵育于37 ℃ 45min,加1.25 ml PBS –Tween,室温静置10min,离心,倾去上清液。(6)生物素标记探针的荧光显示:加200 μl 4×SSC含0.1%Trion X-100 和5%BSA。室温静置10 min后,加20 μl抗生物素标记FITC抗血清15 μg/ml,孵育在37 ℃ 30min,以1.5 ml 4×SSc –0.1% Trion X-100洗1次,加入1.25 ml IBm –Triton X-100,室温静置10~15min,间歇振荡、离心。(7)荧光显微镜观察:将细胞核混悬液稀释于250 μl的IBM-Trion X-100中,轻加振荡混匀。为抗荧光褪色可加等量的抗褪色溶液至载片上的细胞核涂片上,选择适当的激发波长观察。(8)流式细胞计:将750 μl的细胞核混悬液通过流式细胞仪(Flow cytometry, FCM),DEMS处理过的红细胞作为对照(Df 530/30nm, Omega ·Optical Inc, Brattleboro, VT)。
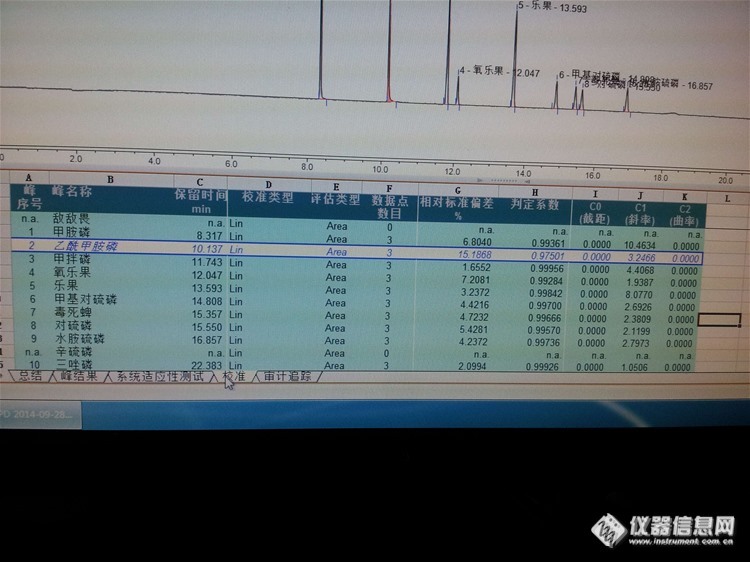
配了10个标样的混标,三个梯度混标的判定系数基本都是99%以上。我现在做四个梯度,反而只有6个是99%,其他是96-98%不等。如果再做五个梯度的话岂不是更低了,是不是得去掉一些偏差略大的峰来优化判定系数?标曲是否对梯度的个数有要求?还有啥其他要求呢。。。加标回收一般做些什么农药?回收率有什么要求?http://ng1.17img.cn/bbsfiles/images/2014/10/201410151535_518482_1645480_3.jpg
各位大侠,有谁知道Class Vp中如何显示分离度,我现在只能手算,抄下来峰宽,那计算器酸,好笨笨啊,请指点。
液相柱中有个参数是碳含量,那碳含量代表什么?对分离有什么影响呢?
岛津 CLASS VP 计算 分离度我的CLASS VP 在报告模板里加入分离度和理论塔板数时表格显示为0[~200092~]
清洗:从液体中分离密度较大且不溶的固体,分离沙和水;过滤:从液体中分离不溶的固体,净化食用水;溶解和过滤:分离两种固体,一种能溶于某溶剂,另一种则不溶,分离盐和沙;离心分离法:从液体中分离不溶的固体,分离泥和水;结晶法:从溶液中分离已溶解的溶质,从海水中提取食盐;分液:分离两种不互溶的液体,分离油和水;萃取:加入适当溶剂把混合物中某成分溶解及分离,萃取水溶液中的碘;蒸馏:溶液中分离溶剂和非挥发性溶质,海水中取得纯水;分馏:分离两种互溶而沸点差别较大的液体,液态空气中分离氧和氮;石油的精炼;升华:分离两种固体,其中只有一种可以升华,分离碘和沙;吸附:除去混合物中的气态或固态杂质,活性炭除去黄糖中的有色杂质。
[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质联用[/color][/url]检测食品包装材料的烷基酚。买了单标和混标,先不安柱子。能单标和混标分别建立质谱方法,在把买的单标和混标配成更混的标液,用之前建立的质谱方法做标曲吗?没安柱子时,是不是要先优化好流动相在做质谱啊?还想问问[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质联用[/color][/url]方法建立的顺序,是先不安柱子,将标液峰分开(需要峰好看吗),建立质谱方法。然后再安柱子,优化流动相,将物质峰分开,峰好看吗?配图1.2是没安柱子时,用的梯度洗脱,有机试剂:水,可能4:6,2:8这种比例。然后用的课上的梯度洗脱,见图三。用的混标但是质谱打的其中一个物质,想问问这个流动相怎么优化啊?3张图都是用纯甲醇冲过系统后测的。[img]https://ng1.17img.cn/bbsfiles/images/2024/02/202402271625316272_3777_6231875_3.png[/img][img]https://ng1.17img.cn/bbsfiles/images/2024/02/202402271625316721_9466_6231875_3.png[/img][img]https://ng1.17img.cn/bbsfiles/images/2024/02/202402271625316643_1975_6231875_3.png[/img]

在下现在在优化液相条件,用流动相为A:10mM甲酸铵水溶液,加0.1%甲酸;B:甲醇:水(95:5),加2mM甲酸铵和0.1%甲酸,测10种氨基酸的混标,试了很多种梯度都不能把样品很好地分开,而且峰形也很差,如图所示是其中一个梯度,实验室师姐之前用过的,我现在做也不行。求大神解答调整流动相梯度有没有什么规律可循,感激不尽!http://ng1.17img.cn/bbsfiles/images/2015/06/201506161802_550372_2979393_3.pnghttp://ng1.17img.cn/bbsfiles/images/2015/06/201506161802_550373_2979393_3.png
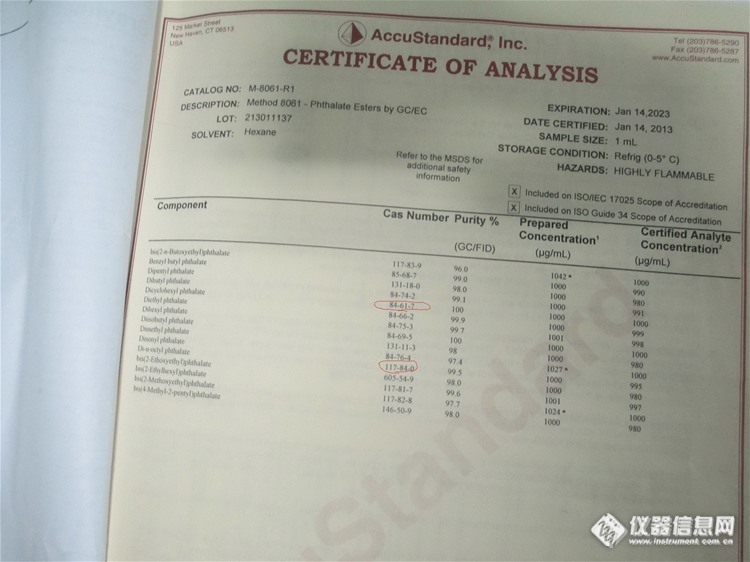
请教各位版友老师,关于邻苯15P混标中DEHP和DCHP峰分离的不好。(直接上图比较清楚)GC-MS 参数参考GB T 22048 2008 塑料中邻苯二甲酸酯增塑剂测定升温程序:150℃(3min)25℃/min 250℃25℃/min290℃(10min)尝试流速降低或者增加均没有什么效果。http://ng1.17img.cn/bbsfiles/images/2014/09/201409120945_513558_2190021_3.bmphttp://ng1.17img.cn/bbsfiles/images/2014/09/201409120944_513557_2190021_3.bmphttp://ng1.17img.cn/bbsfiles/images/2014/09/201409120943_513556_2190021_3.bmp标准溶液证书http://ng1.17img.cn/bbsfiles/images/2014/09/201409120943_513555_2190021_3.jpg
测羰基化合物,但是只跑了混标,想知道如何去区分这些混标中的各个单标?谢谢
混在纳米氮化硼中的α-氧化铝有什么好的分离办法吗?在知网的论文中没有查到资料,各位大神有好的办法吗?
各位,我配置了200μg/l的混标液,不知道能用多久啊?想用这个做加标和配标曲,这样合适吗?标曲和加标能用同一份母液吗?先谢谢大家啦
碳是有机世界的“主角”,在地球上按重量计算,占地壳中各元素总重量的0.4%,按原子总数计算不超过0.15%。而元素碳是一种十分神奇的物质,像碳纤维是比钢轻而抗拉强度高于钢7-9倍的材料。尤其是近20年纳米级大小的碳(富勒烯,碳纳米管,石墨烯等)人们给以前所未有的重视。 在利用各种吸附剂进行混合物分离发展的早期,人们就利用各种形态的碳做吸附剂用于分离各种混合物,现在人们又把目光投向富勒烯,碳纳米管,石墨烯等纳米级材料做新型分离材料用作固相萃取的吸附剂。 1. 活性炭作固相萃取吸附剂 活性碳是最早使用的固相萃取吸附剂,开始主要使用工业级别的活性碳,但是,使用了一段时间以后,吸附性能不能令人满意,就把它改性,以适应萃取分离的要求。在制备活性碳当中,要得到所需要的性能,碳化和活化过程的参数中最重要的是原料的选择和预处理。活性碳的基本性质取决于所用原料,使用的原料有自然的木头、泥炭、煤、果核、坚果的外壳以及人工合成物质——主要是聚合物。在没有空气和化学品条件下的碳化过程中,首先是大多数非碳元素(氢、氧和微量硫和氮)由于裂解的破坏而分解挥发了,这样元素碳就留下来,形成结晶化的石墨,其结晶以无规则方式相互排列,而碳则无规律地存在于自由空间里,这一空间是由于滞留在这里的物质被沉积和分解而形成的。进行碳化的目的是使之形成适当的空隙并形成碳的排列结构,碳化过程使碳吸附剂具有较低的吸附容量,使其比表面只有几个 m2/g,使之没有过高的吸附性。为了得到高空隙度和一定的比表面积,碳化还要进行活化过程。从天然原料制得的活性碳要比从合成物制得的活性碳具有较高的灰分,从合成化合物制得的活性碳几乎没有灰分,并且具有很好的机械性能,不易压碎和被磨损。由天然原料制得的活性碳其吸附性能受到它表面化学结构的影响,而其表面性质又决定于与其键合在一起各种杂原子(如氧、氮、氢、硫、氯等)的种类,活性碳是没有特殊选择性,或选择性很小的吸附剂。制备良好的活性碳为多孔结构,主要是各种直径的微孔和介孔,其比表面可达1000 m2/g到2m2/g,或者更高一些,使其具有高的吸附容量。活性碳表面具有很高的化学和几何不均一性,特别是工业用活性碳尤为严重。 固相萃取(SPE)使用活性炭始于上世纪 50 年代初,Braus 等人使用活性碳做吸附剂,在铁管中装1200-1500 g 碳纤维,用以富集水中的污染物,之后用索氏萃取器提取被吸附的有机物,包括水中的有机氯农药。(Anal Chem,1951,23:1160)。萃取管长91.44 cm,直径在10.16 cm,装填1200-1500 g 颗粒状活性碳,通过 5000 gal - 7500 gal 地表水吸附有机氯氯农药。 由于活性碳的缺点妨碍其使用,即吸附性不均一,重复性不好,有过高的吸附性,有不可逆活化点,回收率低。所以从上世纪 60 年代末到80 年代初,一直在寻找更为合适的适应性更强的 SPE 填料。 2. 碳分子筛作固相萃取吸附剂 在上世纪 70 到 80 年代,在研究聚合物吸附剂和键合有机物硅胶的同时,再次使用了性能改进的碳吸附剂——碳分子筛。这是由于当时的碳吸附剂结构改进、材质均一、性能稳定,同时它对极性化合物的吸附有好的选择性。碳分子筛的性能与 XAD-4 大孔树脂(以苯乙烯和丙烯酸酯为单体、乙烯苯为交联剂进行聚合)相同。 1968年 Kaiser 制备出一种碳吸附剂叫“碳分子筛”,国外的商品名是 Carbosieve B,它是用偏聚氯乙烯小球进行热裂解,得到固体多孔状的碳,其比表面为1000 m2/g,平均孔径为 1.2 nm 。这种吸附剂用于气-固色谱的固定相,我国称之为碳多孔小球(TDX),自然可以用作固相萃取的吸附剂。早年我国上海高桥化工厂、中科院化学所和天津试剂二厂相继研制成功这类碳分子筛,商品名叫做:碳多孔小球(Tan Duokong Xiaoqiu,TDX), 具体的牌号有 TDX-01;TDX-02。它们的堆积密度为 0.6 g/mL,比表面为 800 m2/g。碳多孔小球的特点是:非极性很强,表面活化点少,疏水性强,耐腐蚀、耐辐射,寿命长。表1列出国外厂家的碳分子筛的性能。表 1 商品碳分子筛的性能吸附剂商品名厂家比表面/(m2/g)孔径/nm堆积密度/(g/mL)Carbosieve BSupelco10001-1.20.226Carbosieve SSupelco5601-1.20.5-0.7Carbosieve S-II*Supelco5480.5-0.70.55-0.60Carbosieve G*Supelco 2040.5-0.70.25-0.28SpherocarbFoxboro12001.50.5+0.05CarbosphereChrompack10001.3 3 近年用碳纳米材料作固相萃取吸附剂 自从1991年日本学者饭岛澄男(Sumo Iijima)发现了碳纳米管(CNTs)之后,改变了人们过去对碳的三种形态(金刚石、石墨和无定形碳)的认识,对碳纳米管不断进行研究,并竞相把这种新奇的材料用在各个领域。在2004年又出现了另外一种有趣的碳物质——石墨烯,G),CNTs和G是碳的两种同素异形体,它们具有sp2杂化网络,但是结构不同,CNTs具有管状纳米结构,由石墨烯片卷成管状,形成准一维结构,而G是打开纳米管形成的平面二维薄片。CNTs可分为单壁碳纳米管(SWCNTs)和多壁碳纳米管(MWCNTs),石墨碳家族的各种形态如图1所示。http://img1.17img.cn/17img/images/201603/insimg/bcb66e42-ef71-4d27-964f-3618bb6e1ce4.jpg图 1 碳家族的各种形态(TrAC,2016, 77:23-43) (1) 富勒烯及其衍生物作固相萃取吸附剂 自从1990年Huffman 和 Kratschmer发表了能大量制备富勒烯(C60)之后,对这类物质进行大量研究,对这类化合物的制备和性能有不少文章和综述发表,日本的 Jinno Kiyokatsu研究组对富勒烯进行了大量研究(Anal. Chem., 1995, 67:2556),把富勒烯键合到硅胶上用作HPLC的固定相,分离多环芳烃。Gallego等揭示了C60作为吸附剂在分离富集金属离子的潜力(Anal Chem,1994, 66:4074),它对金属离子的分离富集能力优于常规萃取剂——键合烷基硅胶和活性炭。例如超痕量镉在C60富勒烯微柱上进行分离, 形成中性配合物,用200mL对甲基异丁基酮洗脱吸附的镉,用原子吸收光谱进行测定。用双螯合试剂,即吡咯烷铵(APDC)和8-羟基喹啉,在一个流路中进行检测。APDC和C60富勒烯对镉进行选择性吸附,与含有的铜、铅、锌、铁中分离出来。与其他方法对比, C60和APDC的方法得到更为准确的结果(J Anal Atom Spectrom, 1997, 12: 453-457)。 2000年M Valcárcel等使用一个简单的流动注
混合气体可主要含有一氧化碳、二氧化碳,少量氧气、氮气,只想把二氧化碳分离出来,其他气体为一个峰,什么色谱柱分离效果好?
最近在做一个实验,使用的机子是PE 8000,配制的混标10个元素,浓度分别是1,5,10ppm,在测标曲结束后,线性很好,基本都是至少999以上,但在回测标曲1ppm,有其中4个元素浓度低于1ppm,回测5ppm还是这4个元素低于5ppm,只有到最后回测10PPM时才基本差不多,但有1,2个元素值在9.86,9.9左右,算行不行,各位大虾支支招,怎么保证回测3个标准点都是相应的值?
用气相分离有机磷的混标,但却检测到很多农药中间体,这个实验结果让我很困惑,请教一下这个是什么情况?
1:杂质明确,能够得到,且能保障供给的,应首选该杂质与主成分按适当比例混合,组成分离度用溶液。2:杂质明确,能够得到,但不能保障供给的,可选一易得的化合物,通过试验找到该杂质、主成分、化合物三者之间的关系,在制定标准时,通过对主成分与化合物分离度要求,用化合物与主成分的混合液作为分离度用溶液。3:未知杂质或杂质无法得到的情况下,可根据工艺、主成分的特性分析,尝试将主成分经合适条件(光、热、酸碱等)降解后,如果能够得到一组降解物溶液,其组分固定,且基本稳定,并能够与主成分组成比例合适的也可以作为分离度用溶液。4:当上述方法均不适用时,可考虑选用一种与主成分结构非常相似的化合物,与主成分按适当比例混合,组成分离度用溶液。
请问用UPLC碳18柱能分离DNA吗?如果能可以用什么样的流动相。会不会度柱子
求教mass hunter怎么显示分离度和理论塔板数之类的数据啊,现在是只能添加column,没法显示出数值
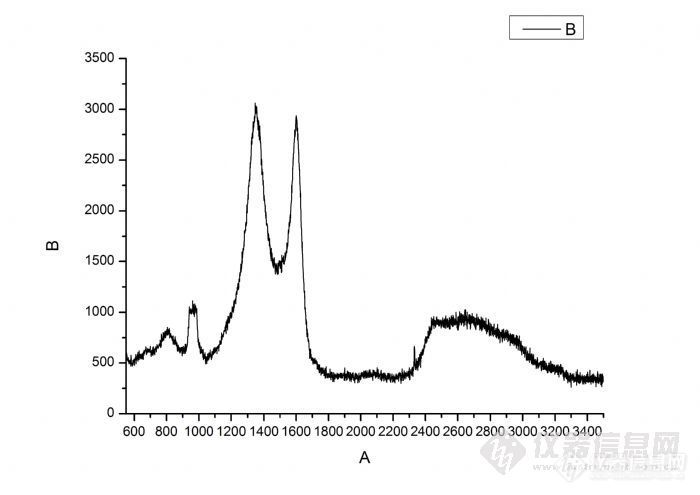
PAN基碳纳米纤维的拉曼光谱如下(纤维放在Si(n)片上的,碳化温度是900度)问题:为什么在940到990波数出现很宽的峰(不管升温速度怎么变,这个现象都会出现)?如果碳化温度是600度,则在940到990波数不会出现峰。为什么会发生这种情况?有谁能解释一下吗?)http://ng1.17img.cn/bbsfiles/images/2012/04/201204170927_361789_2112012_3.jpg
请教各位: 我将丙酮和丁酮混标分别注入FFAP、TVOC、,均能得到较好的分离条件,但做职业卫生样品时发现FFAP柱子在丙酮或丁酮出峰时间出的峰很大,有的时候甚至能超标,但同一个样品用TVOC柱子不会有这种情况,测得的值较低。请问这是怎么回事? 现在我又将丙酮和丁酮混标 打入KB-WAX柱子,峰形也很好,会不会再出现上述情况?与柱子极性有关系吗?
求助谁知道哪卖阴离子交换介质呀!--用于分离生物基质中的蛋白混合物颗粒最好小些,5微米左右的,孔径大于300埃的至少300埃。不要柱子,只要填料。
近期考核做了一个钾钠钙镁混合标样,要求是直接吸取10ml样品用蒸馏水稀释至250ml容量瓶测定。因之前做过钾、钠、钙、镁单标考核样,如果直接用蒸馏水定容而不加遮蔽剂的话根本不准,但钾钠遮蔽剂为硝酸铯,钙镁遮蔽剂为硝酸镧,头疼~后来做的时候把样品分成3份:1.10ml样品用蒸馏水定容至250ml。(含5%硝酸铯)2.10ml样品用蒸馏水定容至250ml。(含2%硝酸镧)3.10ml样品用蒸馏水定容至250ml。以下是实验数据:K:0、0.2、0.4、0.6、0.8、1.6对应吸光度为0.001、0.039、0.073、0.108、0.145、0.277,曲线为y=0.172x+0.004,r=0.9997结果为1的钾浓度为1.395,2的结果为1.072,3的结果为1.040;同理,1的钠浓度为0.810,2的钠浓度为0.732,3的浓度为0.702;1的钙浓度为4.052,2的钙浓度为4.041,3的钙浓度为3.475;1的镁浓度为0.218,2的镁浓度为0.211,3的镁浓度为0.177。发现钙镁加硝酸镧或硝酸铯浓度基本一致,而钾钠则差距很大。后得知混标样品真值为K:1.23-1.35Na:1.14-1.24Ca:3.78-4.10Mg:0.189-0.231。。。。求大侠解释,钾钠钙镁混标到底怎么做?(我标准液按大气降水中钾钠、钙镁的测定方法配置)
提高回弹法检测混凝土抗压强度精确度的探讨回弹法检测混凝土抗压强度在我国使用已达四十余年,因其简便、灵活、准确、可靠、快速、经济等特点而倍受工程检测人员的青睐,是我国目前工程检测中应用最为广泛的检测仪器之一。当对工程结构质量有怀疑时,均可运用回弹法进行检测。但回弹法在使用过程中还是出现了较多的操作不规范、随意性大、计算方法不当等问题,造成了较大的测试误差。如何保证检测精度,使其在监督检验结构工程和混凝土质量中发挥应有的作用,已成为众多工程建设者所关注的话题。要提高回弹法的检测精度,应综合考虑以下几个方面因素。 1 注意回弹法检测的适用条件 回弹法是通过回弹仪检测混凝土表面硬度从而推算出混凝土强度的方法,当出现标准养护试件数量不足或未按规定制作试件 对构件的混凝土强度有怀疑 或对试件的检验结果有怀疑时,可按《回弹法检测混凝土抗压强度技术规程》(JGJPT2322001) (以下简称《规程》) 进行检测。必须注意回弹法的使用前题是要求被测混凝土的内外质量基本一致,当混凝土表层与内部质量有明显差异,如遭受化学腐蚀、火灾、冻伤,或内部存在缺陷时,不能直接采用回弹法检测混凝土强度。 2 测试前必须进行回弹仪的率定试验回弹仪的质量及测试性能直接影响混凝土强度推定的准确性,只有性能良好的回弹仪才能保证测试结果的可靠性。回弹仪的标准状态应是在洛氏硬度HRC 为60 ±2 的标准钢砧上,垂直向下弹击三次,其平均率定值应为80 ±2 ,否则回弹仪必须进行调整或校验。在单个构件检测中,一般只需测试前进行率定即可,但在大批量检测时,由于受现场灰粉及回弹仪自身稳定性等因素的影响,随着工作时间的延长,回弹仪的工作状态逐渐低于标准状态。有时一个批量检测项目检测前后回弹仪率定值的差异较大,从而导致测试结果偏低。因此,在大批量检测时,应随身携带标准钢砧,以便随时进行率定检测,适时更换,从而保证检测结果的精确性。 3 测区选择要正确 检测构件布置测区时,相邻两测区的间距应控制在2 m以内,测区离构件端部或施工缝边缘的距离不宜大于0. 5 m且不宜小于0. 2 m 测区应选在使回弹仪处于水平方向检测混凝土浇筑面,并选在对称的两个可测面上,如果不能满足这一要求时,也可选在一个可测面上,但一定要分布均匀,在构件的重要部位及薄弱部位必须布置测区,并应避开预埋件。当遇到薄壁小构件时,则不宜布置测区,因为薄壁构件在弹击时产生的振动,会造成回弹能量的损失,使检测结果偏低。如果必须检测,则应加以可靠支撑使之有足够的约束力时方可检测。 4 测试动作要规范,切忌随意操作 回弹法本身是一种科学的操作方法,国家也专门制定了相应的规程,不容许操作人员随意操作。回弹的精度也取决于操作人员用力是否合适和均匀,是否垂直于结构或构件的表面,是否规范操作。但实际检测中却很少有人严格按照标准规定的技术要求进行检测操作,责任心不强,敷衍了事,这样的检测将带来较大的测试误差,无法保证回弹质量,为此,应加强检测人员的职业道德素养,提高检测责任心,也只有如此,才能真正提高回弹法的检测精度。 5 消除测试面因素的影响 《规程》规定:用于回弹检测的混凝土构件,表面应清洁、平整,不应有疏松层、浮浆、油垢、蜂窝、麻面。我们在检测时经常遇到麻面或有浮浆的构件,回弹前必须有砂轮磨平,否则结果偏低。在测试面达到清洁、平整的前提下,还需注意混凝土表层是否干燥,混凝土的含水率会影响其表面的硬度,混凝土在水泡之后会导致其表面硬度降低。因此,混凝土表面的湿度对回弹法检测影响较大,对于潮湿或浸水的混凝土,须待其表面干燥后再进行测试。建议采用自然干燥的方式。禁止采用热火、电源强制干燥,以防混凝土面层被灼伤,影响检测精度。 6 注意碳化深度的测试取值 碳化深度值的测量准确与否与回弹值一样,直接影响推定混凝土强度的精度。在碳化深度的测试中,注意其深度值应为垂直距离,而非孔洞中呈现的非垂直距离。孔洞内的粉末和碎屑一定要清除干净之后再测量,否则将难以区分已碳化和未碳化的界线,造成较大的测试误差。测量碳化深度值时最好用专用测量仪器,不能采用目测方法。还有一种情况应特别注意,在检测已用粉刷砂浆覆盖的构件碳化深度时,由于测试面受水泥砂浆的充填渗透影响,其表层含碱量较高,而用于碳化测试的酚酞酒精溶液遇碱即变红,极易使人产生视觉误差,认为其碳化深度值很小。如果认真观察测试孔,可发现外表层颜色较深,而孔内混凝土所变的颜色较浅,这颜色较浅部分的厚度即为混凝土实际的碳化深度。这一点细微的差别,检测人员一定要注意区分。 7 注意混凝土回弹值的修正 近年来,随着城市泵送混凝土使用的普及,采用回弹法按测区混凝土强度换算值表推定的测区混凝土温度值将明显低于其实际强度值。这是因为泵送混凝土流动性大,粗骨料粒径较小,砂率增加,混凝土的砂浆包裹层偏厚,表面硬度较低所致。因此在运用回弹法检测混凝土强度时,必须要事先了解到施工单位浇注混凝土的方式,并注意修正。另外,当检测时回弹仪为非水平方向且测试面为非混凝土侧面时,一定要先按非水平状态检测时的回弹值进行修正,然后再按角度修正后的回弹值进行不同浇筑面的回弹值进行修正,这种先后修正的顺序不能颠倒,更不能用分别修正后的值直接与原始值相加或相减,否则将造成计算错误,影响对混凝土强度的推定。 8 测试异常时,需与钻芯法配合使用现行的工程施工中,普遍采用胶合板面的大模板,此种模板密闭性能极好但不透气,振捣过程中产生的气泡聚集在混凝土表面和大模板之间,不易排出,致使拆模后在混凝土表面存在大量的微小气孔,使混凝土表面不是很密实,如果混凝土养护跟不上,混凝土表面将不能有效地进行水化反应,不仅有粉化现象,而且混凝土碳化深度较大,造成混凝土表面强度低。如我市某一框架结构商住楼,在使用回弹仪抽检三层剪力墙混凝土时发现,全部抽检构件混凝土表面强度都比较低,只达到原设计强度等级的67 %。经查施工技术资料,该工程的混凝土配合比以及使用的原材料均不存在问题,施工单位混凝土搅拌后的管理也比较到位,遂用钻芯法取样复检,芯样上观察,混凝土表层10 mm 较疏松。内层较为坚硬,芯样检测结果是实际混凝土抗压强度符合原设计强度等级,从而避免了一次误判。 9 建立本地区的专用测强曲线 国家标准虽给出了全国通用回弹法检测的测强曲线并由此得到测定混凝土强度值换算表,但全国统一曲线仅综合考虑到全国各地的原材料使用情况,没有把碎、卵石普通混凝土区分开来,而实际上回弹法检测碎、卵石普通混凝土强度是有很大差异的。而地区测强曲线正是充分考虑本地区的混凝土原材料、气候条件和成型养分护工艺,通过试验、校核、修正所建立的曲线,与通用测强曲线相比较,该曲线比通用测强曲线更接近实验数据,能更好的推算本地区混凝土的实际强度。因此,建立本地区的专用测强曲线,能有效地提高回弹法的检测精度。
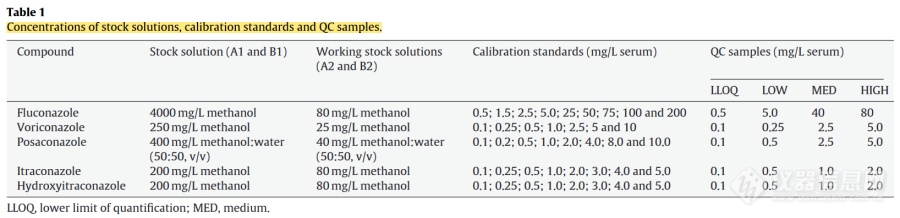
这篇文献里用的是不同浓度的甲醇或者甲醇-水配制,意思是各自从储备液稀释到系列工作溶液了,再各取100μ配个混标溶液吗?配制溶液的时候用不同浓度的甲醇或者甲醇-水不会很麻烦吗?还是说这个calibration standards的浓度是混标溶液里算出来该药物的浓度?怎么算呢?我对混标溶液的理解是,各自配1 mg/ml的储备液,然后从这各自的储备液里就各取100μ,配制混标,稀释得到系列工作溶液,再继续稀释到标曲的浓度[img=,690,167]https://ng1.17img.cn/bbsfiles/images/2022/10/202210161045239449_1230_5833917_3.png!w690x167.jpg[/img][img=,690,411]https://ng1.17img.cn/bbsfiles/images/2022/10/202210161045240738_2760_5833917_3.png!w690x411.jpg[/img]
Sepax CNT Size Exclusion Phases用于分离碳钠米管和碳纳米纤维 分离纳米管的先驱 质量最优产品概述 利用独特的表面技术, Sepax CNT SEC固定相由特殊涂布的多孔硅胶物质组成。硅胶纯度高,且具有增强的机械稳定性。 Sepax CNT SEC经过革新后,特别对纳米管(如纳米碳管和纳米碳纤维)的分离具有最高的分辨率及最大回收率。 Sepax 独特的表面技术使柱与柱之间具有很好的重现性及稳定性。 Sepax CNT 体积排阻柱固定相颗粒均匀,球形颗粒孔径有 300Å , 500Å , 1,000Å , 和 2,000Å ,孔体积为 1.0 mL/g 。 Sepax CNT SEC 固定相 用特殊技术填充,使其均一稳定,从而具有最高柱效。 Sepax CNT SEC 柱主要用于缓冲溶液和普通有机溶剂(如乙腈、甲醇和四氢呋喃)中纳米管的分离。 应用 根据长度分离碳纳米管 根据长度分离纳米纤维 根据直径分离纳米粒子 分析、半制备、制备型分离 详情请查询:www.sepax-tech.cn
用四氯化碳萃取碘后,那碘和水混合,变为碘与四氯化碳混合,不是没有分离碘单质吗》
最近要用气质考核这个三氯甲烷和四氯化碳,给的浓度范围是三氯甲烷是100-200mg/L,四氯化碳是30-80mg/L,我应该是把线陪在未知样周围,对吧但是对于混标以前没做过,所以有些疑问,请各位高手帮忙解释一下,先谢谢了一、怎么能把标液的浓度分别均为1000mg/L的三氯甲烷和四氯化碳配成混标? 现在有个棘手的问题,没有那么小的容量瓶来定容。我设定的是分别取0.75mL的三氯甲烷和0.24mL的四氯化碳定容在3mL,可是没有这么小的啊二、安捷伦的自动进样瓶可以作为定容的容器吗?先想到这里,希望没睡的兄弟姐妹们帮帮忙吧







 我要推广仪器
我要推广仪器
 下载APP
下载APP