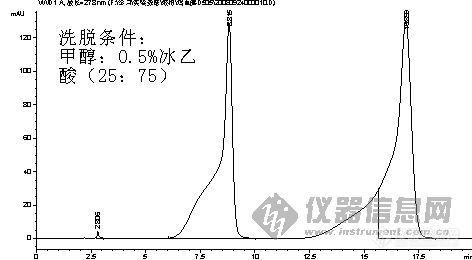
请教各位:我现在想做个药材中儿茶素的含测,但是在做对照品的时候,儿茶素出现前延峰。我的洗脱条件是甲醇:0.5%乙酸(25:75),请教各位如何解决这个前延问题啊? 我做了很多针都这样呢,还有购买对照品需要注意些什么问题呢 ?我是在药检所买的,他有义务为我提供一张图谱么?是不是小弟的话不清楚啊?我们可以共同讨论一下的 ,希望不要见怪!不好意思 刚想起来添加一张儿茶素、表儿茶素混合对照品的图谱 敬请高手指点,是对照品问题还是流动相问题 拜谢了![img]http://ng1.17img.cn/bbsfiles/images/2008/09/200809251040_110105_1628023_3.jpg[/img]
表儿茶素(L-Epicatechin)分子式:C15H14O6分 子 量:290.27白色结晶性粉末。儿茶素又称儿茶精,茶单宁。为黄烷醇的衍生物。分子式:C15H14O6分子量:290.27儿茶素最初由儿茶中提取出。为无色结晶形固体,能溶于水。其水溶液受热或在无机酸存在下,容易聚合成无定形鞣质。 和咖啡因同属茶叶中的两大重要机能性成分,但是又以儿茶素为茶汤中最主要的成分。临床实验调查显示,儿茶素可以通过血液循环进入全身,加强新陈代谢,增强脂肪的氧化和能量消耗从而达到抑制肥胖的作用,尤其是对内脏脂肪的抑制作用,能达到理想的减肥效果。以下为使用资生堂色谱柱对儿茶素和表儿茶素检测得到的谱图,请参考。http://ng1.17img.cn/bbsfiles/images/2017/01/201701191701_669681_2222981_3.jpghttp://ng1.17img.cn/bbsfiles/images/2016/11/201611221550_01_2222981_3.jpghttp://ng1.17img.cn/bbsfiles/images/2016/11/201611221550_02_2222981_3.jpg【色谱条件】色谱柱:CAPCELL PAK C18 S5; 4.6×150流动相:(N,N-二甲基酰胺/四氢呋喃=4:1)/0.04moL/L枸橼酸溶液=13/87流 速 : 1.0mL/min检 测 : UV280nm注:文献中所用液相方法与《中国药典(2015版)》中小儿泻速停颗粒检测方法一致。
https://www.docin.com/p-1041417404.html [b][size=24px][color=#333333]表10 绿茶精加工生产记录单 word 版本[/color][/size][/b]
哪位老师有儿茶素,表儿茶素标准拉曼光谱图谱,希望大家帮助一下,谢谢啦
【作者】 邢淑艳; 郭娇娜;【机构】 吉林省白城市食品药品检验所;【摘要】 目的建立HPLC法测定脑络通胶囊中原儿茶醛的含量。方法采用Diamonsil C18 5μm 250mm×4.6mm色谱柱。以甲醇-1%磷酸溶液(8∶92)为流动相,流速为0.7mL/min,柱温为30℃,检测波长为230nm。结果原儿茶醛线性关系良好,回归方程Y=6334.9X+1.1137,r=0.9999;精密度RSD=0.14%;稳定性RSD=1.16%;重现性试验RSD为1.69%(n=5);加样回收试验本方法的平均回收率为93.69%,3次测定结果的RSD为1.69%;阴性干扰试验表明,阴性对照液不干扰原儿茶醛的含量测定。结论本方法专属性强、准确度高、重现性好。 更多还原【关键词】 高效液相色谱法; 脑络通胶囊; 原儿茶醛; 含量;
用纯甲醇溶解(+)-儿茶素对照品,以乙腈(A)-0.1%磷酸水(B)为流动相,洗脱程序为0-2min 5%A,2-5min 5-17%A,5-12min 17-18%A。在上面的条件下,进样量的差别非常大,5ul的时候会出现分叉的峰,8ul的时候会出现两个峰,请问各位大佬这个是什么原因?[img]https://ng1.17img.cn/bbsfiles/images/2021/11/202111251934277564_3512_3242650_3.png[/img]
作者:偶志红,徐丹丹,郭薇 来源:中国论文下载中心【摘要】 目的 建立薄层层析-紫外分光光度法测定香丹注射液中原儿茶醛的含量。方法 以原儿茶醛为对照品,展开剂为二氯甲烷-乙酸乙酯-甲酸(8∶5∶0.8)分离得到原儿茶醛,采用紫外分光光度法,检测波长281nm,测定原儿茶醛的含量。结果 原儿茶醛在0.55~4.95μg/ml 范围内线性关系良好,r=0.9998。平均含量为0.174mg/ml,加样回收率为97.3%~102.3%,RSD为1.6%。结论 实验结果表明,薄层层析-紫外分光光度法测定香丹注射液中原儿茶醛的含量,方法操作简便,准确度高,精密度好,可作为样品的检测方法。 【关键词】 香丹注射液;原儿茶醛;薄层层析-紫外分光光度法;含量测定
作者:徐晓明1 徐大勇2纪晓芳2 邢俊波3(1.吉林省梅河El卫生职工中等专业学校,吉林梅河口,135000;2.吉林省梅河L1市医院爱民医院药剂科,吉林,梅河口,135000;3.总后卫生部药品仪器检验所,北京,100071)摘要:目的:建立测定复方儿茶胶囊中儿茶素的HPLC定量法。方法:采用高效液相色谱法。以DiamonsilCl8(250Ⅻx4.6mm,5¨Jn)为色谱柱;流动相:甲醇一水(21:79);流速:1.0ml·min~;检测波长:280nm;结果:儿茶素在0.1728一1.728199范围内。进样量与峰面积线性关系良好(r=O.9998)。平均回收率为:99.62%(RSD=O.94%)。结论:方法简便、快速准确、灵敏度高,重现性好。可作为复方儿茶胶囊的含量测定法。谱图:无
表儿茶素H-NMR氢谱图,溶剂是氘代甲醇?请问谁有?若能给我传一份,感激不尽啊,linmuyuying@163.com
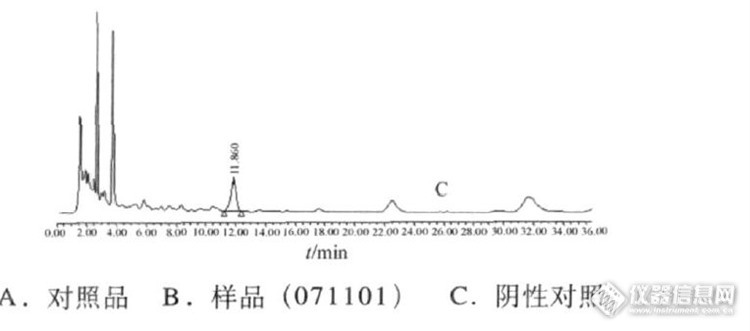
作者:王丽彦, 张竹( 长春市食品药品检验所,长春 130012)摘要:目的:探索康复灵栓中儿茶的含量测定方法。方法:用迪马钻石ODS C18色谱柱,以0.04 mol.L-1枸橼酸溶液-N,N二甲基甲酰胺-四氢呋喃(45∶8∶2)为流动相,检测波长为280 nm。结果:儿茶素在0.148 7~1.487μg范围内,表儿茶素在0.1081~1.081μg范围内线性关系良好(r=1.000),平均回收率为97.4%(n=6)。结论:用HPLC测定法检测康复灵栓中儿茶含量方法简便,准确可靠。谱图:http://ng1.17img.cn/bbsfiles/images/2012/07/201207161422_377869_1606903_3.jpg
茶多酚中总儿茶素的[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱[/color][/url]分析摘 要:采用[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱[/color][/url]分析技术对茶多酚中儿茶素进行了光谱分析,结果表明该技术能够解析出儿茶素中各主要基团在近红外波段的吸收特性,并且结合定标过程定量快速检测总儿茶素在茶多酚中的含量,分析结果在很大的样品浓度范围内给出了很高的精度, SEC = 2. 15% ,相关系数( r)为0. 9947。关键词 茶多酚,儿茶素,[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱[/color][/url]1 引 言 茶多酚( tea polyphenol)是从茶叶中分离提取出的一类多酚类化合物复合体,其主要的成分是儿茶素。研究证明,茶多酚不仅是一类优良的天然抗氧化剂,还具有多种保健功能和药理作用] ,如抗菌、抗病毒、抗毒素、防癌抗癌及降血压等,其中起主要作用的是儿茶素类物质。茶多酚中总儿茶素含量的高低直接影响着茶多酚的品质和价格。总儿茶素的常规定量检测方法是高压液相色谱(HPLC)法。该方法分析精度较高,但是分析过程复杂,速度较慢,且对样品有化学污染,无法满足生产过程中在线成分含量监控的要求。[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱[/color][/url]技术[ 4 ]是一种快速无损的分析技术,可以在不到1 min的时间内即可完成物质多组分的检测。[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱[/color][/url]是物质中官能团CH、OH及NH等在中红外区能量较高的基频吸收的倍频、合频和差频吸收谱叠加而成的。由于自然界中许多物质含C、H、O、N,所以该技术目前被广泛应用于农业、化工、医药、纺织、煤炭等多个领域。茶多酚中儿茶素的主要成分有E[url=https://insevent.instrument.com.cn/t/Mp]gc[/url]G( ep igallo-catechin gallate) , E[url=https://insevent.instrument.com.cn/t/Mp]gc[/url] (ep igallocatechin) , ECG( ep icatechin gallate) , EC ( ep icatechin)及catechin,其中含有大量的OH、CH、OH 、CH2 、CH OH 、COROH 等官能团,在[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱[/color][/url]区有明显的吸收(见图1) 。本实验采用[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱[/color][/url]分析技术定量分析茶多酚中总儿茶素的含量,给出了较高的分析精度。通过对总儿茶素的[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱[/color][/url]进行解析,找到了其主要官能团在近红外区的最大相关吸收谱区,为建立稳定的定标模型奠定了基础。2 实验部分 NicoletNEXUS 870傅里叶变换光谱仪(美国Thermo Electron公司)以及配套的TQ Analyst V6化学计量分析软件。实验样品:具有一定总儿茶素含量梯度的茶多酚粉末样品46份,成分浓度范围8% ~98% ,从中选取具有代表性40个样品作为定标集, 6个样品作为预测集,总儿茶素的化学值由标准HPLC法测定。茶多酚光谱数据:茶多酚的[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱[/color][/url]是在Thermo Nicolet NEXUS 870傅里叶变换光谱仪上测定。测量方式为漫反射式,数据采集分辨率为32 cm- 1 ,扫描平均次数为30次。3 结果与讨论3. 1 红外光谱解析 近红外区的倍频吸收谱是其对应的中红外能量较高的基频吸收的整数倍,合频和差频则分别对应于不同官能团基频之和或差。但由于各基团化学组成环境及内部结构的不同而有所偏移。在中红外波__段各官能团和基团都有各自的特征吸收谱线,吸收峰相互分离且较为尖锐,易于判别,而在近红外波段(12500 ~4000 cm- 1 )倍频、合频和差频吸收峰较宽且相互重叠,所以很难直接解析其所对应的吸收峰位置。通过对[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱[/color][/url]进行导数处理来提高光谱分辨率可以找到各官能团所对应的吸收峰位置。然而导数阶数越高噪声影响越显著,所以要求用于采集原始光谱数据的光谱仪具有很高的信噪比。图2中给出了茶多酚的[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱[/color][/url]及其二阶导数光谱。二阶导数光谱中的极小值对应于原始吸收谱的吸收峰位置,通过与近红外经验吸收光谱[ 5 ]相对照,确定儿茶素中主要官能团的吸收位置。3. 2 定标波长的优选 依据朗伯-比尔定律,[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱[/color][/url]数据与样品浓度数据之间要求满足严格的线性关系,这就要求实际测量数据(包括光谱数据、成分化学分析数据)的采集过程中应尽量减小引入如噪声、颗粒散射、光源光谱特性的漂移、探测器响应特性等。导致非线性影响的因素。在定标的过程中采用散射校正、导数光谱、光谱数据除法数学运算等软修正方法及延长光谱仪稳定时间、选取光谱特性线性较高的光源和探测器等硬修正方法。在非线性影响较小时,建立稳健的定标模型的关键在于选择恰当的定标波长,通过波长选择建立样品吸光度数据和成分化学浓度数据之间的最优相关关系。图1和图3分别为茶多酚在10000~4000 cm- 1范围内经过散射校正的近红外吸收谱及其相关光谱。经过散射校正后的光谱能最大限度的保留各样品[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱[/color][/url]之间由于成分浓度的线性变化而产生的光谱变化,很大程度上消除了由于散射所导致的非线性效应。相关光谱是通过在整个波长范围内每个经过散射校正的波长点处所有定标样品的吸光度数据与成分浓度数据进行简单的线性回归得到相关系数,然后将各个波长点计算所得的相关系数依次绘图获得的。通过对照比较,相关光谱反映了每个波长与茶多酚样品中总儿茶素之间的相关关系。相关系数的变化与茶多酚中儿茶素能量较强的主要基团的吸收峰严格对应。如图2中儿茶素在6896 cm- 1、5208 cm- 1、4672 cm- 1处的3个强吸收峰分别为OH的一级倍频、C O 的二级倍频、CH的合频吸收峰在相关光谱图中都有很高的相关系数。比较而言, 5208 cm- 1处的相关较弱而且相关峰很尖锐,在9000~5200 cm- 1处由于CH的一级倍频吸收而形成了较宽且较高的吸收峰,这段区域为较为理想的定标波长选择区域。依据相关光谱所给出的信息,通过实验比较,发现选取6000~5200 cm- 1波长范围内的光谱数据点作为定标波长结合偏最小二乘回归方法给出了很低的定标标准差( SEC) , SEC = 2115% ,相关系数( r)为0. 9947,定标结果散点图如图4所示。3. 3 标定结果 总而言之,通过对茶多酚中儿茶素的[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱[/color][/url]进行二阶导数光谱分析并结合儿茶素中各主要基团在近红外波段的经验吸收相比照,找到了其倍频及合频的吸收峰位。对儿茶素的[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱[/color][/url]特性与化学浓度在各个波长处进行相关分析,得到的相关光谱与散射校正光谱相对比发现,儿茶素1450、1920和2140 nm 处的3个强吸收峰在对应的相关光谱中给出了较高的相关系数。而这3个波长处的相关峰的展宽及强度又有所差异,这对于起始最优定标波长选择提供了依据,展宽较宽且相关较高的吸收峰是最佳的定标波长选择区域。依据相关光谱选取6000~5200 cm- 1波长范围内的光谱数据点作为定标波长,并结合偏最小二乘回归方法给出了很高的定标精度。
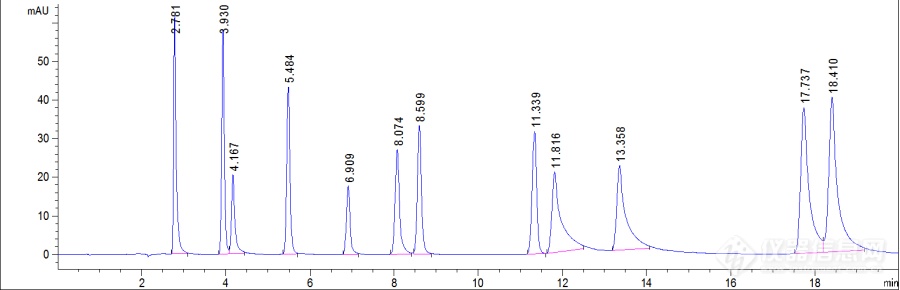
用安捷伦Poroshell 120 SB-C18的新柱子测定儿茶素和生物碱的标品,后面4种物质拖尾严重和参考资料的图谱相差很多,向各位老师请教一下有什么方法可以改善拖尾。流动相为乙腈和0.2 %的甲酸。[img=,690,222]https://ng1.17img.cn/bbsfiles/images/2019/04/201904181944037493_2590_3893580_3.png!w690x222.jpg[/img]
儿茶素的标液,表儿茶素和表没食子儿茶素没食子酸酯分不开,流动相什么,都是照国标做的,只做了儿茶素的五个混标[img=,690,517]https://ng1.17img.cn/bbsfiles/images/2021/12/202112181438294998_8078_4208728_3.png[/img]
关于《GB/T8313--2008茶叶中茶多酚和儿茶素类含量的检测方法》有很多问题需要向各位请教。 一、标准只规定五种儿茶素的标准工作液浓度范围,是不是国标法只能测这五种儿茶素的含量啊?其它几种儿茶素类的标准工作液浓度范围是多少呢? 二、 名称GA+EGC+C+EC+EGCG+ECGRRFstd0.8411.243.583.671.721.42表中只有五种儿茶素相对咖啡因的校正因子,其它几种儿茶素类相对咖啡因的校正因子各是多少呢?
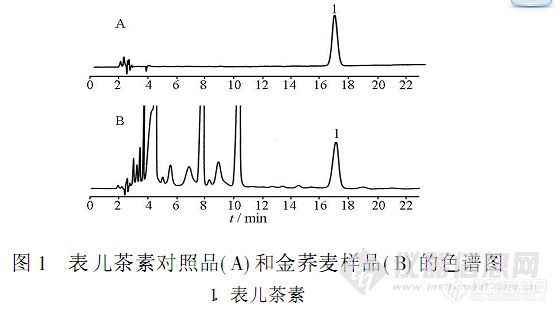
【作者中文名】辛敏通; 杨滨; 李化; 黄璐琦;【作者英文名】XIN Min-tong; YANG Bin; LI Hua; HUANG Lu-qi (Institute of Chinese Materia Medica; China Academy of Chinese Medical Scienes; Beijing 100700; China);【作者单位】中国中医科学院中药研究所; 中国中医科学院中药研究所 北京;【摘要】目的:建立可测定金荞麦饮片中表儿茶素含量的高效液相色谱-电化学检测法。方法:采用DiamonsilC18(4.6 mm×250 mm,5μm)柱,流动相甲醇-0.1 mol.L-1磷酸盐缓冲液(1∶3,pH 2.5),流速1.0 mL.min-1,柱温35℃,参比电极ISAAC(in-situ silver/silver chloride),工作电极玻碳电极,检测电位+600 mV。结果:表儿茶素的线性范围为0.004 875~1.56μg,r=0.999 9;平均回收率为100.7%,RSD 1.9%(n=5)。9批不同市售来源的金荞麦饮片中表儿茶素的含量存在差异。结论:该方法重复性好,可以用来测定金荞麦饮片中表儿茶素的含量。http://ng1.17img.cn/bbsfiles/images/2012/08/201208131754_383602_2379123_3.jpg
我是从茶叶中提取有效成分,然后冷冻干燥出儿茶素L-EGCG单体,可由于从西格玛公司购买的儿茶素L-EGCG单体用完,重新买又买不起(会被老板吐沫淹死)。请问前辈们有什么方法能检测出该单体的纯度?液体和固体都有,最好能得到专业检测机构的认可,因为我还想要他们的检测报告。谢谢!
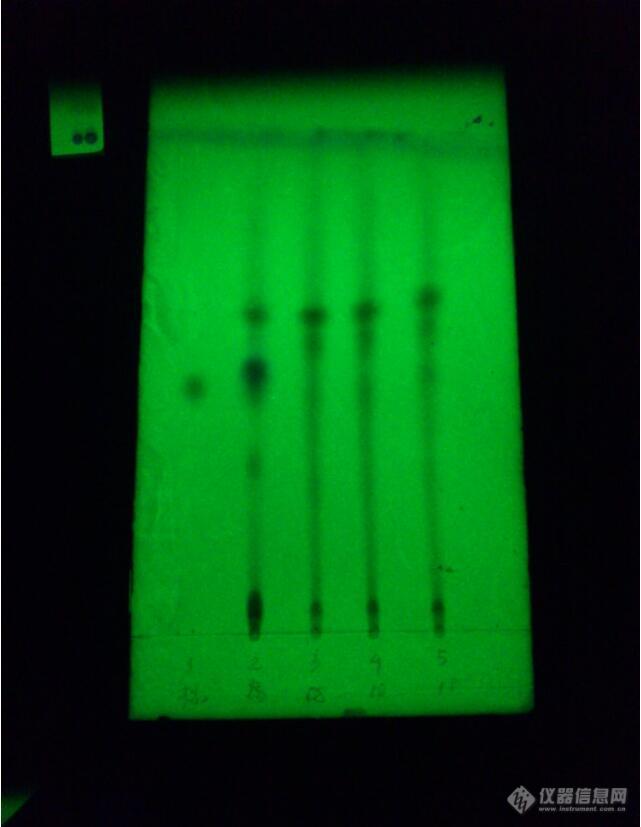
按照药典规定的,展开剂:氯仿-醋酸乙酯-丙酮-甲酸=6:2.5:2.5:0.21为原儿茶酸对照品2药材对照品3、4、5均为制成的中药制剂供试品。我已经增大浓度试过了,色谱图无差异[img=,641,827]https://ng1.17img.cn/bbsfiles/images/2019/08/201908140947125804_8826_1791505_3.jpg!w641x827.jpg[/img]
建立液相色谱-串联质谱(LC-MS/MS)测定各种植物油中儿茶素的方法。植物油经水或甲醇液-液萃取,采用Zorbax SB C18柱,以乙腈–0.1%甲酸为流动相进行梯度洗脱,用多反应监测扫描方式对表没食子儿茶素、儿茶素、表儿茶素、表没食子儿茶素没食子酸酯和没食子儿茶素没食子酸酯等6种儿茶素类茶多酚进行检测。方法的最低检出限均为5~20pg,平均回收率为79.6~99.9%,精密度为0.97%~5.35%。用所建立的方法测定市售植物油中儿茶素类茶多酚的含量。
比如儿茶素,7个组分,但是我买的对照品全是单一组分的,CAF就只是100%的CAF,EGCG就只是100%EGCG。我不能单一的跑。我想把7个单一组分的对照混合,做一个浓度为1mg/ml的对照出来,然后就跑一个图谱,上面就是7个对照峰。是先全部各称50mg,各自分别溶到50ml的容量瓶里面,在各吸几毫升出来混合好一点。还是直接各称一点,反正7个对照平均一下凑齐50mg放到一起,一个容量瓶,然后溶到50ml容量瓶里好一点
大家好!我准备做儿茶素的测定,才刚接触到标准,有不明白的请大家指点,感谢!按国标GB/T 8313-2008, 儿茶素类总量=(EGC含量+C含量+EC含量+EGCG含量+ECG含量),可用儿茶素类标准物质外标法直接定量,也可用儿茶素类与咖啡碱的相对较正因子来定量,问题如下:1.如果使用外标法直接定量,是否只用上述五种标准溶液的工作溶液即可?如果还要用+GA和咖啡碱,作用是什么?2.如果选用儿茶素类与咖啡碱的相对较正因子来定量,GA是否必要,有什么作用?
问题: 有谁做儿茶素类的?标准品有个+C的cas号谁知道是多少?

指示剂3:儿茶酚紫指示剂的CIE 1976(L*,a*,b*)色空间数字化特征 前言 儿茶酚紫(Pyrocatechol Violet)是常用指示剂,也称邻苯二酚紫、邻苯二酚磺酞、邻苯二酚紫、邻苯二酚咪、兒茶酚紫、二茶酚紫、邻二酚磺酞,英文名:Catechol violet;Pyrocatecholsulfonphthalein;Pyrocatechimsulfonphthalein violed;Pyrocatechol Violet;Catecholsulfonphthalein。分子式C19H14O7S,分子量386.38,CAS号115-41-3。红棕色结晶粉末,有金属光泽。易溶于水和含水的醇中,微溶于冷无水乙醇,不溶于非极性的有机溶剂(如醚、苯、二甲苯),熔点185℃,溶解度(H2O)1 mg/mL,水溶液呈黄色稳定,但易被氧化剂氧化。用作络合滴定指示剂,特别是用作金属络合滴定指示剂(滴定铋、镉、钴、铜、铁、镓、铟、镁、铅、锰、镍、钍、锌;用于铋、硼、铬(III)、铜、铌、锑、锡、锆、钍的光度测定)。工业生产上是由邻苯甲酰磺酰亚胺与邻苯二酚反应而得。其水溶液呈黄色,当酸度增加时则由黄变红,其水溶液在pH值1.5~7呈黄色,pH值7~9呈紫色,pH值9~11呈红紫色。http://ng1.17img.cn/bbsfiles/images/2017/01/201701191701_670043_1722582_3.jpg 图1. 儿茶酚紫的化学结构式1. 实验部分 0.5 mol/L H2SO4溶液,0.5 mol/L NaOH溶液,儿茶酚紫溶液(儿茶酚紫0.1 g,加乙醇溶解、定容至100 ml)。光光度计,色度测量系统(自研)。测量条件:光谱范围380 nm~780 nm,△λ5 nm,10 mm光程,CIE 1976(L*,a*,b*)色空间,D65,以水为空白。1.2 实验内容1.2.1 儿茶酚紫指示剂溶液的吸收峰 将儿茶酚紫指示剂溶液滴入不同pH值的溶液中,在分光光度计测量其吸收峰,见表1、图2。http://ng1.17img.cn/bbsfiles/images/2017/10/2016091714532813_01_1722582_3.jpg 图2. 儿茶酚紫指示剂在pH环境的吸收曲线 图2显示,儿茶酚紫在不同pH值的溶液中的最大吸收峰是不同的。当pH值增加时,吸收峰向短波长方向移动。在pH1、pH2时,吸收峰的波长是510 nm;在pH3时,吸收峰向短波长方向移动,最大吸收峰在480 nm;pH4以后,最大吸收峰在450nm左右不变,说明分子结构在pH3和pH4之间发生的变化已经趋于稳定,不在变化。表1. 不同PH环境下儿茶酚紫指示剂最大吸收峰的变化波长PH1PH 2PH 3PH 4PH 5PH 6PH 7PH 8PH 9PH 10PH 11PH 124500.876980.782051.490273.030534.234312.125991.694172.598341.978752.922852.965893.263804802.410231.852111.726422.284923.316821.480041.172041.822341.386252.115912.142422.401735103.776352.785151.664580.898921.233780.379810.285610.490930.364550.655730.655760.77114 1.2.2 儿茶酚紫指示剂在不同pH值的色度值测定 在不同pH溶液中,儿茶酚紫指示剂的色度值变化见表2。表2. 儿茶酚紫指示剂的色度值变化PHL*aba*b*C* H*0.0 1.00 0.00 0.00 0.000 0.000 0.4 [/t
儿茶素中EGCG,EC,EGC,ECG的测定,仪器方法,流动相比例,分峰,求学习

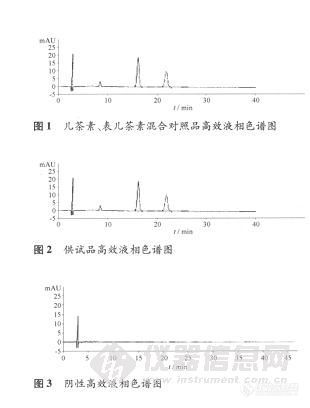
这是一个心酸的历程!http://simg.instrument.com.cn/bbs/images/default/em09509.gif 话说为了配合高校学术研究,公司领导要求我们能够检测茶叶中儿茶素组分。前提是现有资源的情况下,一星期内完成方法开发(此处挖一坑- -!)。于是马上行动翻阅标准,发现用国标做以不现实,因为除了设备条件符合,标样具备外,其他试剂药品基本不全。没办法另行他道,还好其他方法中所用试剂种类较少,都是常用试剂,现有资源都满足,可以开工嘞。http://simg.instrument.com.cn/bbs/images/default/em09502.gif 第一步:配制标样。 茶叶中儿茶素组分含量一般在1%-12%,每种单标纯物质只有25mg,考虑到最后做的线性浓度比较大,直接配制混标(此处又一坑- -!)。混标中包括7中成分(儿茶素:GA、EGC、C、EGCG、EC、ECG及咖啡碱)。 第二步:液相方法的建立 根据相应文献,设定了检测波长、进样量、柱温、流速、流动相梯度。一切都是那么的自然,顺利!开始走样。图谱如下:http://ng1.17img.cn/bbsfiles/images/2016/08/201608260827_606811_2900054_3.jpg 怎么比标准中出峰时间提前这么多?打开柱温箱一看http://simg.instrument.com.cn/bbs/images/default/em09501.gifhttp://ng1.17img.cn/bbsfiles/images/2016/08/201608260938_606837_2900054_3.jpg 4.6×150mm、忘记确认柱子长度了,标准中用的是250mm的柱子。这就合理了,提前就提前只要分离效果好就OK,瞬间释然http://simg.instrument.com.cn/bbs/images/default/em09503.gif。开始数峰,1,2,3,4,5,6,7,很好刚好七个峰。自然的开始进行重现性和线性的测试,重现性很好,线性除了第一个峰差点其他基本都能达到三个九。自我感觉很良好,于是乎开始走单标确认每种成分的保留时间。(杯具即将上演) 2.246(GA)、5.193(EGC)、6.515(C)、11.001(CAF)、12.574(EGCG)、12.576(EC)、16.290(ECG)。咦,什么鬼,EGCG和EC怎么在一起?1.535到底是什么鬼?难道EGCG和EC中我只配了一个? 于是EGCG和EC重新配过再进,结果还是这样!http://simg.instrument.com.cn/bbs/images/default/emyc1010.gif莫不是标准品错了?好吧当我没问。。。再想想,我好像忽略了什么,于是进了针空白,1.535是溶剂,是溶剂,是溶剂。我居然忘了溶剂峰,如此低级的错误,瞬间感觉自己变成了http://simg.instrument.com.cn/bbs/images/brow/em35.gif。 既然确定了1.535这鬼,也就是说EGCG和EC并没有分离开,必须想办法让两个峰分离。http://simg.instrument.com.cn/bbs/images/default/emyc1010.gif 方法一:调节流动相比例。各种调企图达到分离的目的,结果:失败。 方法二:降低流速,延迟分析时间,结果:失败。 方法三:降低柱温+流速+流动相比例,结果:失败。 方法四:更换流动相,将乙腈改为甲醇,结果:咦,有门http://simg.instrument.com.cn/bbs/images/default/em09511.gif。如图:http://ng1.17img.cn/bbsfiles/images/2016/08/201608260831_606812_2900054_3.jpg 于是开始心里各种总结,甲醇粘性比乙腈大,相同流速下虽然柱压变高,出峰时间推后,但能起到很好的分离效果。http://simg.instrument.com.cn/bbs/images/default/em09503.gif,然而http://simg.instrument.com.cn/bbs/images/default/em09504.gif,为毛线还是6个峰。我的内心是崩溃的。再进单标确认,what?这次变成EGC和C重叠在14.679。我想静静。。。 用上面的三个方法进行调试,企图达到分离的效果,但还是被残酷的现实打败了。 用乙腈前面两个分离效果好,用甲醇后面两个分离效果好http://simg.instrument.com.cn/bbs/images/default/emyc1010.gif。那么我前半用乙腈后半用甲醇呢?马上测试,上图:http://ng1.17img.cn/bbsfiles/images/2016/08/201608260831_606813_2900054_3.jpg 哇哈哈!天才http://simg.instrument.com.cn/bbs/images/default/em09503.gif。再用前面的方法一,方法二,方法三进行调戏,哦不,调试!最终达到分离的效果。如图:http://ng1.17img.cn/bbsfiles/images/2016/08/201608260834_606814_2900054_3.jpg 1,2,3,4,5,6,7七个小矮人,http://simg.instrument.com.cn/bbs/images/default/em09503.gif一个都不少,重新测试重现性及线性。重现性良好,线性轻轻松松三个九、四个九。 整个过程历时5天,在领导规定的时间内完成,瞬间感觉如释重负! PS:处子作献给广大仪友。http://simg.instrument.com.cn/bbs/images/default/em09511.gif
我按照国标法GB/T 8313-2018测定茶叶中儿茶素类遇到问题,还请大佬们帮帮忙看一下,感恩!我的流动相A是9%乙腈,2%乙酸和0.2%EDTA-2Na,流动相B是80%乙腈,2%乙酸和0.2%EDTA-2Na。梯度洗脱程序:0-10min,100%A;10-25min,68%A;68%A和32%B保持10min,最后100%A我在单独流动相跑空白,后十分钟出现鬼峰,基线漂移不稳,请问一下我该怎么调呢,初碰液相,还请大佬们指点,万分感谢[img=,690,517]https://ng1.17img.cn/bbsfiles/images/2019/06/201906040852054727_2306_3925358_3.png[/img]
儿茶酚胺类试剂大家是怎么称取测量的?说明书上有的光热水空气敏感,这该怎么称啊?这个空气要怎么弄?
各位大侠,小女子最近用电化学检测器来测定人血浆中的儿茶酚胺,期见碰到了几个难题。(1)儿茶酚胺很不稳定,可以采取什麽措施来保证他的稳定性?(2)儿茶酚胺是内源性物质,如何得到不含儿茶酚胺的空白血浆用于标准曲线的制备?希望各位大侠提供些建议,不胜感激。
临床检测儿茶需要将病人抽的血添加稳定剂吗?发表观点的源处在哪?谢谢
请问各位大神有没有做过尿儿茶酚胺,我拿尿液直接离心过滤还是没有儿茶酚胺类物质,请问是不是样品处理有问题呢
做茶多酚的检测中,请问儿茶素里所含的几种物质的稳定性。如:EC,EGC,GC ,EGCG,ECG







 我要推广仪器
我要推广仪器
 下载APP
下载APP