最近有单位来让帮忙检测四聚乙醛样品中的烟酰胺和三聚乙醛的含量,他们提供了厂家制定的标准,我查了相关文献,附件中发表在分析化学2000年28卷第10期1313页上的文章就是该厂的标准,里面的[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]条件让我非常纳闷,理论上讲进样口的温度通常要大于被测样品中最高沸点的组分的沸点,但是该标准进样口温度才100度,而待测样品中三聚乙醛沸点128度,四聚乙醛176度,烟酰胺150度,我比较纳闷待测样品都没有气化如何测得的数据,希望各位高手能帮忙解决这个问题。另外还有一个问题,四聚乙醛只溶于氯仿,常规溶剂都不溶,我试过丙酮、THF、乙腈、乙醇、乙酸乙酯、DMF等,而烟酰胺却不溶于氯仿,仅溶于乙醇、甘油等,而文献中在溶解样品时加入一小粒氢氧化钠,不知加碱是什么意思,从结论上来看文献中都能得出烟酰胺的含量,难道氯仿中加入一小粒氢氧化钠就能溶解烟酰胺。还有[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]能测含氢氧化钠的样品?我的机子是HP6890 HP101柱子,FID。希望各位老师能帮忙解决上述问题,出于对仪器考虑,我没有敢帮他测,等待大家的支持,谢谢!
有谁做过乙醛?需用什么色谱柱和条件,乙醛是不是易挥发,用乙酰胺做溶剂可以吗?
[table=100%][tr][td]苯胺与乙醛在甲酸存在下反应,目的是生成分子量为119的席夫碱,用质谱检测结果,存在有少量的119,有未反应的苯胺峰,还有比较强的分子量为146的峰,求有机大神们帮忙分析分析,这是什么产物?谢谢[/td][/tr][/table]
检测乙醇溶残,乙醛和乙缩醛超标 对照乙醛峰面积很小 仪器7890A 柱子DB-624(和乙醇厂家用的一样的)分离度也特别不好,我想问下大家有没有碰到过类似问题 怎么解决的?
乙醇挥发性杂质检测,对照溶液b中的乙醛峰不明显,感觉和甲醇没有分开
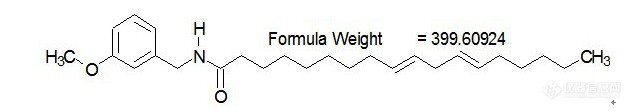
公司玛咖研究团队专心致力于我国玛咖产业发展,团队核心成员多年来一直致力于玛咖在我国的产业化推进,尤其是通过长期的研究玛咖药效物质基础和作用机制,发掘出多种具有医药健康产品开发潜力的玛咖生物活性成分,如:玛咖酰胺,玛咖烯,芥子油苷等,并开展玛咖生物活性成分含量及基础营养成分分析——标准建立、玛咖品质评价、深加工产品开发、玛咖活性物质基础研究——玛咖提取物及深加工产品开发、医药产品开发。 现应部分玛咖科研机构及深加工企业需求,提供部分玛咖酰胺标准品,信息如下:[color=#ff00ff] 中文名:[/color][color=#ff00ff]间-甲氧基苄基-[/color][color=#ff00ff]亚麻酰胺[/color];N-(3-甲氧基苄基)-(9Z, 12Z, 15Z)-十八碳三烯酰胺 英文名:N-(3-methoxybenzyl)-(9Z,12Z,15Z)-octadecatrienamide 分子式:C26H39NO2 [color=#ff00ff]CAS:[url=https://detail.1688.com/offer/565181222786.html?spm=a2615.7691456.0.0.46631c53wILZes#][color=#ff00ff]883715-23-9[/color][/url][/color] 分子量:397 贮存条件: -20℃冷藏、密封、避光 推荐溶剂:甲醇、乙腈 纯 度: ≥ 95% 用 途: 用于含量测定/鉴定/药理实验等。 结构式:[img=,633,111]http://ng1.17img.cn/bbsfiles/images/2018/03/201803121640131086_801_3030551_3.jpg!w633x111.jpg[/img] 规 格:20mg 含 量:98% 储存温度:0℃~8℃[img=,690,490]http://ng1.17img.cn/bbsfiles/images/2018/03/201803121640324296_653_3030551_3.jpg!w690x490.jpg[/img] 公司玛咖研究团队专心致力于我国玛咖产业发展,团队核心成员多年来一直致力于玛咖在我国的产业化推进,尤其是通过长期的研究玛咖药效物质基础和作用机制,发掘出多种具有医药健康产品开发潜力的玛咖生物活性成分,如:玛咖酰胺,玛咖烯,芥子油苷等,并开展玛咖生物活性成分含量及基础营养成分分析——标准建立、玛咖品质评价、深加工产品开发、玛咖活性物质基础研究——玛咖提取物及深加工产品开发、医药产品开发。[img=,690,370]http://ng1.17img.cn/bbsfiles/images/2018/03/201803121640463341_1492_3030551_3.jpg!w690x370.jpg[/img] 玛咖研发技术服务: 玛咖研究中心引领我国玛咖产业发展——玛咖核心技术研究——玛咖良种培育——玛咖标准化种植——采收及深加工——玛咖新型健康产品开发和推广。 核心技术:玛咖良种培育——玛咖标准化种植——玛咖中生物活性成分含量检测分析——玛咖中活性成分浓缩提取制备技术——玛咖生理活性物质作用机理研究——玛咖深加工过程中生物活性成分稳定性研究等。[img=,690,387]http://ng1.17img.cn/bbsfiles/images/2018/03/201803121641093953_8734_3030551_3.jpg!w690x387.jpg[/img] 开展多项技术服务: 玛咖研究中心实现了玛咖中生物活性成分物质,如:玛咖酰胺、玛咖烯、玛咖咪唑生物碱、玛咖苄基芥子油苷、异硫氰酸苄酯、腺苷类物质等玛咖特征活性物质的结构解析、含量测定、制备技术。 玛咖样品检测分析: 玛咖生物活性成分:1 、玛咖酰胺;2、 玛咖烯;3、 总芥子油苷;4、 挥发油(主要检测异硫氰酸苄酯类物质等);5 、玛咖咪唑生物碱;6 、甾醇 ;7、 皂苷;8、 腺苷等 玛咖基础营养成分:1 、蛋白质;2、 氨基酸(氨基酸总量及水解17种氨基酸比例);3 、膳食纤维等 [color=#3333ff] [b]玛咖研发中心(武汉)面向全国提供玛咖检测服务[/b][/color] 玛咖是一种原产南美安第斯山脉海拔3800米处的高原珍稀药食两用植物。在秘鲁,当地人把玛咖奉为“安第斯皇后”,古印加帝国的“皇室贡品”,从20世纪60年开始,多国科研人员对玛咖作用进行临床试验,证明:玛咖含有多种丰富,均衡合理的营养成分及多种生理活性物质,通过调节内分泌系统平衡,达到提高生育力、抗疲劳的目的,被誉为天然的“荷尔蒙发动机”,因为玛咖能够温和的调理身体机能,这些独有的功能是其它药剂所不能替代的。 2005年前后玛咖在我国形成一定规模。由于玛咖品种的差异、种植技术的差异、玛咖产地的差异、加工方式的差异等,我国玛咖原料和产品的营养成分和活性成分也就有一定差异,这就影响了玛咖的食用效果和产品品质稳定性,因此无论是玛咖种植基地、原料生产企业还是玛咖产品深加工企业,都有必要对玛咖品质进行全面的检测分析。[img=,690,339]http://ng1.17img.cn/bbsfiles/images/2018/03/201803121641268751_4701_3030551_3.jpg!w690x339.jpg[/img] 玛咖品质优劣主要包括生物活性成分和营养成分两部分,玛咖生物活性成分则应该关注:玛咖酰胺、玛咖烯、玛咖芥子油苷等成分,营养成分中应该关注:玛咖粗蛋白质、17种氨基酸含量及比例、膳食纤维和矿物质等,这些成分的品质分析及含量检测对于一些普通企业来说有较大困难,尤其是活性成分检测需要用到[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质联用[/color][/url](HP[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url])、[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质联用[/color][/url](HPLC-GS)等高新技术手段。为方便玛咖种植基地、原料供应商及玛咖产品深加工企业全面了解玛咖品质,本公司现面向全国提供玛咖品质分析及检测服务。 [img=,690,349]http://ng1.17img.cn/bbsfiles/images/2018/03/201803121641415015_6891_3030551_3.jpg!w690x349.jpg[/img] 武汉华士特玛咖研发中心科研人员长期致力于玛咖研究与产业化开发工作,已经为秘鲁( Perú )利马、玻利维亚、厄瓜多尔;云南丽江、云南香格里拉、云南昭通、云南宣威;西藏林芝、西藏曲水、西藏昌都;新疆塔什库尔干、喀什、四川阿坝;宁夏、青海;吉林抚松等多地玛咖种植基地、原料供应商及产品深加工企业提供了玛咖检测服务,年均服务玛咖种植基地及玛咖产品开发企业超过300家,接待样品检测量超过1000份,同时建立了玛咖研究中心全球玛咖样本及产品展示中心,同时依托玛咖分析测试中心首次建立了国内、外玛咖品质大数据库,追溯国内外各玛咖产区的品质变迁,为玛咖研究和产业化开发提供技术支撑。同时,玛咖研究中心是目前国内玛咖成分检测服务最全面的单位。[img=,690,304]http://ng1.17img.cn/bbsfiles/images/2018/03/201803121642040581_2553_3030551_3.jpg!w690x304.jpg[/img] 检测服务项目包括:玛咖品种鉴定和玛咖产品真伪辨别、玛咖DNA指纹图谱鉴定、玛咖营养成分分析(总蛋白、氨基酸、膳食纤维、总糖、矿物质等)、玛咖活性成分分析(玛咖酰胺、玛咖烯、玛咖芥子油苷、玛咖挥发油、玛咖甾醇、玛咖咪唑生物碱、腺苷等)、各类玛咖产品活性成分分析(玛咖根、玛咖干粉、玛咖片、玛咖胶囊、玛咖片剂、玛咖口服液、玛咖提取物、玛咖饮料、玛咖酒)、玛咖中常见违禁物添加筛查(可对玛咖产品里添加的西地那非、他达那非及其类似物进行分析)、其它检测项目。 检测费用:通过电话咨询,提供专业性的检测技术服务,同时可与种植基地和产品开发企业签订长期检测技术服务协议,为种植基地玛咖品质和玛咖产品开发企业提供优质的品控服务; 检测周期:7-30日,具体需根据检测项目和数量确定; 检测咨询网址:[url]https://huaster.1688.com/[/url] QQ:422450190 电话:15926423062
乳及乳制品中舒巴坦敏感β-内酰胺酶类药物检验方法指定检验方法4.乳及乳制品中舒巴坦敏感β-内酰胺酶类药物检验方法杯碟法1、范围本标准规定了乳及乳制品中舒巴坦敏感β-内酰胺酶类药物的检验方法。本标准适用于乳及乳制品中舒巴坦敏感β-内酰胺酶类物质的检验。本方法的检出限为4U/mL。2、原理该方法采用对青霉素类药物绝对敏感的标准菌株,利用舒巴坦特异性抑制β-内酰胺酶的活性,并加入青霉素作为对照,通过比对加入β-内酰胺酶抑制剂与未加入抑制剂的样品所产生的抑制圈的大小来间接测定样品是否含有β-内酰胺酶类药物。3、设备和材料除微生物实验室常规灭菌及培养设备外,其他设备和材料如下:3.1 抑菌圈测量仪或测量尺。3.2恒温培养箱:36℃±1℃。3.3 高压灭菌器。3.4 无菌培养皿:内径90 mm,底部平整光滑的玻璃皿,具陶瓦盖。3.5 无菌牛津杯:外径(8.0士0.1) mm,内径(6.0士0.1) mm,高度(10.0士0.1) mm。3.6 麦氏比浊仪或标准比浊管。3.7 pH计。3.8 无菌吸管:1mL(0.01mL刻度值),10mL(0.1mL刻度值)。3.9 加样器:5μL~20μL,20μL -200μL及配套吸头。4、培养基和试剂 除另有规定外,所用试剂均为分析纯,水为GB/T6682中规定的三级水。4.1 试验菌种:藤黄微球菌(Micrococcus luteus) CMCC(B) 28001,传代次数不得超过14次。4.2 磷酸盐缓冲溶液:按附录A中A.1规定。4.3生理盐水(8.5 g/L):按附录A中A.2规定。4.4 青霉素标准溶液:按附录A中A.3规定。4.5 β-内酰胺酶标准溶液:按附录A中A.4规定。4.6 舒巴坦标准溶液按附录A中A.5规定。。4.7 营养琼脂培养基:按附录A中A.6规定。4.8 抗生素检测用培养基Ⅱ:按附录A中A.7规定。5、操作步骤5.1 菌悬液的制备将藤黄微球菌接种于营养琼脂斜面上,经36士1℃培养18h-24 h,用生理盐水洗下菌苔即为菌悬液,测定菌悬液浓度,终浓度应大于1×1010 CFU/mL,4 ℃保存,贮存期限2周。5.2 样品的制备将待检样品充分混匀,取1 mL待检样品于1.5 mL离心管中共4管,分别标为:A、B、C、D,每个样品做三个平行,共12 管,同时每次检验应取纯水1 mL加入到1.5 mL离心管中作为对照。如样品为乳粉,则将乳粉按1:10的比例稀释。如样品为酸性乳制品,应调节pH值至6-7。5.3 检验用平板的制备取90mm灭菌玻璃培养皿,底层加10 mL灭菌的抗生素检测用培养基Ⅱ,凝固后上层加入5 mL含有浓度为1×108 CFU/mL藤黄微球菌的抗生素检测用培养基Ⅱ,凝固后备用。5.4 样品的测定按照下列顺序分别将青霉素标准溶液、β-内酰胺酶标准溶液、舒巴坦标准溶液加入到样品及纯水中:A 青霉素5 μL。B 舒巴坦25 μL、青霉素5 μL。C β-内酰胺酶25 μL、青霉素G5 μL。D β-内酰胺酶25 μL、舒巴坦25 μL、青霉素5 μL。混匀后,将上述A~D 试样各200 μL 加入放置于检验用平板上的4个无菌牛津杯中,36士1℃培养培养18~22 h ,测量抑菌圈直径。每个样品,取三次平行试验平均值。5.5 结果报告纯水样品结果应为:(A)、(B)、(D)均应产生抑菌圈;(A)的抑菌圈与(B)的抑菌圈相比,差异在3 mm以内(含3 mm),且重复性良好;(C)的抑菌圈小于(D)的抑菌圈,差异在3 mm以上(含3 mm),且重复性良好。如为此结果,则系统成立,可对样品结果进行如下判定:7.1 如果样品结果中(B)和、(D)均产生抑菌圈,且(C)与(D)抑菌圈差异在3 mm以上(含3 mm)时,可按7.1.1、7.1.2 判定结果。7.1.1(A)的抑菌圈小于(B)的抑菌圈差异在3 mm以上(含3 mm),且重复性良好,应判定该试样添加有β- 内酰胺酶,报告β- 内酰胺酶类药物检验结果阳性。7.1.2(A)的抑菌圈同(B)的抑菌圈差异小于3 mm,且重复性良好,应判定该试样未添加有β- 内酰胺酶,报告β- 内酰胺酶类药物检验结果阴性。7.2 如果(A)和(B)均不产生抑菌圈,应将样品稀释后再进行检测。附 录 A(规范性附录)培 养 基A.1 磷酸盐缓冲溶液(pH6.0)无水磷酸二氢钾8.0 g无水磷酸氢二钾2.0 g蒸馏水加至1000 mLA.2 生理盐水(8.5 g/L)氯化钠8.5 g蒸馏水1000 mL121℃高压灭菌15 min。A.3 青霉素标准溶液准确称取适量青霉素标准物质,用磷酸盐缓冲溶液溶解并定容为0.1mg/mL的标准溶液。当天配制,当天使用。A.4 β-内酰胺酶标准溶液准确量取或称取适量β-内酰胺酶标准物质,用磷酸盐缓冲溶液溶解并定容为16000 U/mL的标准溶液。当天配制,当天使用。A.5 舒巴坦标准溶液准确称取适量舒巴坦标准物质,用磷酸盐缓冲溶液溶解并定容为1 mg/mL的标准溶液,分装后-20 ℃保存备用,不可反复冻融使用。A.6 营养琼脂蛋白胨10 g牛肉膏3 g氯化钠5 g琼脂15-20 g蒸馏水1000 mL将上述成分加入蒸馏水中,搅混均匀,分装试管每管约5~8 mL,120℃高压灭菌15 min,灭菌后摆放斜面。A.7 抗生素检测培养基Ⅱ蛋白胨10 g牛肉浸膏3 g氯化钠5 g酵母膏3 g葡萄糖1 g琼脂14 g蒸馏水1000mL将上述成分加入蒸馏水中,搅混均匀,120 ℃高压灭菌15 min,其最终pH 值约为6.6。
乳及乳制品中舒巴坦敏感β-内酰胺酶类药物检验方法杯碟法1、范围本标准规定了乳及乳制品中舒巴坦敏感β-内酰胺酶类药物的检验方法。本标准适用于乳及乳制品中舒巴坦敏感β-内酰胺酶类物质的检验。本方法的检出限为4U/mL。2、原理该方法采用对青霉素类药物绝对敏感的标准菌株,利用舒巴坦特异性抑制β-内酰胺酶的活性,并加入青霉素作为对照,通过比对加入β-内酰胺酶抑制剂与未加入抑制剂的样品所产生的抑制圈的大小来间接测定样品是否含有β-内酰胺酶类药物。3、设备和材料除微生物实验室常规灭菌及培养设备外,其他设备和材料如下:3.1 抑菌圈测量仪或测量尺。3.2恒温培养箱:36℃±1℃。3.3 高压灭菌器。3.4 无菌培养皿:内径90 mm,底部平整光滑的玻璃皿,具陶瓦盖。3.5 无菌牛津杯:外径(8.0士0.1) mm,内径(6.0士0.1) mm,高度(10.0士0.1) mm。3.6 麦氏比浊仪或标准比浊管。3.7 pH计。3.8 无菌吸管:1mL(0.01mL刻度值),10mL(0.1mL刻度值)。3.9 加样器:5μL~20μL,20μL -200μL及配套吸头。4、培养基和试剂 除另有规定外,所用试剂均为分析纯,水为GB/T6682中规定的三级水。4.1 试验菌种:藤黄微球菌(Micrococcus luteus) CMCC(B) 28001,传代次数不得超过14次。4.2 磷酸盐缓冲溶液:按附录A中A.1规定。4.3生理盐水(8.5 g/L):按附录A中A.2规定。4.4 青霉素标准溶液:按附录A中A.3规定。[size=1
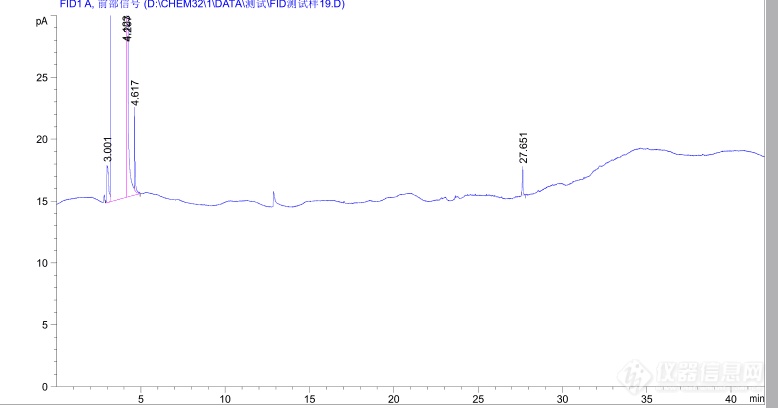
乙醇挥发性杂质,按药典配的对照品,柱子是DB-1301,升温程序:40℃12分钟,10℃到240℃,分流比10:1,流速1ml/min。乙醛和甲醇的响应信号的特别小,基线是好的!这是什么原因?[img=,690,361]https://ng1.17img.cn/bbsfiles/images/2019/07/201907161113098116_7300_1815404_3.png!w690x361.jpg[/img]
各位大侠 帮忙! 标准 YBB00102002 药用聚酯瓶的测乙醛残留含量 不得超过千分之二 如何计算?是对照品和样品的峰面积之比吗?
大家有没有做过甲酰胺的检测,药典中其限度是0.022%,沸点200多度,而我的供试品浓度为50mg/ml,进了对照,发现甲酰胺出峰高度约为检测限高度,降低分流比后甲酰胺峰形变差,峰高仍然小于定量限高度,大家有没有同样的经历或者有什么好的检测方法,谢谢
[color=#444444]最近使用[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]参照药典方法测定乙醇中挥发性杂质,柱子是KB-624,对照品、样品每次都是新处理的,但是每次测定的结果都是一样:乙醛峰面积逐渐增加,一针一针都是增加(包括对照品b,它大于对照品a,小于对照品c)。按理说应该除了对照品b,其它都是没有的或者是一样的,但现在一直是这种情况。请有经验的老手们给点建议,谢谢!![/color]
1、直接从氨基酸生成丙烯酰胺。比如,天门冬酰胺(Asn)在受热之后,脱掉一个CO2和一个NH3,即可转化为丙烯酰胺。凡是富含天门冬酰胺的食物,都非常容易产生丙烯酰胺。比如土豆、麦类、玉米等都是富含天门冬酰胺的食品。 2、氨基酸和淀粉类食物中的微量小分子糖在加热条件下发生美拉德反应,生成丙烯酰胺。在食品中,只要是含淀粉的食品,一般都会同时含有一些蛋白质,比如所有的主食、所有的薯类、所有的淀粉豆类。不过,各种氨基酸合成丙烯酰胺的“能力”有所不同。其中还是以天门冬酰胺独占鳌头,其次是谷氨酰胺(Gln),再次是蛋氨酸(Met)和丙氨酸(Ala)等。淀粉倒是不产生丙烯酰胺,但淀粉分解产生的糖会产生丙烯酰胺,葡萄糖最有效,后面依次是果糖、乳糖和蔗糖。 3、脂肪和糖降解形成丙烯醛,然后和氨基酸分解产生的氨结合,形成丙烯酰胺。凡是油炸的食品,都会发生油脂热氧化反应,而反应产物之一就是丙烯醛,它是一种挥发性小分子物质和油烟的味道有密切关系。油炸食品特别容易产生丙烯酰胺,这是理由之一。此外,蛋白质氨基酸分解也能产生少量的醛类,其中包括丙烯醛。
在做白酒分析时,我们用乙缩醛标准品配好标准液进样后发现有乙醛峰,比较小,放置一段时间(几天)后,再进样,2个峰都差不多大了,这样怎么定量?我查询了一下好像乙缩醛和乙醛在有乙醇存在的情况下会相会转化,那么乙缩醛的标液是不是配了就要测定,不能保存?是不是高浓度的也不能保存?另外标准上没有写要配制乙醛标准溶液,但是结果却又出现要体现乙醛值的,那是不是还要买乙醛标准品才行?如果同时加入乙醛标准溶液,是不是会抑制乙缩醛转化为乙醛,又或者是乙缩醛转化过来的乙醛会和乙醛标液中的乙醛叠加造成信号偏大?
问下:吸取的对照品无水甲醇、乙醛、乙缩醛是专门的对照品吗?还有用到的50μl、100μl、150μl取样管是微量进样器吗?
1 范围本标准规定了乳及乳制品中舒巴坦敏感β-内酰胺酶类药物的检验方法。本标准适用于乳及乳制品中舒巴坦敏感β-内酰胺酶类物质的检验。本方法的检出限为4U/mL。 2 原理该方法采用对青霉素类药物绝对敏感的标准菌株,利用舒巴坦特异性抑制β-内酰胺酶的活性,并加入青霉素作为对照,通过比对加入β-内酰胺酶抑制剂与未加入抑制剂的样品所产生的抑制圈的大小来间接测定样品是否含有β-内酰胺酶类药物。 3 设备和材料除微生物实验室常规灭菌及培养设备外,其他设备和材料如下:3.1 抑菌圈测量仪或测量尺。3.2恒温培养箱:36℃±1℃。3.3 高压灭菌器。3.4 无菌培养皿:内径90 mm,底部平整光滑的玻璃皿,具陶瓦盖。3.5 无菌牛津杯:外径(8.0士0.1) mm,内径(6.0士0.1) mm,高度(10.0士0.1) mm。3.6 麦氏比浊仪或标准比浊管。3.7 pH计。3.8 无菌吸管:1mL(0.01mL刻度值),10mL(0.1mL刻度值)。3.9 加样器:5μL~20μL,20μL -200μL及配套吸头。 4 培养基和试剂 除另有规定外,所用试剂均为分析纯,水为GB/T6682中规定的三级水。4.1 试验菌种:藤黄微球菌(Micrococcus luteus) CMCC(B) 28001,传代次数不得超过14次。4.2 磷酸盐缓冲溶液:按附录A中A.1规定。4.3生理盐水(8.5 g/L):按附录A中A.2规定。4.4 青霉素标准溶液:按附录A中A.3规定。4.5 β-内酰胺酶标准溶液:按附录A中A.4规定。4.6 舒巴坦标准溶液按附录A中A.5规定。。4.7 营养琼脂培养基:按附录A中A.6规定。4.8 抗生素检测用培养基Ⅱ:按附录A中A.7规定。
小弟最近在编制环境监测类的实验标准操作规程, 由于部分标准内使用的仪器和试剂比较老, 所以想求组各位大侠,有没有内部的关于乙醛、丙烯醛、硝基苯类、苯胺类的作业指导书啊, 麻烦各位大家了,万分感谢!
我按照10版药典的方法用气相检测糖精钠中的甲苯磺酰胺,两种对照品邻甲苯磺酰胺和对甲苯磺酰胺都没有出峰,所有的图只有溶剂峰—二氯甲烷出峰了。色谱柱用的是药典规定的OV-17.有没有人做过这个东西??求解~~
请教大家一下,大家买回来的甲醛和乙醛标准溶液都是怎么存放的啊?是用安剖瓶装的,假如一次配储备液的时候用不完,剩下的怎么保存好啊?有20ml呢!
请问用岛津GC-2010如何测定水中乙醛,丙烯醛?按照国标方法,直接进水溶液,使用FID检测器,HP-INNOWAX的柱子,进样口温度130,柱温76,检测器150,怎么检测不到峰呢?换用RTX-1701和Rtx-1后测定乙醛还是不出峰。请各位帮忙!急!
药材样品溶液和对照品在聚酰胺板跑,但样品与对照总是不一致,这个成分在样品中含量约0.06%左右,样品称的是2g,最后溶解于2ml的EP管中,点样量5μL。后来把样品称到5g,但最后无法全部洗脱于2ml的EP管中,由于太浓稠了,点样量只是点了一下。但还是一样。 药材为叶类药材
用天津光复精细化工研究所的40%乙醛色标,用超纯水稀释后出两个峰,用乙酸和乙醇验证后都不是其中的一个,请问大家是怎么做乙醛的啊,难道非要蒸馏后自己标定吗?
[size=5]超临界流体色谱快速测定烟酰胺的含量[/size] 来源: 作者:郭亚东,马银海,张艳,彭永芳摘要:采用超临界流体色谱快速测定制剂中烟酰胺的含量.在CO2流动相中添加10%的甲醇,于填充柱上分离,检测波长为216nm,在测定范围内,浓度与其峰面积呈良好的线性关系(r=0.9998),峰面积的相对平均偏差(RSD)为1.39%,平均回收率97.3%~101.3%,4min即可完成分析.方法简便,样品前处理简单,可用于制剂中烟酰胺的快速分析。关键词:烟酰胺;超临界流体色谱;含量测定水溶性维生素对人们的生长发育和健康有着重要作用,人们除了从水果和蔬菜中摄取外,还从添加了维生素的食品和复合维生素制剂中补充人体的需要,有必要建立快速,稳定的分析方法用于其含量测定。对烟酰胺的含量测定方法主要是高效液相色谱法,该方法取得了较好的结果,但其分析时间长,样品前处理麻烦。此外,毛细管电泳,胶束电动毛细管电泳,气/质联用等方法也用于它的含量测定.本文探索了用超临界流体色谱测定维生素含量的方法,结果令人满意。1 仪器与试药超临界流体色谱仪:Gihon Model SF3系统(英国),对照品烟酰胺购自Lancaster化学公司(英国),甲醇为高效液相色谱纯,CO2为超临界流体色谱纯.2 实验方法及结果分析2.1 色谱条件色谱柱cyano(5μm,4.6×250mm),柱温50℃,紫外检测器配有高压检测池,其检测波长为216nm,进样装置带有l0μL进样阀的自动进样器,流动相压力20MPa,流动相流速为2.0mL/min。2.2 流动相对分离的影响只用CO2作流动相时,其保留时间太长,且色谱峰拖尾严重;当在流动相中加人10%的甲醇后,其峰形大为改善,保留时间缩短;当流动相流速改变时,保留时间会有小的改变,但对峰形几乎没有影响。2.3 线性关系考查将烟酰胺对照品取适量,精称后用甲醇稀释成5~50μg/mL的标准溶液,取6个不同浓度的对照品溶液按上述实验条件各进样三次,记录其色谱图,以对照品浓度(μg/mL)为横坐标,峰面积值为纵坐标,绘制标准曲线,得回归方程Y=一351.6+147.7X,R=0.9998,可见在所用浓度范围内具有良好的线性关系。2.4 精密度及稳定性试验测定该维生素日内和日间的峰面积,以考查分析方法的精密度和样品的稳定性,在上述实验条件下,连续进样8次,烟酰胺峰面积的相对标准偏差(RSD)为1.11%,放置1d后的RSD为1.39%,表现出良好的精密度和稳定性。2.5 回收率试验精密称取已知含量的同一样品三份,加人不同量的烟酰胺对照品,按样品测定项下的条件进样分析,计算其回收率,烟酰胺的回收率平均值±SD为98.3±1.28%。2.6 样品测定取昆明振华制药厂生产的复合维生素B(批号990701)5片,碾碎后用5mL甲醇溶解并超声提取20min,用0.45μm滤膜过滤,适当稀释后在上述色谱条件下进样分析,以峰面积按标准曲线法计算含量。3 讨论3.1 流动相选择当用CO2加10%甲醇作流动相时,烟酰胺的保留时间为3.7min。可以满足快速分析的要求,且色谱峰形得到改善。3.2 提取溶剂的选择比较了水、甲醇和乙醇作溶剂提取样品,结果以甲醇较好。样品用甲醇溶解并超声提取后,直接进样分析,不需要复杂的前处理.3.3 结果通过测定回收率,精密度并考查其线性关系,表明该方法可以于快速测定烟酰胺含量,能给出满意的结果.[参考文献][1] Hurtado S A,Nogues M T V,Pulido M I et a1.Determination of water—soluble vitamins in infant milk by HPLC[J].chromato A,1997,778:247.[2] Wills R B H,Shaw C G,Day W R.Analysis of water soluble vitamins by PHLC[J].Chromatogr Sci.,1977,15:62.
请教:注射用水溶性维生素中关于烟酰胺、等5项的液相检测方法其标准为烟酰胺、盐酸吡哆辛、硝酸硫胺、泛酸钠、维生素C钠和核黄素磷酸钠 照高效液相色谱法(中国药典1995年版二部附录Ⅴ D)测定。 色谱条件与系统适用性试验 用氨基键合多孔硅胶为填料,以(0.02mol/L)磷酸二氢钾溶液-乙腈(27:73),用10%盐酸溶液调节pH为5.3的溶液为流动相,流速为1.5ml/min,检测波长:烟酰胺、盐酸吡哆辛、硝酸硫胺、泛酸钠、维生素C钠为214nm;核黄素磷酸钠用萤光检测λEX=445nm、λEM=520nm。各组分的分离度应符合要求。 对照品溶液的制备 (1)取烟酰胺对照品约150mg、硝酸硫胺对照品约12mg、盐酸吡哆辛对照品约18mg、泛酸钠对照品约62mg,分别精密称量置50ml量瓶中,加水溶解并稀释至刻度摇匀,精密量取2ml置50ml量瓶中,用流动相稀释至刻度,摇匀,即为对照品溶液(Ⅰ),此溶液置暗处充氮气于零下20℃可保存1个月。(2)取维生素C钠对照品约425mg、核黄素磷酸钠对照品约19mg,精密称定,置50ml量瓶中加水溶解并稀释至刻度摇匀,精密量取2ml置50ml量瓶中,用流动相稀释至刻度,摇匀即为对照品溶液(Ⅱ),此溶液必须临用新鲜配制,并于零下20℃保存,用前放置至室温。 等容混合对照品溶液(Ⅰ)和对照品溶液(Ⅱ)即为对照品溶液。 供试品溶液的制备 取装量差异项下的内容物约2瓶重量,精密称定,置100ml量瓶中,加水溶解并稀释至刻度,摇匀,精密量取15ml置200ml量瓶中,用流动相稀释至刻度。 测定法 取对照品溶液和供试品溶液各10μl,交替注入液相色谱仪,测定,用外标法计算各组分含量,即得。目前存在问题用紫外检测的分不开5种组分,大家有什么好办法,谢谢
顶空气相色谱法测定缬沙坦原料药中残留溶剂,以二甲基乙酰胺(DMA)作溶剂时,单独的对照不出峰,混合对照出峰,急求各位分析这是为什么?如何改进?顶空条件:DANI HSS86.50 顶空进样器,平衡温度100度,进样系统温度120度,传递管路温度120度,加压时间30秒,压力平衡时间5秒,注入时间30秒气相色谱条件:进样口温度200,检测器(FID)温度250,柱温:初始温度40,维持2分钟,以3度/秒升至100,再以30度/秒升温至200,维持2分钟;分流比10:1色谱柱:rtx-5ms,30m,0.25,0.251、以同样的样品(DMA溶解)手动进样出峰,但顶空不出峰。重复前期品种的方法,配置以水溶剂的甲醇、乙酸乙酯混合对照溶液,顶空进样,结果出峰,这是不是说明问题与DMA有关?2、DMA沸点166度,有没有可能是因为它在系统中冷凝?个人将顶空进样系统温度升至180度后,开始几针样品好了(但仍不确定是不是因为温度的原因),但过了一会单独的对照又不出峰了?3、因为中国药典标准中是用二甲基乙酰胺,所以用它了请各位帮忙分析一下,急死了!有什么没有说明白的地方,请指出,非常非常感谢!
请教各位:在乙醇的气相色谱检验中,对照品溶液制备用到的试剂:甲醇、乙醛、乙缩醛、苯、4-甲基-2-戊醇。这些试剂必须用国家标准品吗?还是能用色谱纯的试剂代替,如果能代替使用,需不需要做相关对比试验,又该怎么进行这些对比试验?谢谢各位大神解答。。。。
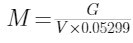
[b][font=微软雅黑]一、原理[/font][/b][font=微软雅黑][font=微软雅黑]用亚硫酸氢钠溶液采样[/font][font=微软雅黑],乙醛与亚硫酸氢钠发生亲核加成反应,在中性溶液中生成稳定的α-羟基磺酸盐,然后在稀碱溶液中共热释放出乙醛,经色谱柱分离,用氢火焰离子化检测器测定。以标准样品色谱峰的保留时间定性,峰高或峰面积定量。[/font][/font][b][font=微软雅黑]二、方法的适用范围[/font][/b][font=微软雅黑][font=微软雅黑]本方法适用于固定污染源有组织排放和无组织排放的乙醛测定。当采样体积为[/font][font=微软雅黑]100L,进样体积为1μl时,乙醛的检出限为4×10[/font][/font][sup][font=微软雅黑][font=微软雅黑]-2[/font][/font][/sup][font=微软雅黑]mg/m[/font][sup][font=微软雅黑][font=微软雅黑]3[/font][/font][/sup][font=微软雅黑],乙醛的定量测定浓度范围为0.14~30mg/m[/font][sup][font=微软雅黑][font=微软雅黑]3[/font][/font][/sup][font=微软雅黑]。[/font][b][font=微软雅黑]三、试剂和材料[/font][/b][font=微软雅黑][font=微软雅黑]除非另有说明[/font][font=微软雅黑],分析时均使用符合国家标准的分析纯试剂和不含有机物的蒸馏水。[/font][/font][font=微软雅黑]①不含有机物蒸馏水的制备:加入少量高锰酸钾的碱性溶液于当通的蒸馏水或去离子水中使呈红紫色,再进行蒸馏即得(在整个蒸馏过程中水应始終保持红紫色,否则随时补加高锰酸钾)。[/font][font=微软雅黑]②丙酮( CH[/font][sub][font=微软雅黑][font=微软雅黑]3[/font][/font][/sub][font=微软雅黑]COCH[/font][sub][font=微软雅黑][font=微软雅黑]3[/font][/font][/sub][font=微软雅黑])。[/font][font=微软雅黑]③三聚乙醛(CH[/font][sub][font=微软雅黑][font=微软雅黑]3[/font][/font][/sub][font=微软雅黑]CHO)[/font][sub][font=微软雅黑][font=微软雅黑]3[/font][/font][/sub][font=微软雅黑]。[/font][font=微软雅黑]④浓盐酸(HCl):ρ=1.19g/ml。[/font][font=微软雅黑]⑤浓硫酸(H[/font][sub][font=微软雅黑][font=微软雅黑]2[/font][/font][/sub][font=微软雅黑]SO[/font][sub][font=微软雅黑][font=微软雅黑]4[/font][/font][/sub][font=微软雅黑]):ρ=I.84g/ml。[/font][font=微软雅黑]⑥无水碳酸钠(Na[/font][sub][font=微软雅黑][font=微软雅黑]2[/font][/font][/sub][font=微软雅黑]CO[/font][sub][font=微软雅黑][font=微软雅黑]3[/font][/font][/sub][font=微软雅黑],基准试剂)。[/font][font=微软雅黑]⑦880[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]担体(酸洗硅烷化硅藻上白色担体)(GCS880AW DMCS),80~100目。[/font][font=微软雅黑]⑧聚乙二醇20000固定液(PEG-20M)。[/font][font=微软雅黑]⑨亚硫酸氢钠吸收液 C=10g/L:称取10.0g亚硫酸氢钠溶于蒸馏水中,并稀释至 1000ml。[/font][font=微软雅黑]⑩碳酸钠溶液C(Na[/font][sub][font=微软雅黑][font=微软雅黑]2[/font][/font][/sub][font=微软雅黑]CO[/font][sub][font=微软雅黑][font=微软雅黑]3[/font][/font][/sub][font=微软雅黑])=2.0mol/L:称取106g碳酸钠溶液溶于蒸馏水中,并稀释至500ml。[/font][font=微软雅黑]?饱和氯化钠水溶液。[/font][font=微软雅黑]?氢氧化钠溶液:C(NaOH)=0.1mol/L。[/font][font=微软雅黑]?羟胺乙醇溶液 C=21g/L:称取 2.1g 盐酸羟胺溶于 10ml 水中,用95%乙醇稀释到100ml。[/font][font=微软雅黑]?溴酚蓝指示剂 C=1g/L:称取0.1g溴酚蓝于100ml 20%乙醇中。[/font][font=微软雅黑]?溴甲酚绿-甲基红指示剂:三份 1g/L 溴甲酚绿乙醇溶液于ー份2g/L甲基红乙醇溶液混合。[/font][font=微软雅黑]?盐酸标准溶液C(HCl)=0.02mol/L:量取浓盐酸1.7ml,用水稀释至1000ml,混匀。[/font][font=微软雅黑][font=微软雅黑]标定方法[/font][font=微软雅黑]:准确称取三份基准无水碳酸钠(预先在270~300℃干燥至恒重)0.0400g,分別放入三个250ml锥形瓶中,各加水5ml,使其溶解,加入溴甲酚绿-甲基红指示剂2~3滴,以配制好的盐酸溶液滴定至溶液由绿色变为暗组色,即为终点。计算如下:[/font][/font][img=,133,43]https://ng1.17img.cn/bbsfiles/images/2022/11/202211070914154975_7921_1623757_3.jpg!w133x43.jpg[/img][font=微软雅黑] [/font][font=微软雅黑][font=微软雅黑]式中[/font][font=微软雅黑]:G——所称碳酸钠的重量,g [/font][/font][font=微软雅黑] V——滴定所消耗的盐酸总体积,ml [/font][font=微软雅黑] 0.05299——1/2mmol/L盐酸标准溶液的浓度,mol/L。[/font][font=微软雅黑]?乙醛标准贮备液:在一个500ml容量瓶(A瓶)中,加入400ml亚硫酸钠吸收液,加入5.00ml新鲜解聚的乙醛,用亚硫酸氢钠吸收液稀释至标线(此乙醛标准贮备液在冰箱中可保存一个月)。与此同时,吸取5.00ml新鲜解聚的乙醛放入已加有400ml重蒸馏水的500ml容量瓶(B瓶)中,用重蒸馏水稀释至标线,用羟按法标定乙醛溶液的浓度。[/font][b][font=微软雅黑][font=微软雅黑]解聚方法[/font][font=微软雅黑]:[/font][/font][/b][font=微软雅黑][font=微软雅黑]在装有分馏柱的蒸馏装置中加入[/font][font=微软雅黑]50ml三聚乙醛和0.5ml浓硫酸,缓慢加热,使乙醛在35℃以下蒸出,用一个冰水冷却的接收器收集解聚新鲜乙醛。[/font][/font][b][font=微软雅黑][font=微软雅黑]标定方法[/font][font=微软雅黑]:[/font][/font][/b][font=微软雅黑][font=微软雅黑]分别吸取[/font][font=微软雅黑]21g/L羟胺乙醇溶液 500ml 和 0.1mol/L NaOH溶液10.0ml于100ml碘量瓶中,然后加入5.00ml乙醛水溶液,塞好磨口玻璃塞,摇匀,在室温放置30min,然后加入3滴溴酚蓝指示剂后,用0.02mol/L盐酸标准溶液滴定至蓝绿色。同时进行空白试验,在5.00ml羟胺乙醇溶液和 10.0ml NaoH溶液中加入2.0ml饱和 NaCl 水溶液及3滴溴酚蓝指示剂,用0.02mol/L盐酸标准溶液滴定至蓝绿色。另取20ml蒸馏水,加入3滴0.1%溴酚蓝指示剂,再滴定至终点。[/font][/font][font=微软雅黑][font=微软雅黑]按下式计算乙醛溶液浓度[/font][font=微软雅黑]:[/font][/font][img=,552,45]https://ng1.17img.cn/bbsfiles/images/2022/11/202211070914226477_4749_1623757_3.jpg!w552x45.jpg[/img][font=微软雅黑] [/font][font=微软雅黑][font=微软雅黑]式中[/font][font=微软雅黑]:V——所取乙醛水溶液样品的体积,ml [/font][/font][font=微软雅黑] M——盐酸标准溶液的浓度,mol/L [/font][font=微软雅黑] A——空白滴定所消耗盐酸标准溶液的体积,ml [/font][font=微软雅黑] B——标定乙醇溶液所消耗盐酸标准溶液的体积,ml [/font][font=微软雅黑] C——蒸馏水滴定所消耗盐酸标准溶液的体积,ml [/font][font=微软雅黑] 44.05——1mol CH[/font][sub][font=微软雅黑][font=微软雅黑]3[/font][/font][/sub][font=微软雅黑]CHO 的克数。[/font][font=微软雅黑]?乙醛标准溶液:临用前,把乙醛标准贮备液用亚硫酸氢钠吸收液逐级稀释成1000mg/L和100mg/L的标准溶液。[/font][b][font=微软雅黑]四、仪器[/font][/b][font=微软雅黑]1)[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱仪[/color][/url]:具氢火焰离子化检测器。[/font][font=微软雅黑]2)色谱柱:长2m,内径3mm的玻璃柱。[/font][font=微软雅黑]3)固定相:20%PEG 20M-GCS880 AW DMCS,80~100目。[/font][font=微软雅黑]4)采样仪器:[/font][font=微软雅黑]①有组织排放监测采样仪器:参考GB 16157-1996 中9.3配置采样仪器。[/font][font=微软雅黑][font=微软雅黑]采样管[/font][font=微软雅黑]:采用不锈钢、硬质玻璃或聚四氟乙烯材质,具有适当尺寸的管料作采样管,并具有可加热至120℃以上的保温夹套。[/font][/font][font=微软雅黑][font=微软雅黑]样品吸收装置[/font][font=微软雅黑]:10ml多孔玻板吸收管。[/font][/font][font=微软雅黑][font=微软雅黑]流量计量装置[/font][font=微软雅黑]:见GB16157—1996中9.3.6。[/font][/font][font=微软雅黑][font=微软雅黑]抽气泵[/font][font=微软雅黑]:见GB16157—1996中9.3.7。[/font][/font][font=微软雅黑][font=微软雅黑]连接管[/font][font=微软雅黑]:聚四氟乙烯软管或内衬聚四氟乙烯膜的硅橡胶管。[/font][/font][font=微软雅黑]②无组织排放监测采样仪器:[/font][font=微软雅黑][font=微软雅黑]引气管[/font][font=微软雅黑]:聚四氟乙烯软管,头部接一玻璃漏斗。[/font][/font][font=微软雅黑][font=微软雅黑]样品吸收装置[/font][font=微软雅黑]:10ml多孔玻板吸收管。[/font][/font][font=微软雅黑][font=微软雅黑]流量计量装置、抽气泵和连接管[/font][font=微软雅黑]:参上述相应部分配置。[/font][/font][b][font=微软雅黑]五、样品采集和保存[/font][font=微软雅黑]1、有组织排放样品采集[/font][/b][font=微软雅黑]①采样位置和采样点:按GB 16157-1996 中9.1.1和9.1.2确定采样位置和采样点。[/font][font=微软雅黑]②采样装置的连接:参考GB16157-1996中9.3图28,按采样管、样品吸收装置、流量计量装置利抽气泵的顺序连接好采样系统,连接管要尽可能知。按GB16157-1996中9.4的要求检查采样系统的气密性和可靠性。[/font][font=微软雅黑]③样品采集:将采样管头部塞适量玻璃棉后,插入排气筒采样点,用一支内装10g/L NaHSO[/font][sub][font=微软雅黑][font=微软雅黑]3[/font][/font][/sub][font=微软雅黑][font=微软雅黑]溶液[/font][font=微软雅黑]5ml的多孔玻板吸收管,以0.3~0.5L/min的流量采样:采样过程中调节夹套温度,以使水汽不在管壁凝结为宜,采样时间视乙醛浓度而定。纪录采样流量、温度、压力及采样时间等。采样结束后,取下吸收管,密封其进、出口,带回实验室。[/font][/font][b][font=微软雅黑]2、无组织排放样品采集[/font][/b][font=微软雅黑]①采样位置和采样点:按GB16297-196中附C的规定确定无组织排放监控点的位置,或按其他特定的要求确定环境空气采样点。[/font][font=微软雅黑]②采样装置的连接:按引气管、样品吸收装置、流量计量装置和抽气泵的顺序连接采样系统,连接管要尽可能短。如无必要,样品吸收装置前可不接引管。按GB16157-1996中9.4的要求检查采样系统的气密性和可靠性。[/font][font=微软雅黑]③样品采集:用一支装 10g/L NaHSO[/font][sub][font=微软雅黑][font=微软雅黑]3[/font][/font][/sub][font=微软雅黑][font=微软雅黑]溶液[/font][font=微软雅黑]5ml的多孔玻板吸收管,在常温下以1.0L/min的流量采样100L以上,同时记录采样温度、压力及采样时间。采样结束后,取下吸收管,密封其进、出口,带回实验室进行分析。[/font][/font][b][font=微软雅黑]3、样品保存[/font][/b][font=微软雅黑][font=微软雅黑]采集好的样品应尽快分析。如不能及时分析[/font][font=微软雅黑],在常温下避光保存,至多可保存6d。[/font][/font][b][font=微软雅黑]六、步骤[/font][font=微软雅黑]1、色谱条件[/font][/b][font=微软雅黑][font=微软雅黑]柱温[/font][font=微软雅黑]:90℃ 气化室温度:140℃ 检测器温度:140℃。[/font][/font][font=微软雅黑][font=微软雅黑]载气[/font][font=微软雅黑]:纯氮(99.99%),流量为20ml/min。[/font][/font][font=微软雅黑][font=微软雅黑]燃气[/font][font=微软雅黑]:纯氢(99.9%),流量为50ml/min。[/font][/font][font=微软雅黑][font=微软雅黑]助燃气[/font][font=微软雅黑]:空气,流量为350ml/min。[/font][/font][font=微软雅黑][font=微软雅黑]进样量[/font][font=微软雅黑]:1μl。[/font][/font][b][font=微软雅黑]2、校准曲线的绘制[/font][/b][font=微软雅黑][font=微软雅黑]乙醛的标准系列[/font][font=微软雅黑]:取七个10ml比色管,按表1配制成乙醛的标准系列[/font][/font][img=,453,123]https://ng1.17img.cn/bbsfiles/images/2022/11/202211070914307977_1129_1623757_3.jpg!w453x123.jpg[/img][font=微软雅黑] [/font][font=微软雅黑][font=微软雅黑]向以上各比色管中加入[/font][font=微软雅黑]2.0mol/L的Na[/font][/font][sub][font=微软雅黑][font=微软雅黑]2[/font][/font][/sub][font=微软雅黑]CO[/font][sub][font=微软雅黑][font=微软雅黑]3[/font][/font][/sub][font=微软雅黑][font=微软雅黑]溶液[/font][font=微软雅黑]0.50ml,摇匀。按所用[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱仪[/color][/url]的操作规程,用微量进样器分别吸取1μl标准系列溶液,在相同色谱条件下测定各标准溶液的色谱峰高,以峰高为纵坐标,乙醛含量为横坐标绘制标准曲线,并计算校准曲线的线性回归方程式。[/font][/font][b][font=微软雅黑]3、样品测定[/font][/b][font=微软雅黑][font=微软雅黑]将吸收管中的吸收液转移至[/font][font=微软雅黑]10ml比色管(带5ml刻度)中,用少量10g/L NaHSO[/font][/font][sub][font=微软雅黑][font=微软雅黑]3[/font][/font][/sub][font=微软雅黑][font=微软雅黑]溶液洗涤吸收管[/font][font=微软雅黑],洗涤液并入10ml比色管中,并定容至5ml刻度,然后加入2.0mol/L的Na[/font][/font][sub][font=微软雅黑][font=微软雅黑]2[/font][/font][/sub][font=微软雅黑]CO[/font][sub][font=微软雅黑][font=微软雅黑]3[/font][/font][/sub][font=微软雅黑][font=微软雅黑]溶液[/font][font=微软雅黑]0.50ml,摇匀。以下按绘制校准曲线相同步骤进行样品测定。[/font][/font][b][font=微软雅黑]七、计算[/font][font=微软雅黑]1、定性分析[/font][/b][font=微软雅黑]按乙醛标准溶液色谱峰的保留时间定性。[/font][font=微软雅黑][font=微软雅黑]若首次分析成分复杂的样品[/font][font=微软雅黑],并对定性结果存有疑虑时,应采用双柱定性。[/font][/font][font=微软雅黑][font=微软雅黑]若用双柱定性后[/font][font=微软雅黑],对结果仍有疑虑,可采用[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]-质谱分析或其他方法作进一步定性。[/font][/font][b][font=微软雅黑]2、定量分析[/font][/b][font=微软雅黑]①校准曲线法:根据测得的乙醛峰高,直接在校准曲线上查得样品溶液中乙醛的含量C[/font][sub][font=微软雅黑][font=微软雅黑]样[/font][/font][/sub][font=微软雅黑],或由回归方程式计算样品溶液中乙醛的含量C[/font][sub][font=微软雅黑][font=微软雅黑]样[/font][/font][/sub][font=微软雅黑],再按下式计算气体样品中乙醛的浓度C。[/font][img=,367,42]https://ng1.17img.cn/bbsfiles/images/2022/11/202211070914379918_1404_1623757_3.jpg!w367x42.jpg[/img][font=微软雅黑] [/font][font=微软雅黑][font=微软雅黑]式中[/font][font=微软雅黑]:C[/font][/font][sub][font=微软雅黑][font=微软雅黑]样[/font][/font][/sub][font=微软雅黑]——样品溶液中乙醛的含量,μg [/font][font=微软雅黑] h——样品溶液中乙醛的色谱峰高,mm [/font][font=微软雅黑] V[/font][sub][font=微软雅黑][font=微软雅黑]nd[/font][/font][/sub][font=微软雅黑]——标准状态下的于采气体积,L [/font][font=微软雅黑] b——回归方程的斜率。[/font][font=微软雅黑][font=微软雅黑]在应用标准曲线法进行定量分析时[/font][font=微软雅黑],任何一次开机分析样品,都应首先绘制校准曲线,然后每分析5~10个样品(根据仪器的稳定情况而定)插入一校准曲线中浓度适当的标准样品,其测值与原先的测值比较,相对偏差应小于15%,否则应重新做校准曲线。[/font][/font][font=微软雅黑]②单点比较法:用单点比较法进行定量分析时,应具备如下条件:[/font][font=微软雅黑]a.标准溶液的响应值应与被测样品溶液的响应值接近 [/font][font=微软雅黑]b.标准溶液与样品溶液同时进行分析,进样体积相同 [/font][font=微软雅黑]c.一个样品连续进样两次,其测定值的相对偏差小5% [/font][font=微软雅黑]d.取两次测定平均值,按下式计算气体样品中乙醛的浓度C:[/font][img=,333,60]https://ng1.17img.cn/bbsfiles/images/2022/11/202211070914438467_1013_1623757_3.jpg!w333x60.jpg[/img][font=微软雅黑] [/font][font=微软雅黑][font=微软雅黑]式中[/font][font=微软雅黑]:h[/font][/font][sub][font=微软雅黑][font=微软雅黑]样[/font][/font][/sub][font=微软雅黑]——样品溶液中乙醛的色谱峰高,mm [/font][font=微软雅黑] h[/font][sub][font=微软雅黑][font=微软雅黑]标[/font][/font][/sub][font=微软雅黑]——标准溶液中乙醛的色谱峰高,mm [/font][font=微软雅黑] C[/font][sub][font=微软雅黑][font=微软雅黑]标[/font][/font][/sub][font=微软雅黑]——标准溶液中乙醛的含量,μg [/font][font=微软雅黑] V[/font][sub][font=微软雅黑][font=微软雅黑]nd[/font][/font][/sub][font=微软雅黑]——标准状态下干采气体积,L [/font][font=微软雅黑] 按GB16157-1996中10.1或10.2计算V[/font][sub][font=微软雅黑][font=微软雅黑]nd[/font][/font][/sub][font=微软雅黑][font=微软雅黑]。如用峰面积定量[/font][font=微软雅黑],峰高h改为峰面积A。[/font][/font][b][font=微软雅黑]3、乙醛有组织排放的“排放浓度”计算[/font][/b][font=微软雅黑][font=微软雅黑]按[/font][font=微软雅黑]GB16157-1996中11.1.2或11.1.4计算乙醛的“排放速率”。[/font][/font][b][font=微软雅黑]4、乙醛有组织排放的“排放速率(kg/h)”计算[/font][/b][font=微软雅黑][font=微软雅黑]按[/font][font=微软雅黑]GB16157-1996中11.4计算乙醛的“排放速率”。[/font][/font][b][font=微软雅黑]5、乙醛的“无组织排放监控浓度值”计算[/font][/b][font=微软雅黑][font=微软雅黑]按下式计算一个无组织排放监控点的乙醛平均浓度[/font][font=微软雅黑]:[/font][/font][img=,128,86]https://ng1.17img.cn/bbsfiles/images/2022/11/202211070914487249_7872_1623757_3.jpg!w128x86.jpg[/img][font=微软雅黑] [/font][font=微软雅黑][font=微软雅黑]式中[/font][font=微软雅黑]:C——一个无组织排放监控点的乙醛浓度平均值 [/font][/font][font=微软雅黑] C[/font][sub][font=微软雅黑][font=微软雅黑]i[/font][/font][/sub][font=微软雅黑]——一个样品的乙醛浓度 [/font][font=微软雅黑] n——一个无组织排放监控点采集的样品数目。[/font][font=微软雅黑] “无组织排放监控浓度值”的计算按GB16297—1996附录C中C2.3计算乙醛的“无组织排放监控浓度值”。[/font][b][font=微软雅黑]八、精密度和准确度[/font][font=微软雅黑]1、统一样品的精密度和准确度[/font][/b][font=微软雅黑][font=微软雅黑]用[/font][font=微软雅黑]10g/L NaHSO[/font][/font][sub][font=微软雅黑][font=微软雅黑]3[/font][/font][/sub][font=微软雅黑][font=微软雅黑]溶液配制的含乙醛分别为[/font][font=微软雅黑]74.0mg/L.(在采样体积为100L时,相当浓度为3.70mg/m[/font][/font][sup][font=微软雅黑][font=微软雅黑]3[/font][/font][/sup][font=微软雅黑])和370.0mg/L(在采样体积为100L时,相当浓度为18.5mg/m[/font][sup][font=微软雅黑][font=微软雅黑]3[/font][/font][/sup][font=微软雅黑])的统一样品,经五个实验室分析,得到方法的精密度和准确度数据见表1。[/font][img=,417,174]https://ng1.17img.cn/bbsfiles/images/2022/11/202211070914536257_5413_1623757_3.jpg!w417x174.jpg[/img][font=微软雅黑] [/font][b][font=微软雅黑]2、实际样品的精密度和准确度[/font][/b][font=微软雅黑][font=微软雅黑]在同一时间、同一采样点统一采集并发放的无组织排放和有组织排放样品[/font][font=微软雅黑],经家实验室分析,相对标准偏差分别为9.5%和10% 加标收率分别为90.8%~126%(均值为103%)和189.0%~118%(均值为98.2%)。[/font][/font][b][font=微软雅黑]九、说明[/font][/b][font=微软雅黑]①在本方法选定的色谱条件下,样品中的甲醛、甲醇、乙醇、丙酮、甲酸、乙酸等有机化合物对醛的测定均无干扰。[/font][font=微软雅黑]②市售乙醛仅仅是400g/L的水溶液(分析纯),而且还有聚合物存在,故不能作为标准样品直接使用,必须标定其准确含量。[/font][font=微软雅黑]③用羟胺法标定乙醛溶液的浓度时,空滴定可作为在滴定样品时观测指示剂终点颜色的对照标准,为了对照准确,必须使在终点时两者溶液的体积相等。所以在样品滴定接近终点之前,应补充加入一定量的蒸馏水,然后将样品溶液继续滴定至终点。同时,由于蒸馏水的pH值比滴定至终点时溶液的pH值高得多,所以必须另取20ml蒸馏水,加入3滴1g/L溴酚蓝指示剂,再滴定至终点。由此可以计算因加入水而消耗的盐酸量的升数。[/font][font=微软雅黑]④乙醛是一种易燃、易挥发的危险品,在制备新鲜乙醛时,应当防止乙醛外溢,应用水浴加热,不能直接加热,而且应用水浴接收装置,将接收管的通入下水道。[/font][font=微软雅黑]⑤当室温较低时,2.0mol/L的Na[/font][sub][font=微软雅黑][font=微软雅黑]2[/font][/font][/sub][font=微软雅黑]CO[/font][sub][font=微软雅黑][font=微软雅黑]3[/font][/font][/sub][font=微软雅黑][font=微软雅黑]溶液会析出[/font][font=微软雅黑]Na[/font][/font][sub][font=微软雅黑][font=微软雅黑]2[/font][/font][/sub][font=微软雅黑]CO[/font][sub][font=微软雅黑][font=微软雅黑]3[/font][/font][/sub][font=微软雅黑][font=微软雅黑]晶体[/font][font=微软雅黑],所以,在冬天需用温水浴加热伙其溶解后方可使用。[/font][/font][font=微软雅黑]⑥在较高气温下,连续长时间采集环境空气中乙醛时,如吸收液体积有明显减 少,需适当补加吸收液,使吸收液的体积维持在5ml左右。[/font][font=Calibri] [/font]
请教个问题,我用40%乙醛色谱纯,纯水做溶剂检测空气中乙醛含量,用的是岛津GC-2010,ffap色谱柱,40%乙醛没有蒸馏,做出谱图出现两个峰,而且高度差不多,现在不知道怎么会有这种情况,不知道哪个是乙醛的峰。是不是和没蒸馏有关系呢?还是乙醛已经氧化了呢
食品中丙烯酰胺的危险性评估丙烯酰胺(CH2=CH-CONH2)是一种白色晶体物质,分子量为70.08,是1950年以来广泛用于生产化工产品聚丙烯酰胺的前体物质。聚丙烯酰胺主要用于水的净化处理、纸浆的加工及管道的内涂层等。在欧盟,丙烯酰胺年产量约为8-10万吨。2002年4月瑞典国家食品管理局(National Food Administration,NFA)和斯德哥尔摩大学研究人员率先报道,在一些油炸和烧烤的淀粉类食品,如炸薯条、炸土豆片、谷物、面包等中检出丙烯酰胺;之后挪威、英国、瑞士和美国等国家也相继报道了类似结果。由于丙烯酰胺具有潜在的神经毒性、遗传毒性和致癌性,因此食品中丙烯酰胺的污染引起了国际社会和各国政府的高度关注。为此,2002年6月25日世界卫生组织(WHO)和联合国粮农组织(FAO)联合紧急召开了食品中丙烯酰胺污染专家咨询会议,对食品中丙烯酰胺的食用安全性进行了探讨。2005年2月,联合国粮农组织(FAO)和世界卫生组织(WHO)联合食品添加剂专家委员会(JECFA)第64次会议根据近两年来的新资料,对食品中的丙烯酰胺进行了系统的危险性评估。1.人体接触途径人体可通过消化道、呼吸道、皮肤粘膜等多种途径接触丙烯酰胺,饮水是其中的一种重要接触途径,为此WHO将水中丙烯酰胺的含量限定为1μg /L。2002年4月斯德哥尔摩大学研究报道,炸薯条中丙烯酰胺含量较WHO推荐的饮水中允许的最大限量要高出500多倍。因此,认为食物为人类丙烯酰胺的主要来源。此外,人体还可能通过吸烟等途径接触丙烯酰胺。2. 吸收、分布及代谢丙烯酰胺可通过多种途径被人体吸收,其中经消化道吸收最快,在体内各组织广泛分布,包括母乳。经口给予大鼠 0.1 mg/kg bw 的丙烯酰胺,其绝对生物利用率为23-48%。进入人体内的丙烯酰胺约90%被代谢,仅少量以原型经尿液排出。丙烯酰胺进入体内后,在细胞色素P4502E1的作用下,生成活性环氧丙酰胺(glycidamide)。该环氧丙酰胺比丙烯酰胺更容易与DNA上的鸟嘌呤结合形成加合物,导致遗传物质损伤和基因突变;因此,被认为是丙烯酰胺的主要致癌活性代谢产物。研究报道,给予大小鼠丙烯酰胺后,在小鼠肝、肺、睾丸、白细胞、肾和大鼠肝、甲状腺、睾丸、乳腺、骨髓、白细胞和脑等组织中均检出了环氧丙酰胺鸟嘌呤加合物。目前,尚未见人体丙烯酰胺暴露后形成DNA加合物的报道。此外丙烯酰胺和环氧丙酰胺还可与血红蛋白形成加合物,在给予动物丙烯酰胺和摄入含有丙烯酰胺食品的人群体内均检出血红蛋白加合物,建议可用该血红蛋白加合物作为接触性生物标志物来推测人群丙烯酰胺的暴露水平。3 丙烯酰胺毒性3.1急性毒性急性毒性试验结果表明,大鼠、小鼠、豚鼠和兔的丙烯酰胺经口LD50为150-180 mg/kg,属中等毒性物质。3.2 神经毒性和生殖发育毒性 大量的动物试验研究表明丙烯酰胺主要引起神经毒性;此外,为生殖、发育毒性。神经毒性作用主要为周围神经退行性变化和脑中涉及学习、记忆和其他认知功能部位的退行性变;生殖毒性作用表现为雄性大鼠精子数目和活力下降及形态改变和生育能力下降。大鼠90天喂养试验,以神经系统形态改变为终点,最大未观察到有害作用的剂量(NOAEL)为0.2 mg/kg bw/天。大鼠生殖和发育毒性试验的NOAEL为2 mg/kg bw/天。3.3 遗传毒性丙烯酰胺在体内和体外试验均表现有致突变作用,可引起哺乳动物体细胞和生殖细胞的基因突变和染色体异常,如微核形成、姐妹染色单体交换、多倍体、非整倍体和其他有丝分裂异常等,显性致死试验阳性。并证明丙烯酰胺的代谢产物环氧丙酰胺是其主要致突变活性物质。3.4 致癌性动物试验研究发现,丙烯酰胺可致大鼠多种器官肿瘤,包括乳腺、甲状腺、睾丸、肾上腺、中枢神经、口腔、子宫、脑下垂体等。国际癌症研究机构(IARC) 1994年对其致癌性进行了评价,将丙烯酰胺列为2类致癌物(2A)即人类可能致癌物,其主要依据为丙烯酰胺在动物和人体均可代谢转化为其致癌活性代谢产物环氧丙酰胺。3.5 人体资料 对接触丙烯酰胺的职业人群和因事故偶然暴露于丙烯酰胺的人群的流行病学调查,均表明丙烯酰胺具有神经毒性作用,但目前还没有充足的人群流行病学证据表明通过食物摄入丙烯酰胺与人类某种肿瘤的发生有明显相关性。4.食品中丙烯酰胺形成、含量和人体可能暴露量4.1食品中丙烯酰胺形成丙烯酰胺主要在高碳水化合物、低蛋白质的植物性食物加热(120°C 以上)烹调过程中形成。140-180℃为生成的最佳温度,而在食品加工前检测不到丙烯酰胺;在加工温度较低,如用水煮时,丙烯酰胺的水平相当低。水含量也是影响其形成的重要因素,特别是烘烤、油炸食品最后阶段水分减少、表面温度升高后,其丙烯酰胺形成量更高;但咖啡除外,在焙烤后期反而下降。丙烯酰胺的主要前体物为游离天门冬氨酸(土豆和谷类中的代表性氨基酸)与还原糖,二者发生Maillard反应生成丙烯酰胺。食品中形成的丙烯酰胺比较稳定;但咖啡除外,随着储存时间延长,丙烯酰胺含量会降低。4.2食品中丙烯酰胺含量既然丙烯酰胺的形成与加工烹调方式、温度、时间、水分等有关,因此不同食品加工方式和条件不同,其形成丙烯酰胺的量有很大不同,即使不同批次生产出的相同食品,其丙烯酰胺含量也有很大差异。在JECFA 64次会议上,从24个国家获得的2002-2004年间食品中丙烯酰胺的检测数据共6,752个,其中67.6%的数据来源于欧洲,21.9%来源于南美,8.9%的数据来源于亚洲,1.6%的数据来源于太平洋。检测的数据包含早餐谷物、土豆制品、咖啡及其类似制品、奶类、糖和蜂蜜制品、蔬菜和饮料等主要消费食品,其中含量较高的三类食品是:高温加工的土豆制品(包括薯片、薯条等),平均含量为0.477 mg/kg,最高含量为5.312 mg/kg;咖啡及其类似制品,平均含量为0.509 mg/kg,最高含量为7.3 mg/kg;早餐谷物类食品,平均含量为0.313 mg/kg,最高含量为7.834 mg/kg;其它种类食品的丙烯酰胺含量基本在0.1 mg/kg以下,结果见表1。由中国疾病预防控制中心营养与食品安全研究所提供的资料显示,在监测的100余份样品中,丙烯酰胺含量为:薯类油炸食品,平均含量为0.78 mg/kg,最高含量为3.21 mg/kg;谷物类油炸食品平均含量为0.15 mg/kg,最高含量为0.66 mg/kg;谷物类烘烤食品平均含量为0.13 mg/kg,最高含量为0.59 mg/kg;其它食品,如速溶咖啡为0.36 mg/kg、大麦茶为0.51 mg/kg、玉米茶为0.27 mg/kg。就这些少数样品的结果来看,我国的食品中的丙烯酰胺含量与其他国家的相近。
[table=100%][tr][td]癸酰乙醛的极性?当进行气—质谱是的溶剂是什么?在进行质谱之前要做什么准备?[/td][/tr][/table]







 我要推广仪器
我要推广仪器
 下载APP
下载APP