本人做液相,测定硫酸阿托品的含量,采用磷酸缓冲盐的流动相,硫酸阿托品拖尾严重,请问有什么办法能有效降低硫酸阿托品拖尾呢?谢谢
标准曲线有更换频率不?怎么才知道什么时候该重新制定标准曲线?拜托拜托求指教
《药品注册现场核查要点及判定标准》课件-11-14版(幻灯片)药品注册现场核查工作方案1106 药品注册现场核查要点及判定标准发文报表7个(附表1药品注册撤回申请表,附表2药品注册申请撤回汇总报表,附表3药品注册自查报告表,附表4药品注册全面现场核查报告表,附表5药品注册全面现场核查汇总报表,附表6药品注册现场核查委托表,附表7药品注册现场核查委托报告表)[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=52224]都在这一个包里了[/url]
如何确定标准品色谱峰和样品中的色谱峰是同一种物质? 标准品由于比较纯,所以随着浓度递增,其出峰时间基本差不多。 对于样品,前后相近的色谱峰好几个,哪我怎么判断哪个色谱峰是与标准品相对应的同一种物质呢?
如题,该如何进行前处理呢?我看文献有些是直接加入高锰酸钾沉淀蛋白,然后离心,去上清液就行了,不知这样能提取出血液中的硫酸阿托品吗?
各会员单位: 近日,国家卫生部和质检总局发布2011年第6号公告,再次重申食品添加剂的标准包括使用安全标准和产品质量规格标准,且均纳入食品安全国家标准管理范围。公告规定,食品添加剂生产企业应依据相应国家标准或指定标准组织生产,生产企业不需要制定食品添加剂产品企业标准,地方卫生行政部门和标准化行政主管部门亦不再对食品添加剂产品企业标准进行备案;对于生产尚没有国家质量规格标准的食品添加剂产品,可依据卫生部等九部门《关于加强食品添加剂监督管理的通知》(卫监督发89 号)的规定,提出参照国际组织和其它国家标准(含质量要求、检验方法)的资料,报中国疾病预防控制中心,作为指定标准的依据。 在我国现行生产监管体制下,国家标准和指定标准是企业组织产品生产的重要技术文件,也是食品添加剂生产企业申领生产许可证的必要条件之一。为此,协会希望各生产企业进一步重视标准方面的工作,对企业所生产的食品添加剂产品的标准进行认真梳理,对于没有国家或行业标准的产品,按照公告的规定,尽快向卫生部委托的中国疾病预防控制中心营养与食品安全所上报参照国际组织和其它国家标准制定产品指定标准(含质量要求、检验方法)的文本和国内外相关标准的资料,便于中国疾病预防控制中心营养与食品安全所组织专家研究,缩短指定标准颁布实施的时间,减少因缺失标准造成对企业正常生产经营的影响。联系方式:一、中国疾病预防控制中心营养与食品安全所地址:北京市朝阳区潘家园南里7 号,邮编:100021电话:010-87776914 传真:010-67711813电邮:GB2760@gmail.com二、中国食品添加剂和配料协会地址:北京市朝阳区朝外大街甲6 号万通中心3 座1402 室,邮编:100020电话:010-59795833 传真:010-59071335电邮:cfaa2003@yahoo.com.cn 二○一一年四月六日
求宝石鉴定标准,加工工艺

LC-MSMS做拉米夫定标准品,不知为啥一直有个明显肩峰。图谱在附件里http://ng1.17img.cn/bbsfiles/images/2013/09/201309051141_462516_2167114_3.bmp
请教一下各位:中检所是不是不能定标准品了呢?
做外标法时,要配制标准品溶液,如何判定标准品的浓度是否准确?如何才能提高准确度?
请教各位老师,食品中重金属测定的判定标准除了2762还有什么啊?
讨论:各类食品中重金属、有害元素、微量元素等等的测定标准,欢迎大家分享与讨论!大家平时在食品检验工作中测定最多的元素又是哪些呢?
因公司项目停止,购买的法定标准品(EP和USP)现全部闲置,都是现行批号,有证书,现全部低价处理,有需要的全部拿走 联系QQ:342832185声明一下,不能开票哦
(1)基准标准品又分为法定基准标准品和内部基准标准品两类。法定标准品包括:中国药品生物制品检定所(NICPBP)提供的国家标准品(CP 标准品)、美国药典委员会(USP)提供的USP标准品、欧洲EDQM委员会提供的EP标准品、JP标准品、BP标准品及其它WHO、ISA标准品等官方渠道得到的标准品。(2)工作标准品包括:企业内部标定的工作标准品、科研单位或合同实验室标定的工作标准品、其他厂家在认证实验室标定的标准品。(3)杂质标准品分为定性用杂质标准品及定量用杂质标准品,来源于法定标准品或自制杂质对照品。【来源:实验与分析】
现急需能量色散x荧光校准或鉴定标准(办法),因新设备已安装调试完成,需要验收但国家现在没有标准,望各位大虾能帮忙,本人邮箱540887927@qq.com.谢谢
药品GMP认证检查评定标准
请问目前有气质联用的国家鉴定标准吗?
流动相:0.05molL-1磷酸二氢钠-甲醇(60∶40) (其中含0.01molL-1庚烷磺酸钠,用磷酸调pH2.9 我按照上述条件配制流动相,只是PH值用试纸调的,不是太准,这样是不是会影响峰型?我怕由于在做标准系列的检测,出的峰峰型太宽,而且脱位严重,有没有比较好的解决方法?我要检验的样品比较多,流动相每次都要调节PH值太麻烦,能不能直接用双蒸水和甲醇配置,如果替代会不会不出峰啊?盼各位老师同仁告之,不胜感激!
近日,农业部发布新修订的《巴氏杀菌乳和UHT灭菌乳中复原乳的鉴定》标准(以下简称《标准》),标准号NY/T 939-2016,代替农业行业标准NY/T 939-2005,自2016年4月1日起实施。该《标准》的修订出台,完善了我国复原乳鉴定标准,为监管违规添加复原乳提供了科学依据,对维护消费者知情权,促进奶业健康发展将起到积极的推动作用。http://img1.17img.cn/17img/images/201603/insimg/fc5afcfd-3cfe-4e02-83aa-42bea773c637.jpg 该《标准》由中国农业科学院北京畜牧兽医研究所、农业部奶及奶制品质量监督检验测试中心(北京)修订,增加了超高效液相色谱测定糠氨酸的方法、修改了原有乳果糖的测定方法,有效地缩短了检测时间,提高了检测效率。经多家检测机构验证,该《标准》能够确保检出结果的准确性。 专家组介绍,《标准》选取的标示物--糠氨酸和乳果糖,均为生乳中含量极低的物质。糠氨酸是牛奶热加工过程中出现的副产物,乳果糖是牛奶在加热过程中乳糖发生碱基异构的产物。作为乳品工业的一种乳原料,奶粉在复原之后至少还得再经过一次商业性热杀菌,总体上复原乳制品所经受的热伤害程度强于以生鲜乳为原料的乳产品。《标准》主要原理是根据生鲜乳、巴氏杀菌乳、UHT灭菌乳和奶粉在生产过程中糠氨酸和乳果糖变化的规律显著不同,通过测定糠氨酸和乳果糖的含量并结合其比值建立模型,来判定巴氏杀菌乳和UHT灭菌乳中是否添加了复原乳,因此修订后的标准可以准确鉴定复原乳。
哪位有傅立叶变换红外光谱仪的国家鉴定标准?谢谢了
乳与乳制品中蛋白质测定标准发布有助于三聚氰胺等非蛋白含氮物不再被误判为蛋白质本报讯 近日,作为我国农业行业标准,《乳与乳制品中蛋白质的测定 双缩脲比色法》(NY/T 1678-2008)标准发布并实施。 该标准的发布,标志着在利用其进行乳与乳制品中蛋白质测定的前提下,三聚氰胺等非蛋白含氮物将不会再被误判为蛋白质,也不会再出现蛋白质测定值虚高的问题。 该标准由中国农业大学食品学院牵头,农业部乳品质量监督检验测试中心、农业部奶及奶制品质量监督检验测试中心(北京)、农业部食品质量监督检验测试中心(上海)共同完成。 据悉,早在2004年,中国农业大学研究人员就开展了鉴别在食品中非法添加“非蛋白含氮物”的研究,奠定了研发此项技术的基础。中国农业大学食品学院侯彩云教授表示,新标准的出台,为堵塞乳与乳制品中恶意造假行为提供了必要的技术支撑,但乳品质量安全保障还需要标准体系的进一步完善,以及食品安全长效保障机制的进一步健全。(记者赵陕雄) 信息来源:中国质量报 发布时间:2008-11-21
[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=34469]药品GMP认证评定标准及发展变化[/url]看看吧
供参考。新标准更严了感觉上借鉴了些 欧洲GMP的要求[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=78840]药品GMP认证检查评定标准[/url]
一般分析方法可以分成两大类,一是绝对的方法,另外就是相对的方法。绝对的方法是不需要使用标准品的。可是相对的分析方法就需要使用标准品了。 借着与标准品在同一条件下的比较,就可以完成对样品的定性与定量。所以如何来决定标准品自然就成为很重要的课题。一般的标准品可以分成几类,分别讨论如下。对於一个崭新的分子(new molecular entity),我们首先是需要完成纯化的制备,然后完成分析分子的物理特征(physical characterization)。纯化的制备方法可根据分子而使用不同的方法。譬如小分子而言,可以使用一般的再结晶方法。当然最理想还是使用制备色谱。不管使用何种纯化的方式,在做物理特征定性之前,我们必须取得高纯度的样品。而一般纯度就是使用高效液相色谱来做初步的决定。换句话说就是取得色谱纯度(chromatographic purity,也就是我们说的homogeneity)。要决定色谱纯度,自然需要开发出色谱的方法。一般可以使用梯度或等度方法,唯一的要求就是要能够检测到所有可能存在0.1%以上的杂质。有了这种方法,我们必须调整进样量,使我们能够检测到0.1%的杂质。根据峯面积的百分比,我们可以求得主峰的色谱纯度。一般的要求就是要达到99.9% 以上。换句话说就是只有单一主峰的存在。这也是使用制备色谱纯化标准品的主要原因。这一类的标准品,我们叫做原始标准品(primary reference standard)。取得此类标准品后,就需要做一系列的化学构造鉴定。一般使用UV/Vis, FT-IR, NMR, MS, X-Ray等等来确定分子的结构式。然后就需要使用绝对的分析方法来做化学定量。对小分子可以使用DSC, 酸碱滴定,NMR, 电化学,也可以使用元素分析等。使用元素分析就是做C, H, 等等元素的分析。然后根据分子的理论值来计算标准品的化学纯度。当然,我们还需要其他的无机成分(如水,阴阳离子等)以取得标准品含量的物质平衡(material balance)。如此,我们就可以知道这个原始标准品的化学纯度(chemical purity)。一般原始标准品的制备是很花时间,所以不需过多的量。一般有个20-30毫克就足够使用了。主要的用途就是用於鉴定化学构造及纯度。一旦有了原始的标准品,我们就可以拿这个标准品,用色谱方法来定量了。另外一类的标准品就是所谓药典标准品 (compendial standard)。这种标准品相当的昂贵。其实这类标准品就可以视为上述的原始标准品。只是我们可以直接使用,而不需要经过上列的构造鉴定程序。一般可向美国的USP, 欧洲的EP采购。每一个标准品都带有使用的手续。譬如贮存的情况,是否在使用前需要干燥,干燥的时间等等。这些标准品都是假设是100%纯度。换句话说我们如果制备1.00微克每毫升,那相对的浓度就是1.00。这类的标准品的保存期是一直到下一个批号的标准品上市以前。也就是说一旦新的一个批号标准品出来后,前一个批号的标准品就自然的作废了。前面提到这类的标准品相当的昂贵,为了有充足的标准品做一般实验,因此就必须选择所谓的工作标准品 (working standard)。这一类的标准品也可以称为二级标准品(secondary reference standard)。工作标准品,可以选择一般的原料药,或着市面上出售的化学药品。一般我们都是选用某一个批号的化学原料药。可以取出100克的药量,发配一个标准品的批号。然后使用高效色谱的方法,用前述的原始或药典标准品来定量工作标准品的纯度。这个纯度就是化学纯度。有一点需要注意的就是这个工作标准品的纯度可能不会到99.9%,他的色谱纯度也许也不高。但是我们仍旧可以使用,只要我们能够从正确的色谱方法取得有重复性的纯度。当然这个标准品的稳定性,保存方法,保存期是可以参考化学原料药来处理。最后一类的标准品就是色谱工作液标准品 (working standard solution)。
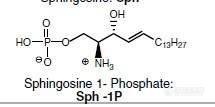
http://ng1.17img.cn/bbsfiles/images/2013/10/201310152015_471106_2803904_3.jpghttp://ng1.17img.cn/bbsfiles/images/2013/10/201310152015_471108_2803904_3.jpghttp://ng1.17img.cn/bbsfiles/images/2013/10/201310152015_471109_2803904_3.jpghttp://ng1.17img.cn/bbsfiles/images/2013/10/201310152016_471110_2803904_3.jpghttp://ng1.17img.cn/bbsfiles/images/2013/10/201310152016_471111_2803904_3.jpg为什么标准品浓度不一样?峰型也不一样? 峰拖尾严重如何处理?标准品怎么会有两个峰?我的用的是C18拄,色谱条件是甲醇:水(0.1%甲酸,10mm的乙酸铵90:10,等度洗脱。做所得是2种磷脂分子,分子式如上

[center]产品销售与回收《药品GMP认证检查评定标准》08版[/center][img]http://ng1.17img.cn/bbsfiles/images/2009/04/200904030735_142125_1626679_3.jpg[/img][img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=142126]产品销售与回收《药品GMP认证检查评定标准》08版[/url]
有没有冶标炭素制品中灰份、挥发份测定标准最新版的。请用附件形式发给我,谢谢
[color=red][size=4]假如你是制定标准的人,你该如何制定---《食品防护计划建立与实施指南 食品生产企业》的国家标准?[/size][/color]《食品防护计划建立与实施指南 食品生产企业》的国家标准目前正在征求广大用户的意见~欢迎广大的仪器人和关心食品的人前来PK~也许您的意见会被制定标准的人采纳噢~有好的建议和意见的人,论坛将给予一定积分奖励~希望大家积极参与~
想鉴定某种植物里某种化合物的含量,前期有分离出该种化合物的纯品,但是后期做定量时,不同浓度的标准品的出峰时间有细微差别,且样品中的该化合物的峰要么是出峰时间与标准品不一样,要么就是分不开,是什么原因呢?还有基线总是跑不平(如图所示),流动性是乙腈比水为4:6,
脂肪酸同分异构体的鉴定,买了C16:1的标准品,出了一个峰是C16:1 7c,但是实验目的是想要C16:1 5c,是要重新买标准品吗?







 我要推广仪器
我要推广仪器
 下载APP
下载APP