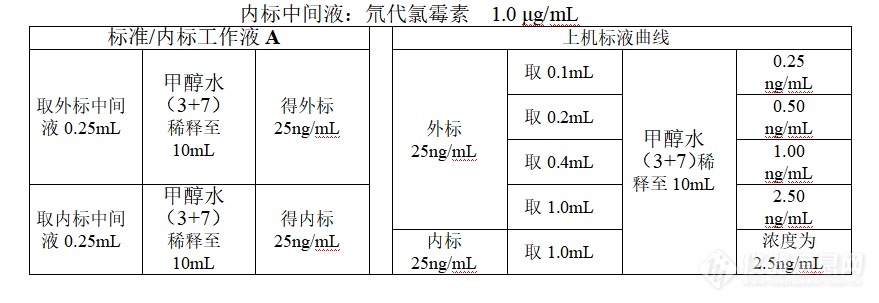
动物源性食品中氯霉素、甲砜霉素、氟苯尼考(氟甲砜霉素)的测定色谱柱:菲罗门 kinetex C18柱(100×2.1mm 2.6um)试剂溶液: 甲醇水(3+7):300 mL甲醇与700 mL去离子水混匀;乙腈饱和正己烷:乙腈与正己烷等比例混合,取上层;[b]说明:下列所述标准工作液A、B为氯霉素、氟苯尼考、甲砜霉素混合标液[/b]标准中间液:氯霉素、氟苯尼考、甲砜霉素1.0 μg/mL内标中间液:D-CAP 1.0 μg/mL标准工作液[b]A[/b](25 ng/mL):吸取1.0 μg/mL标准中间液0.25 mL用30%甲醇水定容至10 mL,混匀(该溶液临用现配);内标工作液[b]A[/b](25 ng/mL):吸取1.0 μg/mL内标中间液0.25 mL用30%甲醇水定容至10 mL,混匀(该溶液临用现配);标准工作液[b]B[/b](1 ng/mL):吸取标准工作液[b]A [/b]0.4 mL用30%甲醇水定容至10 mL,混匀 (临用现配)。上机标准曲线:分别移取0、0.1、0.2、0.4、1.0mL标准工作液[b]A[/b](25 ng/mL),1.0mL内标工作液[b]A[/b](25 ng/mL)于5个10mL容量瓶中,30%甲醇水定容至刻度,得标液浓度为0、0.25、0.5、1.0、2.5 ng/mL,内标溶度为2.5ng/mL。附:标准曲线配制: 外标中间液:氯霉素、氟苯尼考、甲砜霉素1.0 μg/mL[img=,690,237]https://ng1.17img.cn/bbsfiles/images/2018/10/201810310617262550_9107_2166779_3.png!w690x237.jpg[/img]样品前处理:准确称样5.00 g(精确至0.01 g)于50 mL离心管中,添加氯霉素内标[sup][/sup],加入0.5mL氨水,加入5 g 无水Na[sub]2[/sub]SO[sub]4[/sub][sup][/sup]和20 mL乙酸乙酯,用均质器以10000 r/min的速度均质1 min,15 mL乙酸乙酯[color=#ff0000]清洗均质头[/color],加盖振摇[sup][/sup],3500 r/min离心5 min,上清液转移至150 mL的棕色梨形瓶中,在40 ℃减压旋转蒸发至干,加入2 mL甲醇水(3+7)和2 mL 乙腈饱和正己烷溶解洗脱,高速离心分层,取下清液,用0.2 μm微孔[color=#ff0000]有机[/color]滤膜过滤,上机测试。[img=,690,393]https://ng1.17img.cn/bbsfiles/images/2018/10/201810310619102674_6914_2166779_3.png!w690x393.jpg[/img]氯霉素、甲砜霉素、氟苯尼考(氟甲砜霉素)的测定涉及标准汇总、比较见表1:[img=,690,247]https://ng1.17img.cn/bbsfiles/images/2018/10/201810310621041150_5153_2166779_3.png!w690x247.jpg[/img][b]线性范围、线性方程[/b]分别对氯霉素、甲砜霉素、氟苯尼考标准系列工作溶液进行测定,以标准溶液的浓度(ng/mL)与氘代氯霉素浓度的比值为横坐标,以标准溶液中被测组分峰面积与氘代氯霉素峰面积比为纵坐标,绘制标准曲线或计算回归方程,具体结果见表2。[img=,673,185]https://ng1.17img.cn/bbsfiles/images/2018/10/201810310636522800_5170_2166779_3.png!w673x185.jpg[/img]氯霉素(0.25ng/mL)、甲砜霉素(2.5ng/mL)、氟苯尼考(氟甲砜霉素)(2.5ng/mL)MRM图:[img=,690,399]https://ng1.17img.cn/bbsfiles/images/2018/10/201810310626212579_611_2166779_3.png!w690x399.jpg[/img][img=,690,434]https://ng1.17img.cn/bbsfiles/images/2018/10/201810310626315649_9067_2166779_3.png!w690x434.jpg[/img][img=,657,417]https://ng1.17img.cn/bbsfiles/images/2018/10/201810310626401955_677_2166779_3.png!w657x417.jpg[/img]鸡肉样品测定MRM图:[img=,689,363]https://ng1.17img.cn/bbsfiles/images/2018/10/201810310629369622_5025_2166779_3.png!w689x363.jpg[/img][img=,666,443]https://ng1.17img.cn/bbsfiles/images/2018/10/201810310629457983_2475_2166779_3.png!w666x443.jpg[/img][img=,655,416]https://ng1.17img.cn/bbsfiles/images/2018/10/201810310629536961_3288_2166779_3.png!w655x416.jpg[/img]鸡肉样品加标氯霉素(0.25ng/mL)、甲砜霉素(2.5ng/mL)、氟苯尼考(氟甲砜霉素)(2.5ng/mL)MRM图:[img=,690,361]https://ng1.17img.cn/bbsfiles/images/2018/10/201810310633092540_6864_2166779_3.png!w690x361.jpg[/img][img=,682,443]https://ng1.17img.cn/bbsfiles/images/2018/10/201810310633202300_3044_2166779_3.png!w682x443.jpg[/img][img=,671,425]https://ng1.17img.cn/bbsfiles/images/2018/10/201810310633330468_2212_2166779_3.png!w671x425.jpg[/img][img=,673,185]https://ng1.17img.cn/bbsfiles/images/2018/10/201810310638525567_679_2166779_3.png!w673x185.jpg[/img][img=,690,437]https://ng1.17img.cn/bbsfiles/images/2018/10/201810310639003515_512_2166779_3.png!w690x437.jpg[/img][img=,690,455]https://ng1.17img.cn/bbsfiles/images/2018/10/201810310639089904_8264_2166779_3.png!w690x455.jpg[/img][img=,690,455]https://ng1.17img.cn/bbsfiles/images/2018/10/201810310639089904_8264_2166779_3.png!w690x455.jpg[/img][img=,658,289]https://ng1.17img.cn/bbsfiles/images/2018/10/201810310641135748_6268_2166779_3.png!w658x289.jpg[/img]鸡肉样品加标0.1ug/kg氯霉素提取定量子离子151.9的信噪比:[img=,690,290]https://ng1.17img.cn/bbsfiles/images/2018/10/201810310644563180_387_2166779_3.png!w690x290.jpg[/img]鸡肉样品加标1.0ug/kg甲砜霉素提取定量子离子184.9的信噪比:[img=,685,303]https://ng1.17img.cn/bbsfiles/images/2018/10/201810310646330148_451_2166779_3.png!w685x303.jpg[/img]鸡肉样品加标1.0ug/kg氟苯尼考提取定量子离子336.0的信噪比:[img=,690,294]https://ng1.17img.cn/bbsfiles/images/2018/10/201810310648135100_953_2166779_3.png!w690x294.jpg[/img]注意事项:1、[color=#ff0000]每批样品检测应包含:[/color]标液曲线、试剂空白、样品空白、样品加标、样品。2、加标回收:分别添加1 ng/mL标准工作液[b]B [/b]0.5 mL于空白样品中,氯霉素、氟苯尼考、甲砜霉素相当于0.1 μg/kg水平;3、负离子模式,上机前如响应不够需清洗离子源[color=#ff0000]或增加仪器平衡时间[/color];4、初测选适用基质的任一样品加标,如遇特殊基质(如饲料、血粉等粉末样品)称1.0 g,其余步骤同肉制品,定量下限与加标水平同时提高为氯霉素1.0 μg/kg,特殊基质应选用该样品做加标回收,复测时做平行样品,如初测浓度过高,复测时应加大曲线范围或者将样品稀释后上机,保证样品浓度在曲线范围内。
问题: 请问大家伙氯霉素、甲砜霉素、氟甲砜霉素的[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url]方法里液相条件是什么?谢谢回复: 甲醇和水作为流动相
我想问下大家,我用三甲基硅烷基重氮甲烷衍生青霉素,然后加入甲醇做催化剂,空白里没有青霉素,可是从峰上看,出现苯环结构,想问问有没有大神知道原因呢,求告知
最近做这三种,标准上甲砜霉素出峰时间最后,可做起来感觉在前面,那位有做过的,请指教,最好附温度程序

10,抽取5个版友);中奖名单:m3071659(注册ID:m3071659)馨语(注册ID:huangdm)玲儿响叮当(注册ID:jshbhh)捌道巴拉巴巴巴(注册ID:v3082413)zengzhengce163(注册ID:zengzhengce163)http://ng1.17img.cn/bbsfiles/images/2017/03/201703131618_01_708_3.jpghttp://ng1.17img.cn/bbsfiles/images/2017/03/201703131618_02_708_3.jpg【注意事项】同样的答案,每人只能发一次PS:该贴浏览权限为“回贴仅作者和自己可见”,回复的版友仅能看到版主的题目及自己的回答内容,无法看到其他版友的回复内容。下午3点之后解除,即可看到正确答案、获奖情况及所有版友的回复内容。=======================================================================动物源性食品中氟苯尼考、氟苯尼考胺和甲砜霉素的测定方法:SPE/HPLC基质:动物组织及内脏应用编号:103020化合物:氟苯尼考 氟苯尼考胺 甲砜霉素固定相:ProElut PLS色谱柱/前处理小柱:ProElut PLS 60mg / 3ml 50/pkg样品前处理:样品准备/提取(1) 称取试样1g(精确到0.01g)置于50ml 具塞离心管中,加入15ml 乙腈 +乙酸乙酯+氨水(80+15+5)(2) 均质1min,6000 r/min,离心5 min,取出上清液。(3) 残渣按照上述步骤重复提取一次,合并两次上清液。(4) 加入5 mL 正丙醇,在45℃以下水浴减压浓缩至近干(务必蒸干)。(5) 依次加入0.5ml 甲醇,10 ml 4%NaCl 溶液溶解残渣,震荡3 min,转入50ml 具塞离心管中,加入10 正己烷,震荡3 min, 10000r/min 离心5min,弃 去正己烷层。(6) 重复(5)的操作。,然后加入0.5 mL 氨水。混匀,作为上样液待净化。SPE柱净化——ProElut PLS 60 mg/3 mL(Cat.#68003)(1)活化: 依次加入3 mL 甲醇,3 mL 水,流出液弃去。(2)上样: 将待净化液加入柱中,流出液弃去。(3)淋洗: 依次用3 mL 水,5 mL 5%氨水(10%甲醇水)溶液淋洗,流出 液弃去,然后将PLS 柱抽干。(4)洗脱: 3 mL10%氨水甲醇洗脱,流出液收集。(5)重新溶解:* 将洗脱液在45 ℃减压浓缩至近干,然后用流动相重新定容至1mL。色谱条件:色谱柱: Diamonsil C18(2) 250×4.6 mm,5 μm(Cat.#:99603) 流速: 0.7 mL/min 检测器:* 激发波长:225 nm 发射波长:285 nm 柱温: 30 ℃ 进样量: 20 μL 流动相: A(庚烷磺酸钠9 g/L,磷酸二氢钠1.54 g/L):B(乙腈)=81:19文章出处:迪马科技应用实验室关键字:氟苯尼考 氟苯尼考胺 甲砜霉素 动物源食品 SPE ProElut PLS谱图:http://www.dikma.com.cn/Public/Uploads/images/dongwu(1).GIF
各位老师,我最近在摸索固相萃取的条件,药物是氯霉素、甲砜霉素、氟苯尼考。用水复溶上样的。用了几个柱子都不是很理想,想和大家讨论一下固相萃取柱的选择和萃取条件的选择?HLB的柱子用纯甲醇洗脱的回收率都不高?峰型也不好看。安捷伦C18的柱子上样的时候会有样品流出来。
氯霉素在弱酸性和中性溶液中较安定,煮沸也不见分解,遇碱类易失效。国标GB/T 20756-2006中提取氯霉素、甲砜霉素、氟苯尼考加氨水的作用是什么啊?
各位老师帮我分析下,我做22338国标,甲砜霉素 氯霉素 氟苯尼考时,用的是乙腈+0.1%甲酸水单独走每个物质都有出峰,唯独甲砜霉素响应值低,其他两个峰不错,但当我把他们方法合在一起,就不醒了,我看了下合在一起只是驻留时间变小了,其他无变化,驻留时间能影响这么大吗?我觉得不应该啊,还是因为他们的母离子太接近了,而且出峰时间有差不多,在扫描时发生混乱了???求助
氯霉素进[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url],负离子模式,如果新配制的话同位素峰就是321/323位3:2,但是配制的氯霉素放在冰箱一晚后,峰型就是323比321高很多,不知道大家有没有遇到过这类问题,氯霉素应该是很稳定的呀,还有就是我的仪器负离子进样甲醇空白,负离子很容易出现325.08这个干扰峰,也不知道哪里引来的干扰,这两个问题求助下打击。
最近在做四环素类试验,OTC、TC和CTC做的都正常,就是强力霉素进空白样一直都出峰,甲醇水也出峰,流动相用的是甲醇:甲酸水。已排除系统污染,梯度洗脱程序也换过了,可那个峰死活就是跟着强力霉素,保留时间一模一样。有人碰到过吗?
请问哪位有农业部958号公告-13—2007——水产品中氯霉素、甲砜霉素、氟甲砜霉素残留量的测定[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法的标准,我在网上找的只有14-2007,也就是[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质联用[/color][/url]法的标准,我只有[url=https://insevent.instrument.com.cn/t/Mp]气相[/url],没质朴,只能用13-2007的方法,哪位有,能不能发一份给我,谢谢!
亲们,求助一下,用GB/T 20764-2006 《可食动物肌肉中土霉素、四环素、金霉素、强力霉素残留量的测定 液相色谱-紫外检测法》做抗生素,仪器:WATERS e2695,检测器:2698 DAD,柱子:Sunfire C18 250*4.6,5,流动相:乙腈+甲醇+0.01mol/L草酸溶液(2:1:7),流速:1.0mL/min,柱温:30度,检测波长:350nm,进样量:20微升,用标曲最大点进样(200ng/mL),四种都不出峰,我们试了更大浓度的也一样。请问问题出在那里呢?有推荐的方法和柱子吗?
混标,氯霉素,甲砜霉素和氟甲砜霉素这三种。加标回收,氯霉素绝对回收接近100%,另外回收两种很低,我全部是按标准操作的,不知道是哪里出问题了。请问有没有前辈碰到过这个问题,有没有经验可以说说?多谢啦!

10,抽取5个版友);中奖名单:WUYUWUQIU(注册ID:wulin321)馨语(注册ID:huangdm)dahua1981(注册ID:dahua1981)吕梁山(注册ID:shih20j07)999youran(注册ID:999youran)http://ng1.17img.cn/bbsfiles/images/2016/08/201608231506_606297_1610895_3.pnghttp://ng1.17img.cn/bbsfiles/images/2016/08/201608231506_606298_1610895_3.png积分奖励:所有回答正确的版友奖励10个积分(幸运奖获得者除外)。【注意事项】同样的答案,每人只能发一次PS:该贴浏览权限为“回贴仅作者和自己可见”,回复的版友仅能看到版主的题目及自己的回答内容,无法看到其他版友的回复内容。下午3点之后解除,即可看到正确答案、获奖情况及所有版友的回复内容。=======================================================================蜂蜜中的氯霉素的测定方法:SPE基质:蜂蜜应用编号:101238化合物:氯霉素固定相:ProElut PLS色谱柱/前处理小柱:ProElut PLS 60mg / 3ml 50/pkg样品前处理:1、提取 称取10 g 蜂蜜于50 mL 离心管中,加入10 mL 水,于涡旋混合器上混合3 min,待样品完全溶解后加入20 mL 乙酸乙酯,高速涡旋混合5 min,3000rpm 下离心5 min,上层溶液转入100 mL 烧瓶中。下层溶液再加入20 mL 乙酸乙酯,高速涡旋混合5 min,3000 rpm 下离心5 min,合并两次上层液于同一烧瓶中,减压浓缩至干,用3×1 mL 20% 的甲醇水溶液溶解残渣,待净化。 2、净化 a 活化: 依次用3 mL 甲醇和3 mL 水活化ProElut PLS 60 mg/3 mL (Cat.#68003),流出液弃去; b 上样: 将待净化的样品液加入柱中,流出液弃去; c 淋洗: 用3 mL 20% 甲醇水溶液淋洗,淋洗液弃去,并将PLS 柱抽干; d 洗脱: 用3 mL 甲醇洗脱,收集洗脱液; e 重新溶解:在40 ℃ 下用氮气将洗脱液吹至近干,然后用1 mL 40% 甲醇水溶液定容,供HPLC 分析色谱条件:色谱柱:Diamonsil C18(2) 250 x 4.6 mm ID, 5 μm (Cat. #99603) 色谱柱:Diamonsil C18(2) 250 x 4.6 mm ID, 5 μm (Cat. #99603) 流动相:甲醇/ 水=40/60 (V/V) 流速:1 mL/min 进样量:20 μL 柱温:30 oC 检测器:UV 280 nm文章出处:P109关键字:蜂蜜,氯霉素,SPE,ProElut PLS摘要:适用于蜂蜜中氯霉素的测定。谱图:http://www.dikma.com.cn/Public/Uploads/images/lvmeisu%20copy.png图例:1. 氯霉素
甲砜霉素(Thiamphenicol,TAP)是一种广谱抗菌素,其结构类似于氯霉素,能抑制革兰氏阳性细菌、阴性细菌和一些厌氧微生物,广泛应用于水产养殖当中并且会造成残留。因此,研究和建立检测方法成了当务之急。本研究以多克隆抗体和免疫分析技术为基础,根据酶联免疫原理,建立了快速简便的水产品中的酶联免疫检测方法。另外,还建立了相应的仪器检测方法。首先通过混合酸酐法合成甲砜霉素与牛血清蛋白偶联物,构建甲砜霉素完全抗原。通过紫外扫描、SDS-聚丙烯凝胶电泳和免疫动物实验,证明了产物合成成功。通过免疫新西兰白兔,得到抗 TAP 的抗血清,抗血清效价高达 1×106。此抗血清分别经过饱和硫酸铵沉淀法、离子交换法和反向免疫亲和层析法纯化后,得到单一特异性的抗体,平均亲和常数为 7.8×10-9mol/L。以此抗体为基础,设计了间接竞争两步酶联免疫检测方法,并优化了缓冲液浓度、pH、吐温 20 浓度等参数。此酶联免疫检测试剂盒检测限为 0.06 ng/mL,血液和组织回收率为 80~110%。通过重复性、检测灵敏度、准确性等指标来评估此方法,并与气相色谱法进行了比较,结果表明,此间接竞争酶联免疫检测方法各项参数均能满足实际检测的需要。建立和优化了甲砜霉素残留的高效液相色谱法(HPLC)、高效液相色谱-质谱法(HPLC/MS)和气相色谱-质谱法(GC/MS)的检测方法。方法的回收率分别为:HPLC 为78.6~95.4%;HPLC/MS 为 72.6~99.8%;GC/MS 为 85.6~101.5%,相对偏差均小于10% ,检出限分别为:HPLC 为 8.3μg/kg;HPLC/MS 为 0.1μg/kg;GC/MS 为 0.1μg/kg。结果表明,这些方法都能满足实际检测的需要。以高效液相色谱法(HPLC)为检测方法,研究了甲砜霉素在鲈鱼血液和可食性组织里的消除规律。鲈鱼单次口服 5 mg/kg(鱼体)和 30 mg/kg(鱼体)的甲砜霉素药物后,药物在血液中的达峰时间分别为 6 小时和 8 小时,药物平均滞留时间的分别为 11.1 小时和 11.9 小时;甲砜霉素在鲈鱼血液中的经时过程符合一级吸收二项指数方程:C=10.812(e-0.087t– e-0.274t),药物在鲈鱼血液中的吸收半衰期为 1.997 h,消除半衰期为 5.589h;连续 5d 口服 15 和 30 mg/kg(鱼体)的甲砜霉素,药物的吸收速率是一个常数,它在血液中的消除速率也是一个常数,这和单次口服结果一致,其消除速率方程分别为C血液=0.849e-0.494t ;C肌肉=1.461e-0.682t,药物在鲈鱼血液和组织中的的消除半衰期分别为1.016d 和 1.403d;通过 EMEA/CVMP/036/95 所规定的统计方法计算休药期,根据 95%的忍受限和最大残留限量,确定了5d和6d分别作为服用低剂量和高剂量药物的休药期。缩写符号说明ELISA 酶联免疫吸附测定法TAP 甲砜霉素TMS 四甲基硅烷BSTFA N,O-双(三甲基硅烷基)三氟乙酰胺TMCS [
一、实验目的掌握GC—MS检测蜂蜜中氯霉素残留量的原理和方法。二、实验原理蜂蜜样品用水溶解后,用乙酸乙酯提取样品中残留的氯霉素,提取液浓缩后再用水溶解,经Oasis HLB固相萃取柱净化并硅烷化后,以GC—MS仪测定,外标法定量。三、仪器与试材1.仪器与器材气相色谱质谱仪(配有化学源)、固相萃取装置、自动浓缩仪、氮气吹干仪、振荡器、液体混匀器、真空泵(真空度应达到80kPa)、离心机等。2.试剂除特别注明外,实验中所用试剂均为分析纯,水为去离子水或蒸馏水。(1)常规试剂与溶液:甲醇、吡啶、乙腈、乙酸乙酯、正己烷、六甲基二硅氮烷、三甲基氯硅烷、乙腈水溶液(1+7,体积比)。(2)Oasis HLB固相萃取柱(60mg,3mL):使用前分别用3mL甲醇和5mL水预处理,保持柱体湿润。(3)硅烷化试剂:将9份吡啶、3份六甲基二硅氮烷和1份三甲基氯硅烷混合。(4)氯霉素标准储备溶液(0.1mg/mL):称取适量的氯霉素标准物质(纯度≥99%),以甲醇配制,于4℃储存,可使用两个月。(5)氯霉素标准工作溶液:用空白样品提取液将氯霉素标准储备溶液分别配成氯霉素浓度为0.5ng/mL、1.0ng/mL、5.0ng/mL、l0ng/mL、50ng/mL和l00ng/mL的标准工作溶液,于4℃保存,可使用1周。3.买验材料2~3种蜂蜜,各l00g。四、实验步骤1.样品提取(1)取5.00g蜂蜜样品,置于50mL具塞离心管中,加入5mL水,于液体混匀器上快速混合lmin,使蜂蜜完全溶解。(2)加入15mL乙酸乙酯,在振荡器上振荡l0min,以3000r/min离心10min,吸取上层乙酸乙酯12mL,转入自动浓缩仪的蒸发管中,用自动浓缩仪在55℃减压蒸干,加入5mL水溶解残渣,得样品提取液。2.样品净化(1)将样品提取液以小于等于3mL/min的流速通过Oasis HLB固相萃取柱,待溶液完全流出后,用l0mL水分两次洗蒸发管和储液器并过柱,再用5mL,乙腈一水溶液洗柱,弃去全部淋出液,在65kPa的负压下减压抽干5min。(2)再以5mL乙酸乙酯洗脱,收集洗脱液于l0mL刻度离心管中。于50℃用氮气吹干仪吹干,待硅烷化。3.硅烷化处理加50μL硅烷化试剂于上述净化样品中,混合0.5~lmin,立即用正己烷定容至lmL。4.测定(1)将氯霉素标准工作溶液硅烷化处理后,于GC—MS仪上分析,以m/z 466为定量离子,以工作溶液浓度为横坐标,峰面积为纵坐标,绘制标准工作曲线。(2)同时,对样品净化液进行分析,并根据标准曲线查出样品净化液中氯霉素的浓度。(3)参考气相色谱一质谱条件:色谱柱为DB一5MS石英毛细管柱(30m×0.25mm,0.25μm);载气为氮气,纯度≥99.999%;流速为1.0mL/min;柱温,初始温度为70℃,然后以25℃/min程序升温至250℃,保持5min;进样量为lμL;进样方式为无分流进样;进样口温度为280℃;接口温度为280℃;负化学源为150eV;离子源温度为150℃;反应气为甲烷,纯度≥9.99%;反应气流量为40%。(4)平行试验和空白试验(除不称取试样外,均按上述步骤进行)。5.计算按下式计算样品中氯霉素的残留量:x=cV/m式中,x为样品中氯霉素的残留量,μg/kg;c为样品净化液中氯霉素的浓度,ng/mL;V为样品净化液的定容体积,mL;优为样品净化液所代表样品的质量,g。五、注意事项1.计算结果应扣除空白值。2.在用标准工作曲线对样品进行定量时,样品溶液中氯霉素的响应值均应在仪器测定的线性范围内,否则应对样品溶液进行稀释或浓缩。3.实验步骤中的色谱一质谱条件仅供参考,实际分析过程中应根据仪器的型号和实验条件,依据说明书,通过预备实验进行调整。4.在本实验的参考色谱条件下,氯霉素衍生物的保留时间为12.31min。
机器:岛津LC–2030c色谱柱:C18(4.6mm*150mm,5μm)流动相:0.01M草酸:乙腈:甲醇=7:2:1盐酸金霉素这两天购入,ups级,纯度97%,用纯甲醇溶解制成1000mg/L,用流动相稀释到100mg/L,上样后出现四个峰,均随浓度的增大而增大,空白只有溶剂峰。(图片明天补)
甲砜霉素不知道什么原因不出峰。另外以前做氯霉素标准物质的衍生效挺好的,这次不知道为什么峰下降剧烈,至少下降100倍,操作步骤没变,下降后的信号与浓度仍然成比例,恳请各位指教
林可霉素,盐酸克林霉素,大观霉素,3种药物的ASE和手动2种方式提取大观霉素回收率低,药物极性强,易溶于水,溶于甲醇。乙腈,甲醇,水,甲酸水都已试过,求各位大神指教。
按GB/T20764-2006的方法检测土霉素,用甲醇+乙腈+0.01mol/L草酸水溶液=10+20+70,Symmetry C18柱,发现土霉素出峰时间在2.9min左右,而且标峰和溶剂峰分不开,换了150cm的柱子,在3.4min出峰,但是存在保留时间一直前飘得问题,请问是我选的柱子问题,还是?而且一直想知道国标流动相中甲醇的作用,是一定要加的吗?
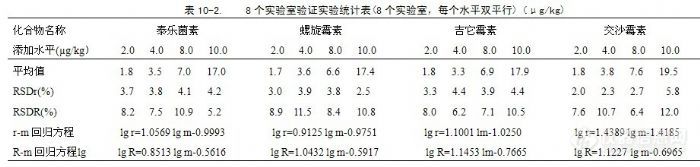
0.9940。河豚鱼中的8种抗生素和鳗鱼中林可霉素、红霉素、泰乐菌素、吉它霉素方法检出限(LOD)为2.0 μg/kg;鳗鱼中螺旋霉素、竹桃霉素、交沙霉素、替米考星方法检出限(LOD)为5.0µg/kg。回收率在75.4%~124.0%之间。八种大环内酯类抗生素重复性相对标准偏差(RSDr)在1.34%~5.79%之间,再现性相对标准偏差(RSDR)在6.20%~15.27%之间。可以用于河豚鱼和鳗鱼中8种大环内酯类抗生素残留检测的高效液相色谱-串联质谱方法的定性和定量。河豚鱼和鳗鱼在中国有着悠久的食用历史,营养丰富。由于目前中国的河豚鱼和鳗鱼主要是养殖的,在养殖过程中不可避免使用抗生素用于保护鱼体正常生长。养殖用药主要是林可霉素、竹桃霉素、红霉素、替米考星和泰乐菌素等,该类抗生素通过羟基以苷键与去氧氨基糖或二甲氨基糖缩合成碱性苷,作用于细胞核糖体50 S亚单位,阻碍细菌蛋白质合成,有较强的抗菌活性。曾广泛应用于食用动物作为预防和治疗用药,而通过食用途径进入人体,导致中毒,甚至死亡。世界各国对抗生素药物残留均有严格的限量要求,欧盟已限制在供食用动物中的饲料中使用,中国也有相应的要求。近年来,日本、韩国针对河豚鱼以药物残留为借口相继对中国河豚鱼实行贸易技术壁垒,限制和排斥中国河豚鱼出口。因此,为破解该类壁垒、促进出口,一种高灵敏度的测定河豚鱼和鳗鱼中大环内酯类抗生素的多残留方法是十分必要的。林可霉素等抗生素的吸收光谱多在紫外末端区,缺乏可用的特征紫外吸收区位。已见报道的文献中,主要分析方法有微生物法、荧光光度法、紫外分光光度法、气相色谱、薄层谱法、高效液相色谱法、毛细管电泳法(CE)和液质联用技术LC-MSn法法。在近期的文献报道中,测定大环内酯类残留,样品前处理大多采用缓冲溶液提取合固相萃取技术分析技术,采用液相色谱-串联质谱方法检测。这种技术灵敏度高、选择性和特异性好,能够对低浓度的样品进行很好的定性确认,已经成为食品和环境中污染物定性、定量分析的重要手段。文献报道的测定大环内酯类分析方法多应用于食品和动物产品,未见到同时适用于河豚鱼、鳗鱼的相关检测研究。在参考以上文献的基础上,建立用Tris缓冲溶液提取河豚鱼和鳗鱼中8种大环内酯类抗生素残留,Oasis HLB固相萃取柱萃取、净化,罗红霉素为内标,LC-MS-MS检测河豚鱼和鳗鱼中8种大环内酯类抗生素的新方法。该方法经过4年的推广使用,提取操作简单、回收率稳定、灵敏度高、选择性好,未发现不良反应,林可霉素、红霉素、泰乐菌素、吉它霉素检出限达到2.0µg/kg,鳗鱼中的螺旋霉素、竹桃霉素、交沙霉素、替米考星检出限达到5.0µg/kg,低于国际上该类药物残留限量的检测要求。1 实验过程1.1 主要试剂水,符合GB/T 6682,一级。甲醇、乙腈,色谱纯。甲醇溶液(2+3)。定容液:0.01 mol/L乙酸铵溶液+乙腈(17+3)。tris溶液:依次溶解12.0 g三羟甲基氨基甲烷(tris)和7.35 g氯化钙(CaCl2·2H2O)于1000 mL水中,用盐酸调节pH值为9。标准物质:林可霉素(CAS 7179-49-9)、竹桃霉素(CAS 7060-74-4)、红霉素(CAS 59319-72-1)、替米考星(CAS 108050-54-0)、泰乐菌素(CAS 74610-55-2)、螺旋霉素(CAS 8025-81-8)、吉它霉素(CAS 1392-21-8)、交沙霉素(CAS 16846-24-5)和内标物质罗红霉素(CAS 80214-83-1),纯度≥95%。2.0 μg/mL标准工作溶液:依次准确称取每种标准物质适量,用甲醇溶解至浓度为1.0 mg/mL的标准储备溶液;将标准储备溶液用甲醇逐步稀释为2.0 μg/mL的标准工作溶液。1.0 μg/mL内标标准溶液:准确称取罗红霉素适量,用甲醇溶解为浓度1.0 mg/mL的内标储备溶液;将内标储备溶液用甲醇逐步稀释为1.0 μg/mL内标标准溶液。测定河豚鱼用基质标准混合工作溶液系列:分别吸取1.0 μL、2.0 μL、5.0 μL、25.0 μL浓度为2.0 μg/mL的标准工作溶液,依次加入到相应的试剂瓶中,再分别加入20.0 μL内标工作溶液,用河豚鱼样品空白提取液定容至1.0 mL。配成内标物浓度均为20 ng/mL,林可霉素、竹桃霉素、红霉素、替米考星、泰乐菌素、螺旋霉素、吉它霉素和交沙霉素分别为2.0 ng/mL、4.0 ng/mL、10.0 ng/mL、50.0 ng/mL的四个浓度水平的测定河豚鱼用基质标准混合工作溶液系列。测定鳗鱼用基质标准混合工作溶液:分别吸取浓度为2.0 μg/mL的林可霉素、红霉素、泰乐菌素、吉它霉素标准工作溶液各1.0 μL、2.0 μL、5.0 μL、25.0 μL和螺旋霉素、竹桃霉素、交沙霉素、替米考星标准工作溶液各2.5 μL、5.0 μL、10.0 μL、25.0 μL,依次加入相应的试剂瓶中,再分别加
最近在做抗生素。可是用[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url]ESI+没有办法电离青霉素V和青霉素G。不知道为什么?标准品用甲醇溶解的找不到分子量+1的峰,(国家标准上的)。MSscan出来的也是没有联系的。后来改用70%水30%ACN,还是不行。不知道是不是标准品有问题还是其他原因。四环素类抗生素就电离的很好。请高手指教。
我在用酶联免疫法测蜂王浆中的氯霉素含量,但是总是有干扰,测出来连空白都是阳性的,所用试剂都重新配过,仪器也用甲醇洗过,但还是没用啊!所以求助,看有没有人知道原因的!!
求助,waters做甲砜霉素,出不来,求液相色谱条件,和质谱调节,
各位大虾,我用GB/T21317-2007液相方法检测四环素和金霉素,流动相是甲醇,乙腈和10mmol的三氟乙酸,C8柱,但是出了四个峰,做了不同浓度,峰面积和浓度的变化一致,请问大家是否遇到类似问题,如何解决啊
有没有在做红霉素液相的大神,最近做红霉素液相一点进展都没有,试了各种液相条件,都没有达到理想的效果,要不然就是不出峰,要不然就是浓度和峰面积不成比例。又跑去分析中心测,最后的结果说在210nm处没有吸收峰。分析中心的用的流动相是:A0.1%的甲酸+10mmol的乙酸铵,B甲醇,以梯度洗提的方式A:B=95:5到5:95.最后在210处没有任何峰了,,问问题可能出在了哪里
(一)食品中氯霉素的限量1999年9月我国农业部发布了《动物性食品中兽药最高残留限量》,规定了氯霉素在所有食品动物的可食用组织中不得检出。2002年3月被我国农业部关于发布《食品动物禁用的兽药及其化合物清单》列为禁止使用的抗生素。欧盟、美国等国家规定动物源性食品中氯霉素的残留限量标准为“零容许量”,即不得检出。(二)测定氯霉素残留的方法①酶联免疫法。采用间接竞争ELISA筛选法,在酶标板微孔条上包被偶联抗原,样本中残留的氯霉素和微孑L条上包被的偶联抗原竞争抗氯霉素抗体,加入酶标二抗后,加入底物显色,样本吸光度值与其残留物氯霉素的含量成负相关,与标准曲线比较再乘以其对应的稀释倍数,即可得出样品中氯霉素的含量。该方法测定低限为氯霉素0.05/g/kg。⑦GB/T 22338--2008动物源性食品中氯霉素类药物残留量测定——气相色谱一质谱法和液相色谱一质谱法测定水产品、畜禽产品和畜禽副产品中氯霉素、氟甲砜霉素和甲砜霉素残留量。气相色谱一质谱法是样品用乙酸乙酯提取,4%氯化钠溶液和正己烷溶液液分配净化,再经弗罗里硅土(Florisil)柱净化后,以甲苯为反应介质,用N,O-I双(三甲基硅烷)三氟乙酰胺一三甲基氯硅烷(BSTFA+TMCS,99+1)于70℃硅烷化,用气相色谱/负化学电离源质谱测定,内标工作曲线法定量。该方法测定低限为氯霉素0.1/μg/kg,氟甲砜霉素和甲砜霉素0.5μg/kg。液相色谱一质谱法是针对不同动物源性食品中氯霉素、氟甲砜霉素和甲砜霉素残留量,分别采用乙腈、乙酸乙酯一乙醚或乙酸乙酯提取,提取液用固相萃取柱进行净化,液相色谱一质谱/质谱仪进行测定,氯霉素采用内标法定量,氟甲砜霉素和甲砜霉素采用外标法定量。该方法对氯霉素的测定低限为0.1μg/kg;氟甲砜霉素和甲砜霉素为O.1/μg/kg。
SCIEX API 4000做负离子模式测PPCPs及其代谢物,流动相用的是常规的甲醇-5mmol醋酸铵,色谱柱是[b]Phenomenex [b][color=#0000cd]Gemini C18[/color][/b],[/b]配标和复溶上机用的都是20%甲醇水,进样量10μL,走标准曲线和样品时发现布洛芬及其代谢产物羟基布洛芬、羧基布洛芬的出峰时间越来越提前,起初怀疑是[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相[/color][/url]没有平衡好,但带着一起做的氯霉素类的出峰时间却一直很稳定,感觉不是[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相[/color][/url]系统的问题,是物质的问题。想求助一下各位大佬这类含有羧基的物质出峰时间不断往前飘的产生原因,后期是应该调整复溶液还是调整流动相或是换柱子呢?
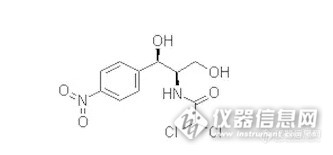
随着现代化市场经济的迅猛发展,人们的生活条件得到了改善,对动物源性食品的需求也越来越多,其安全问题也随之越来越重要,其中,氯霉素残留是较为突出的因素之一。那么,氯霉素是什么?其在动物源性食品中的现状又是怎样的呢?动物源性食品 动物源性食品是指来源于动物,可供人类食用的动物产品,包括肉、脂肪、脏器、血液、蛋、奶等。随着国民经济的发展,我国城乡居民的生活水平和健康消费观念的提高,动物源性食品在我国居民食品结构中占有比例越来越大,需求量不断增大,拉动了畜牧业的快速发展。然而,近几年发生的“瘦肉精”“掺假羊肉”“病死猪肉”“三鹿婴幼儿奶粉”等事件使消费者谈“肉”色变,动物源性食品的安全状况已成为当今广受关注的社会话题。氯霉素http://ng1.17img.cn/bbsfiles/images/2015/08/201508251507_562769_2984502_3.jpg 氯霉素为白色针状或微带黄绿色的针状、长片状结晶或结晶性粉末,味苦,易溶于甲醇、乙醇、丙酮,微溶于水,干燥时稳定。氯霉素的化学结构含有对硝基苯基、丙二醇与二氯乙酰胺三个部分, 因其分子中含有一个不游离的氯,故命名氯霉素,其抗菌活性主要与丙二醇有关。 氯霉素是由委内瑞拉链丝菌产生的一种抑菌性广谱抗生素,它通过与核糖体的50s亚单位结合而抑制细菌蛋白质的合成。对革兰氏阳性菌和革兰氏阴性菌均有较好的抑制作用,对立克次体、衣原体也有抑制作用。因其高效廉价,曾在畜牧业中广为应用。然而由于其对造血系统有严重的不良反应,且细菌对氯霉素有发展缓慢的耐药性,所以对其临床应用已经做出严格控制。动物源性食品中氯霉素的现状 国际上对动物源性农产品的兽药残留问题亦广泛关注,世界上许多国家禁止氯霉素使用于生产食品动物,并规定了其在畜产品中的最高残留限量。欧盟、美国等均在其相关法规中规定氯霉素的残留为“零容许量” ,即不得检出。 我国农业部已将氯霉素从2000年版的《中国兽药典》中删除,作为禁用药品。在2002年底的农业部第235号公告《动物源性食品中兽药最高残留限量》中也明确规定氯霉素禁止使用,在动物性食品中不得检出。现在,氯霉素是动物性农产品的必检指标。动物源性食品中氯霉素的检测 关于动物源性食品中氯霉素的检测,主要使用的标准方法有:GB/T22338-2008《动物源性食品中氯霉素类药物残留量测定》、GB/T20756-2006《可食动物肌肉、肝脏和水产品中氯霉素、甲砜霉素和氟苯尼考残留量的测定 液相色谱-串联质谱法》、GB29688-2013《食品安全国家标准 牛奶中氯霉素残留量的测定 液相色谱-串联质谱法》、GB/T18932.19-2003《蜂蜜中氯霉素残留量的测定方法 液相色谱-串联质谱法》等。食品生产企业和销售单位应把动物源性食品中氯霉素的检测作为原料质控和进货采购验证的一项重要指标进行管理,要求供应商出具相关检测报告,必要时可委托有资质的第三方检测机构进行检测。
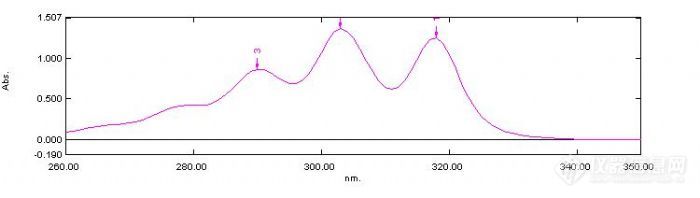
[size=3][font=宋体][/font][/size][size=3][font=宋体]纳他霉素([/font][/size][size=3][font=宋体]Natamycin[/font][/size][size=3][font=宋体])是多稀大环脂类物质,为一种安全、高效、广谱的新型生物防腐剂,对食品中污染的霉菌、酵母等真菌有很强的抑菌效果,广泛应用于乳制品、肉制品、果汁饮料、葡萄酒等食品的防腐[sup][1][/sup].目前已成为[/font][/size][size=3][font=宋体]30[/font][/size][size=3][font=宋体]多个国家广泛使用的一种天然生物性食品防腐剂和抗菌添加剂[sup][2][/sup].纳他霉素在水中和极性有机溶剂中溶解度很低,不溶于非极性溶剂[/font][/size][sup][size=3][font=宋体][3][/font][/size][/sup][size=3][font=宋体],室温下在水中的溶解度30[/font][/size][size=3][font=Times New Roman]~[/font][/size][size=3][font=宋体]100mg/L.[/font][/size][size=3][font=宋体] [/font][/size][size=3][font=宋体]目前纳他霉素的分析方法主要有比色分析法、元素分析法、微生物分析法、色谱分析法、滴定分析法、电泳分析法。本文采用二阶导数光谱法测定酸奶中纳他霉素,方法简便,准确性高等优点。[/font][/size][size=3][font=宋体][/font][/size][size=3][font=宋体]1[/font][/size][size=3][font=宋体]实验部分[/font][/size][size=3][font=宋体]1.1[/font][/size][size=3][font=宋体]主要仪器和试剂[/font][/size][size=3][font=宋体]紫外分光光度计:UV-2550岛津有限公司[/font][/size][size=3][font=宋体]电子天平:AUY220型 岛津有限公司[/font][/size][size=3][font=宋体]离心机:ZN48 三元恒泰有限公司[/font][/size][size=3][font=宋体]醋酸:分析纯 北京化工厂[/font][/size][size=3][font=宋体]甲醇:分析纯 北京化工厂[/font][/size][size=3][font=宋体]以上试剂均为分析纯,实验用水为三级水[/font][/size][size=3][font=宋体]1.2[/font][/size][size=3][font=宋体]标准溶液的制备[/font][/size][size=3][font=宋体]1.2.1[/font][/size][size=3][font=宋体]纳他霉素标准储备液:称取纳他霉素标准品10.0mg ,用冰乙酸+水=5:40的溶液溶解,定容到100ml,其浓度为100ug/ml。[/font][/size][size=3][font=宋体]1.2.2[/font][/size][size=3][font=宋体]纳他霉素标准使用液:吸取纳他霉素标准储备液10ml 于100ml容量瓶中,,用冰乙酸+水=5:40的溶液定容至刻度,其浓度为10ug/ml。[/font][/size][size=3][font=宋体]1.3[/font][/size][size=3][font=宋体]样品溶液的制备[/font][/size][size=3][font=宋体]称酸奶样品2.5g(精确到0.0001g), 加甲醇16mL,加冰乙酸2mL,加水定容于25ml,离心8min后测定。[/font][/size][size=3][font=宋体]1.4[/font][/size][size=3][font=宋体]试验方法[/font][/size][size=3][font=宋体] [/font][/size][size=3][font=宋体]用紫外分光光度计在200nm-350nm区域进行光谱扫描,对扫描的光谱图进行二阶求导,采用峰面积定量。[/font][/size][size=3][font=宋体]2[/font][/size][size=3][font=宋体]结果与讨论[/font][/size][size=3][font=宋体]2.1[/font][/size][size=3][font=宋体]紫外光谱及二阶导数光谱[/font][/size][size=3][font=宋体]图1为1.60ug/kg纳他霉素标准溶液的紫外光谱图,由图1可知,纳他霉素在295,303,311nm处有吸收峰,在303nm有最大吸收峰.为了消除酸奶样品中山梨酸,苯甲酸等物质在此区间的吸收干扰,对吸收光谱进行二阶求导,通过二阶导数有效但是消除了干扰.吸收光谱图如图1-4.[img]http://ng1.17img.cn/bbsfiles/images/2010/04/201004202102_213585_1644065_3.jpg[/img][font=宋体]图[/font][font='Times New Roman']1[/font][font=宋体]标准品谱图[/font][font='Times New Roman'] [img]http://ng1.17img.cn/bbsfiles/images/2010/04/201004202104_213586_1644065_3.jpg[/img]图2样品图[img]http://ng1.17img.cn/bbsfiles/images/2010/04/201004202104_213587_1644065_3.jpg[/img]图3 标准品二阶导数图[img]http://ng1.17img.cn/bbsfiles/images/2010/04/201004202105_213588_1644065_3.jpg[/img]图4样品二阶导数图[/font][/font][/size]







 我要推广仪器
我要推广仪器
 下载APP
下载APP