[font=宋体][size=3]百灵威整合国际资源为烟草行业提供品类齐全的烟草成分分析标准品。产品涵盖烟草分析所用到的各类代谢物、衍生物、农药及香精香料标准品。可以满足烟草行业各类分析实验需求。[/size][/font]
烟草化学成分分析标准品
本法用甲醇-水为流动相,在C18反相柱上建立了陆英中具有乌索酸含量测定的高效液相色谱法,以95%乙醇为溶剂处理样品具有提取率高、色谱干扰小、物质分离效果好的优点,本法简便易行、快速准确,其最小检出限为0.2ug。不同浓度水平检测结果日间、日内相对误差小于4.0%。关键词:陆英草 乌索酸 高效液相色谱 含量测定Analysis of Ursolic Acid in Herba Sambuci Chinensis using HplcThe paper reported the determination of Ursolic Acid inHerba Sambuci Chinensis using Hplc,the separation was achieved by applying an Waters -ODS 150×4.6 mm (5um) column and methanol-water (90:10) as mobile phase at flow rate of 0.8 ml/min. the UV detector was set at 210nm External standard method was applied .The linear range was 150-2000ug/ml with the lower limit of detection of 0.2ug.It was found to be effielent and low interference to extract the samples with ether.陆英(Herba Sambuci Chinensis )系为忍冬科接骨木属植物,生于山坡、路旁、溪边、荒野灌丛中。产于长江以南地区。 据报道[1]陆英草含氯原酸、α-香树脂素棕榈酸酯(α-amyin palmitate)、乌索酸、β-谷甾醇、豆甾醇、油菜甾醇、硝酸钾、黄酮、鞣质等。目前其它中药材乌索酸含量的测定多采用比色法及薄层扫描[2]以及衍生化法[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]检测[3],其操作繁琐,显色及前处理误差大,费事费力。本文采用高效液相色谱法用甲醇-水为流动相,检测波长210nm,在C18反相色谱柱上建立了其含量的测定方法,具有简便可行、准确、快速、物质分离效果好的优点,重现性好,适应范围广,利于对陆英草的准确、深入研究。实验部分一、 仪器设备及试剂Waters 600E Hplc系统,UV-2487 可变波长检测器 ,20ul定量环,M32数据处理工作站。乌索酸对照品:中国医学科学院药物研究所 95%乙醇(AR) 甲醇(AR) 水二、实验方法1、色谱条件 色谱柱:Waters -ODS 150×4.6 mm (5um);流动相:甲醇-水 (90:10); 流速:0.8 ml/minUV波长:210nm ;量程:0.005 AUFS ;温度:25℃数据处理:峰面积外标定量。2、样品处理 (1)将原料(樟树医药公司)的叶粉碎过筛并恒重。(2)称取样品10克,置索氏提取器中,加95%乙醇回流提取8小时,提取液浓缩定容于100毫升的容量瓶中,取上清液约5毫升过针式过滤器过滤,取滤液待测。3、标准曲线的确定分别吸取配好的5.0mg/ml乌索酸标准液0.4、0.8、1.6、2.8、3.6ml置于10ml容量瓶中,用甲醇稀释成 0.2、0.4、0.8、1.4、1.8mg/ml系列标准溶液,在选定的色谱条件下,重复进3次每次20ul,以标准品峰面积与浓度关系得出回归方程:Y=3.35 E+005X+8.74E+003 R=0.99994、样品测定 将预处理好的待测样品液进样20 ul 检测,通过软件操作的得出浓度值。三、结果1、乌索酸对照品与陆英样品的色谱图1 图1 图22、方法重现性 用两个浓度水平进行连续和间隔时间及连续3天测定,考察方法重现性。结果:其最大相对标准偏差小于4.0%。2、加标回收率 吸取5.0mg/ml乌索酸标准液 0.8、1.6、3.6各三份,分别加入原料样品10克,按前法操作,测定结果,计算回收率结果为99.9%-100.8%,平均100.4%,RSD为1.0%。讨论1、迷迭香中的乌索酸是非极性五环三萜类酸,在C18反相柱上有较大的保留值,以甲醇和水二元体系作为流动相,甲醇浓度与极性相近共存物质的分离及峰形有显著影响,本法选定比例是考虑实际样品组分分离确定的,对于其它品种样品可作适当的调整。2、检测波长 我们进行了波长扫描,乌索酸在205nm处有最大吸收峰,本法采用210nm,恒流比流动相洗脱对检测无影响。3、本法检测出陆英草中平均含0.28%的乌索酸。4、本法简便、易行,准确快速。
[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]分析脂肪酸甲酯标准品,怎样处理标准品?用什么溶剂稀释?
食生字06号关于召开“特医食品、保健食品配方研发及生产工艺质量标准实操技术与国内外典型案例分析高级研讨班”通知各有关单位:特医食品和保健食品是我国大健康产业的重要组成部分,新《食品安全法》的颁布实施及相关配套法规《保健食品注册与备案管理办法》、《特殊医学用途配方食品注册管理办法》的出台,对潜力巨大的保健食品、特医食品市场将实现规范与引导,整个行业将得到进一步肃清整顿。有利于帮助消费者科学选择、理性消费保健食品,保证保健产业安全发展。为帮助食品健康营养学、研、产各方面的深入对接合作,提高研发水平,掌握生产工艺质量标准关键控制要点,保证产品在实际生产中的质量控制,明确产品的申报注册、技术审评的操作要点,解决申报过程中遇到的核心问题,顺利通过评审,我单位将于2016年5月20日-22日在郑州市举办“特医食品、保健食品配方研发及生产工艺质量标准实操技术与国内外典型案例分析高级研讨班”届时将邀请经验丰富的特医食品、保健食品审评和研发专家到会,就配方,功能,毒理,生产工艺,卫生学,企标,注册备案等关键问题并进行专题交流研讨,为参会者提供最专业权威的指导,专业有效的咨询服务。请各有关单位积极派员参加,现将有关事项通知如下:一、组织机构: 指导单位:国家食品药品监督管理局主办单位:国家食品行业生产力促进中心 保健食品技术咨询与申报指导服务平台协办单位:待定支持单位:中国营养学会医用食品与营养支持分会 全国医药技术市场协会 中国老年保健医学研究会 北京市营养源研究所 北京联合大学保健食品功能检测中心支持媒体:中国食品报 中外食品 食品科技杂志社 《中外食品工业》杂志社 《食品工业》二、时间、地点: 会议时间:2016年5月20日-22日(20日全天报到)会议地点:郑州市(具体地点会前一周通知报名单位)三、拟邀出席主讲嘉宾:(排名不分先后)嘉宾:国家食品药品监督管理局相关领导专家嘉宾:中国疾病预防控制中心营养与健康所主任 张 坚嘉宾:国家食品安全风险评估中心 徐海滨嘉宾:北京联合大学保健食品功能检测中心主任教授 金宗濂嘉宾:美国外科学院院士(FACS)、中国科学院北京转化医学研究院/ 航空总医院肿瘤医学中心主任、北京康爱营养医学研究院院长、 中国抗癌协会肿瘤营养与支持治疗专委会主任委员 石汉平嘉宾:西安交通大学教授、国家食药总局保健食品审评专家 潘建平嘉宾:北京保健协会副会长、北京中医药大学教授 王林元嘉宾:北京中医药大学中药学院教授/中药新药研究中心主任 国家食药总局保健食品审评专家 张宏桂嘉宾:江西中医药研究院药研所所长、食药总局特医食品审评专家、 澳洲西悉尼大学MINC实验中心教授 熊学敏嘉宾:中国医师协会营养医师专业委员会专家,河南临床营养学会副主任委员、 特医食品临床研究资深专家、郑州大学第一附属医院营养科主任 陈改云四、主要研讨内容:(详见日程安排表)五、参会对象:1、从事食品加工、营养、微生物、植物与医学等研究的科研院(所)、大专院校相关专家学者、食品、卫生、质量监督管理、检测、进出口等机构;2、保健食品、医用食品企业总经理、研发、注册申报、市场、项目、质量管理等部门负责人。申报代理机构相关人员;3、全国医疗机构营养科及养老机构等临床科研人员。六、会议费用:1、会务费:现场交费2200元/人,提前汇款2000元/人;在校学生持学生证1200元/人;会务费包括:场地、研讨、资料及论文集。食宿统一安排,费用自理。2、本次会议为企业及科研机构设立20个产品展示区,每个展位费用为3000元,规格长1.2米款0.8米桌子,两把椅子。诚邀赞助单位、协办单位及新产品、新设备展示单位。有意者请与会务组联系。七、报考条件: 1、参加证书考核者报到时需交三张一寸免冠彩色(蓝底)标准证件照片、身份证复印件(双面复印)一张、学历证明复印件一张。2、凡从事保健食品研发及注册体系人员,均可参加保健食品中高级项目管理工程师的培训及考试(团体培训请致电组委会)。八、证 书: 参加培训的代表,如需要证书者,经考评合格后,颁发由人力资源和社会保障部中国职工教育和职业培训协会颁发的《保健食品项目管理工程师》专业能力证书,证书费用中级1000元,高级1300元(含资料费、考核评审费),全国通用,联网查询,是相关人员上岗、考核和能力评价的重要依据。可在人力资源和社会保障部中国职工教育和职业培训协会官方网站www.zhongguozhixie.com.cn进行查询。(需要申报的代表请提前联系组委会办公室)九、组委会联系方式:联 系 人: 马超 主任 手 机:13240487419传 真:010-51606934 电子邮箱:1683101345@qq.com5月21日一、政策法规解读:1、《特殊医学用途配方食品注册管理办法》(CFDA正式版)解读;2、特殊医学用途配方食品通则及良好生产规范标准;3、《保健食品注册与备案管理办法》(CFDA正式版)解读;4、注册、备案双轨制的不同适用范围与实操要求;5、保健食品功能目录与原料目录解析;6、我国健康产业十三五规划解读;5月21日 星期五 下 午二、特医食品专场1、特医食品的研发报告的编写;2、特医食品生产工艺研究及质量标准;3、特医食品配方设计原则和研发思路与应用群体研究;4、特定全营养配方食品临床试验的开展要点与临床周期要求;5、特医食品现场核查与技术评审原则;6、特医食品、婴幼儿乳品注册要点难点解析;7、特医食品注册、变更延续应提交材料;8、特医食品质量安全管理与安全性评价;9、新《办法》下特医食品标签、说明书的编制要求;10、特医食品配方工艺国际典型案例分析;5月22日 星期六 上 午三、保健食品专场1、保健食品生产工艺研究及存在问题分析;2、保健食品配方设计原则;3、保健食品注册、备案、变更延续注册、变更延续应提交材料;4、保健食品批文转让与名称变更;5、保健食品换证与审批;6、目录外原料的保健食品注册要点;7、保健食品质量安全管理与安全性评价;8、新《办法》下保健食品标签、说明书的编制要求;9、保健食品配方工艺及质量标准国内外典型案例分析。5月22日 下 午人保部中职协《保健食品项目管理工程师》证书考试备 注1、每个主题报告后均有10分钟左右的答疑时间,请提前准备;2、报告日程如有变动,以报到时的会议日程为准。附件二: 报 名 回 执 表发票事宜发票单位名称:发票项目: □培训费 □会务费是否需要保健食品项目管理工程师证书: □是 □否 , □高级 □中级问题提问1、2、3、签名/盖章:日 期:1、请您准确填写上表各项信息,以便我会制作代表证等相关培训资料。2、请您在回传此确认表后3个工作日内办理付款,汇款注明:郑州保健食品注册费。3、请您付款后把汇款底单回传至010-81312425,款到后我们会给您邮寄正式发票。4、我们在培训前一周左右给您发第二轮报到通知。联 系 人: 马超 主任 手 机:13240487419电话/传真:010-51606934 电子邮箱:1683101345@qq.com填表日期: 年 月 日
[font=黑体]根据十一届全国人大常委会第二次委员长会议的决定,全国人大常委会办公厅昨日向社会全文公布食品安全法草案,广泛征求各方面意见和建议。草案明确提出,要建立畅通、便利的消费者权益救济渠道,对消费者的赔偿将提高到10倍。同时,加大处罚力度,对违法案件的处罚,除了援引《刑法》,并可能处以最高多达货值金额20倍的罚款。(4月21日《重庆时报》)[/font] 这是新一届全国人大常委会向社会全文公布、广泛征求意见的第一部法律草案。读完该《草案》全文,我们可以清楚地发现:它比其前身——《食品卫生法》在处罚力度方面明显增强,除了高额的赔偿金、处罚金手段,对《刑法》二百二十五条、一百四十三条、一百四十四条等条例的援引,也不可不谓果决。我们有理由期望,它的面世会给我国食品安全工作带来诸多裨益。但是,笔者也发现了一个问题——或许只是草案的原因——虽然第一章《总则》、第二章《食品安全风险监测和评估》中多次提及,第三章的章名也就叫做《食品安全标准》,可是,究竟“食品安全标准”是什么,除了看到一些不具备操作意义的概念,其具体形态我们依然不得而知——很显然,就该《草案》的整体结构而言,如果《食品安全标准》部门的内容不具备明晰性、确定性的话,那么,接下来的《食品检验》、《食品生产经营》,以及我们普遍叫好的《法律责任》也将随之变得模棱两可、不具备执行效力,而整个《草案》难免有沦为一纸空谈的可能。 其实,“食品安全标准制”不标准,一直是国内相关法律的技术性软肋,也是诸多问题的根源。一方面它未与国际标准接轨致使我们常常被动,比如食品是否含有“苏丹红”,欧盟标准早就有了明确规定,我们的标准却只能“先出事后发布”,标准的预警功能严重缺失。另一方面是标准太多、太乱。有国家标准、部门标准、地方标准、行业标准、企业标准等,使得食品安全标准不协调、不统一,以至多种标准在市场上形成冲突,让生产企业茫然无措。而一些不法企业趁机钻食品标准缺陷空子,让假冒伪劣商品充斥市场,坑害消费者,消费者维权也显得格外无助。
GB/T601-2002草酸标准滴定溶液的不确定度评定,实验中用到高锰酸钾标准溶液----------关于高锰酸钾标准溶液不确定度的是怎样来进行分析的,因为高锰酸钾标准溶液也是配制的(非购买的标准物质),难道由此前配制高锰酸钾标准溶液得出的不确定度直接代入吗?
标定盐酸标准滴定溶液的不确定度分析 作者:吴文英 张春雨 唐惠兰 来源:中华医学研究杂志 在理化分析过程中,一切测量结果都不可避免地具有不确定度。盐酸标准溶液是常用化学定量参比物质,其标定值的准确性直接影响常规分析质量。笔者以GB/T601《滴定分析(容量分析)用标准液的制备》为依据配制并标定盐酸根据JJF1059-1999《测定不确定度评定与表示》分析其测量不确定度。简述由标定过程中得到的不确定度。 1 实验部分 1.1 测定方法[1,2] 准确称量270℃~300℃干燥至恒重的基准碳酸钠(99.95%~100.05%)约0.2g左右,电子分析天平(精度为0.1mg),置于三角瓶中,加入50ml水使之溶解,加指示剂,用盐酸标准液滴定至终点同时作试剂空白实验。 1.2 主要计量仪器与试剂 电了分析天平:AG204;酸式滴定管:50ml A级。 1.3 建立数学模型 C=m (V1-V2)×0.05300 式中 C:盐酸标准滴定溶液的浓度(mol/L);m:基准无水碳酸钠的质量(g);V1:盐酸标准滴定溶液用量(ml);V2:试剂空白实验中盐酸标准滴定溶液用量(ml);0.05300:与1.00ml盐酸标准溶液[C(HCl)=1.000mol/L]相当于以克表示的无水碳酸钠的质量。 1.4 盐酸标准滴定溶液的标定结果 为获得标准溶液重复测量的不确定度分量,对同一标准溶液进行8次独立的标定。测定数据见表1。 表1 盐酸标准滴定溶液的标定结果 略 2 测量不确定度来源 从检测过程和数学模型分析,标定盐酸标准溶液的不确定度主要来源,由四个方面所引起。(1)测量的重复性(A类不确定度);(2)基准无水碳酸钠的纯度;(3)测量使用的电子分析天平及量具;(4)其他相关常数。 3 测量不确定度分析 3.1 A类不确定度的分析 利用表1中的测量结果,按照A类评定测量重复性的标准不确定度。具体计算过程:重复测量的平均值计算式:=1 n∑8 i=1xi=0.09951mol/L 单次测量的标准差按贝塞尔公式计算s(x)为 s(x)=∑8 i=1(xi-)2 n-1=0.0001555mol/L 的标准差s()为 s()=s(x) n=0.000155 8=0.0000548mol/L=5.48×10-5mol/L 由测量重复性引起的相对标准不确定度为U(x):0.0000548/0.09951=0.055%。 3.2 B类不确定度分析 3.2.1 基准碳酸钠的纯度 基准碳酸钠的纯度为1.0000±0.0005,视为矩形分布0.00053=0.00029,则标准不确定度为:由基准碳酸钠的纯度引入的相对不确定度u(p)为:0.029%。 3.2.2 天平称量所引入的标准不确定度 干燥器与天平称量仓内均放置同质硅胶,视为相同湿度,称量时无吸潮。电子天平检定证书标出线性为上0.2mg;可视为矩形分布,则标准不确定度为:因为称量采用的是减量法,故称量的标准不确定度为0.2mg /3=0.12mg:因为称量采用的是减量法,故称量的标准不确定度为:2×0.122=0.17mg,则由称量引入的相对标准不确定度u(m)为:0.17mg/0.2018g=0.084%。 3.2.3 标定体积的不确定度 (1)滴定管的校准:滴定使用50ml酸式滴定管(A级),按照检定规程,其最大允许误差为±0.05ml,相对允许误差为±0.1%,按照矩形分布,则滴定体积的相对标准不确定度u(V)为:u(V)=0.1%/3=0.0577%。(2)环境温度:实验环境在空调条件下,室温近似20℃。温度在20℃左右,标准溶液的温度补正值非常小,对实验结果影响可忽略不计,所以在不确定度分析中不把一温度影响引起的不确定度列入考虑范围。(3)滴定终点的判断:终点时的误差±0.05ml(1滴的体积),两点分布,现由终点分布判断引入的标准不确定度为0.05ml:相对标准不确定度为0.05ml/38.32ml=0.13%标定体积的影响引入相对标准不确定度U(V)为0.0572+0.132=0.142%。 3.2.4 其他常数 基准无水碳酸钠摩尔质量引起的标准不确定度很小,可以忽略。 4 合成标准不确定度 测量重复性、基准无水碳酸钠的纯度、天平称量、标定体积等的不确定度相互独立,故将上述数据合成得盐酸的相对合成标准不确定度U(C)为0.0552+0.0292+0.0842+0.1422=0.176%。 5 扩展不确定度 实验测得盐酸标准溶液浓度为0.09951mol/L,则测量结果的合成标准不确定度U(C)=0.09951mol/L×0.176%=0.000175mol/L。若取包含因子K=2,得测量结果的扩展不确定度U=2U(C)=0.00035mol/L。 6 测量结果的表示 盐酸标准滴定溶液的浓度可表示为:(0.09951±0.00035mol/L,K=2)。 【参考文献】 1 姚正堂,将已峰.奶制品中蛋白质测定的不确定度分析.中华医学研究杂志,2005,5(6):6. 2 国家技术监督局.JJF1059-1999测量不确定度与表示.北京:中国计量出版社,1997,81. 作者单位: 214171 江苏无锡,无锡市惠山区疾病预防控制中心
[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]分析鱼腥草要选?柱子请赐教谢谢!最好提供标准方法?
年后手头上要做新的项目,现来求助给各位达人,分析烟草及烟草制品重金属分析的仪器设备及样品前处理设备(要满足烟草行业检测标准),是否有做过类似的,希望多多指教。谢谢!
对 草酸分析求救贴:http://www.instrument.com.cn/bbs/shtml/20080509/1256137/的回答: 草酸是一种常见的有机酸,分析方法很多,不同的样品和含量有对应不同的检测方法。在色谱法普及前,用酸碱滴定,氧化还原法,以及比色法都是有的,但这些一般适合常量分析,如果有干扰的,分析是很不准确的。从现在分析角度来看,色谱法是最佳的分析方法,分析简单,快速,但不同是色谱方法之间有很大的差异。现在色谱方法很多,都可以分析草酸,但要区别来看。簿层色谱法和纸色谱法是可以分析草酸的,但目前只能用于特定的范围,定量也不准确,操作相对繁琐。 [url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法直接分析草酸有点勉强,因为有二个羧基,沸点高。但可以用衍生的方法来分析草酸,这样的文献也不少,操作也是繁琐一些,也不是最佳的方法。 液相色谱法分析草酸是比较好的手段,根据不同的分离机理,有不同的类型:1反相色谱法:在酸抑制条件下,采用C18柱分析草酸这样的有机酸是有成功的报道,如果用纯水柱则效果更好,波长在210nm,其灵敏度因其有二个羧基响应是很大的,远高于其它有机酸。2离子交换法:采用液相的SAX柱,也可以分析草酸,不过淋洗盐的浓度要比较低,另外在SAX中出峰还是比较快的,复杂样品不是很适合。3离子对色谱法:从理论上讲采用离子对试剂是可以分析草酸的,但由于一般的离子对试剂的纯度问题,低紫外的吸收很高,而且试剂浓度不低,这样基线噪音会很大,对复杂样品不是很理想,而且TBA效果不好,应采用更高碳数的试剂,成本较高,灵敏度低。4离子排斥法:这种方法比较经典,采用专用的有机酸柱(就是离子排斥柱)有很多的文献报道,只是有这种柱子的人很少,我曾试分析过草酸,一般分析时间比C18柱长多了,草酸的保留时间长,但柱效不如C18的柱子,这种柱子用于分析混合有机酸是比较常见的。因为其流动相是很低浓度的稀硫酸,水不溶的样品不适合这种柱子。[url=https://insevent.instrument.com.cn/t/3p][color=#3333ff]离子色谱[/color][/url]法:分析草酸比较方便的方法是用[url=https://insevent.instrument.com.cn/t/3p][color=#3333ff]离子色谱[/color][/url]法,灵敏度高,选择性好。[url=https://insevent.instrument.com.cn/t/3p][color=#3333ff]离子色谱[/color][/url]有碳酸盐体系和OH体系二种柱子,对于复杂化合物用OH体系柱子比较好,可以进行梯度分析,但要注意如果硫酸根离子浓度很高的话,对草酸测定会带来很大干扰。戴安另外还有一种离子排斥的抑制色谱法可测定有机酸。毛细管电泳,也有用于分析有机酸成功的报道,分析速度快,灵敏度高,但目前实用性不强,准确性欠佳。综上所述,草酸可用[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]的衍生法、液相的反相酸抑制法及离子排斥法以及抑制电导[url=https://insevent.instrument.com.cn/t/3p][color=#3333ff]离子色谱[/color][/url]法测定相对比较合适和可靠。
朋友们,帮帮我啊,草酸铜用碘量法做,不知是什么老是干扰终点,兰色老是回头,该选什么样的方法做呢?有没有这方面的分析标准,我怎么找也找不到
最近做小儿七星茶颗粒甘草酸的测定时遇到了很奇怪的问题:对照品有峰,样品却只出杂质峰,开始怀疑是样品没有含量,可是拿以前做过的有含量的样品再做,却没峰了;后来吸一半对照品一半样品进样就出峰了,可是加对照品到样品中一起按标准处理后就又没有峰出来,怎么想都不明白问题出在哪里。所用的试剂换了好几次,也换人配了,结果还是一样没有,会不会是超声引起的呢,因为我们的超声机的功率只有80瓦,不过以前也做得出啊,大家帮帮忙,下面是标准【含量测定】 照高效液相色谱法(中国药典2005年版一部附录VI D)测定。 色谱条件与系统适用性试验 以十八烷基硅烷键合硅胶为填充剂;以甲醇-0.2mol/L醋酸铵溶液—冰醋酸(65:35:1)为流动相;检测波长为250nm。理论板数按甘草酸峰计算应不低于2000。 对照品溶液的制备 取甘草酸铵对照品适量,精密称定,加流动相制成每1ml含16μg的溶液,即得(折合甘草酸为15.672μg)。 供试品溶液的制备 取装量差异项下的本品内容物,混匀,研细,取约7g,精密称定,置50ml量瓶中,加流动相约45ml,超声处理(功率300W,频率40kHz)30分钟,放冷,加流动相至刻度,摇匀,滤过,取续滤液,即得。 测定法 精密吸取对照品溶液与供试品溶液各20μl,注入液相色谱仪,测定,即得。
根据国际日用香料协会(IFRA)的信息通讯(IL 947),今年九月初,欧洲标准化委员会发布了关于“致敏剂分析方法——消费品中致敏性香料的定量分析—气相色谱法”(CEN Standard 16274:2012)的新标准。该标准是在IFRA的草案基础上制定的,日用香料行业组织和资助了此项标准的制定工作。欧洲的相关法规和指令规定,当26种致敏性香料的用量超过指定量时,化妆品、洗涤剂和家用产品等应在标签上对这些香料进行标注。IFRA推荐使用该标准,但是由于版权问题,不能够提供标准文本。标准采用气相色谱—质谱联机(GC-MS),利用两个极性不同的色谱柱结合特定定量和数据处理方法进行分析,该方法适用于对化妆品原料或产品中24种日用香料的分离和定量分析。由于橡苔提取物和树苔提取物属于天然复合物,成分组成复杂,很难通过现有的方法进行定量分析,因此标准未涵盖这两个日用香料。最迟到2013年3月,此标准应被转化为(欧洲各国的)国家标准,不符合此标准规定的国家标准应同时废止。实施这一标准的国家包括:奥地利、比利时、保加利亚、克罗地亚、塞浦路斯、捷克、丹麦、爱沙尼亚、芬兰、马其顿、法国、德国、希腊、匈牙利、冰岛、爱尔兰、意大利、拉脱维亚、立陶宛、卢森堡、马耳他、荷兰、挪威、波兰、葡萄牙、罗马尼亚、斯洛伐克、斯洛文尼亚、西班牙、瑞典、瑞士、土耳其和英国。
食品分析中标准物的管理及其标准溶液的校正一、意义食品分析标准物质是分析方法质控的核心,是定性和定量的依据。标准物质以一定纯度和浓度配制的溶液称标准溶液,其稳定性受其自身降解、化学转化、溶质和溶剂挥发等内在因素的影响,又受其存放条件如温度、湿度、光线照射、存放时间、存放容器及其配制技巧等外界条件的影响。由于影响标准物及其标准液的因素较多,如果条件控制不当或管理不严密,标准物质浓度易发生变化,这是食品分析中难以进行质量控制的主要原因。以上原因也是实验室食品分析测定产生误差的最主要因素,要减小这些产生误差的主要因素,就要特别注重标准物管理,以及进行标准溶液的稳定性观察和校正工作,也是实验室质量控制关键工作之一。当标准溶液发生变化时就要重新配制,并找出变化原因,为分析工作积累经验,并可写入方法注释中。在食品分析质量控制中,准确度和精密度的提高,是以标准溶液稳定性和准确性为前提的,因此对于标准溶液稳定性和准确性的关键技术问题,是质量控制的核心问题。二、标准物质的管理 1、容量分析的基准物是标定其它标准溶液的基准,应购买基准试剂,它可以保证其纯度。另一个因素是水份的影响,用前应充分干燥和恒重,配制时量器要校正。溶剂要纯化,使用的容器要充分洗涤。最终目的都是防止基准物的化合损失。2、用于分光光度分析的标准物,分有机的和无机的标准物,无机标准稳定性好,但使用液浓度低时,极易被容器吸附,并与容器中离子进行交换,因此决定了其稀溶液使用时间短。玻璃容器在碱性介质中易溶出,塑料容器在成型时加入助剂时也含有不同金属杂质,容易溶出如Zn、Ca等金属离子。有机标准最好不放在塑料制容器中,因塑料在成型时加入的有成份比较复杂的助剂和增塑剂。标准使用液应现用现配为最佳。三、标准物存放使用1、无机的标准物要求在干燥并无化学干扰物的条件下存放,选择合适干燥剂如硅胶和分子筛。有机标准物最好分装封入安培瓶中低温避光保存。固体的多环芳烃可配制成溶液,再分装在安瓿瓶中保留溶剂封存,也可把溶剂挥掉后干燥封存,使用时再定容,后一种方法更为稳妥些。配制好一批标准溶液,再一支一支使用也是很方便的。如果液体的标准物特别是几种标准的混合物用于色谱分析同系物如醇类物质,可同时配制一批分装安瓿瓶低温存放,再一支一支使用,能避免溶剂挥发体积变化产生的误差。这种做法更适于实验室间的标准分发和校正工作。最难办的是气体,标准如氯乙烯、氟里昂,最好是钢瓶中存放,或配制钢瓶标准气。这些条件不具备时也可以选择高沸点溶剂,密封溶解这些气体,称量溶质重量,一次性使用。从这一事实出发,气体,测定误差可以稍加放宽,因为标准自身稳定性差。2、用于色谱分析的标准物要求色谱纯,其配剂溶液剂也要求色谱纯,准确配制前要在色谱上进行检查。特别是几种标准进行混合更应慎重,每种要严格检查否则给定性定量带来很多麻烦。如果纯度不够时可以纯化,再结晶或用制备色谱制备。勉强使用是无益的。四、标准溶液的校正1、从安瓿瓶分装标准溶液无论是有机的或是无机的用于校正是很方便的。如原子吸收测定金属,从安瓿瓶中取一定量配制浓度系列,再封存。每隔一段时间(1~2周)再用原溶液配制同样浓度系列,严格控制仪器条件来比较二次标准曲线的斜率,斜率下降时表明有损失。2、相同浓度同时配制的标准的几支安瓿瓶,先用打开的一支标准的测定值与间隔一段时间后打开的另一支标准的测定值进行比较,以此类推最后在一段时间内几支同时测定,其变异程度就是标准在这段时间稳定性变化程度。3、几个实验室用同一标准物分别配制相同浓度标准液,各自进行标准曲线的测定,再按规定交换该标准液再进行测定,比较测定结果差异来观察同一标准的时间和空间变异。如果标准液稳定,配制不准确的实验室很容易查出。配制都准确时,标准液若不稳定时,会使各实验室的测得值都偏低。4、同种标准物来源不同,也应采用分别配制交叉测定的办法来检查标准的纯度及配制是否准确。在食品分析中无论用何种手段分析样品,所使用的标准物应作统一的或确切的规定。例如:过硫酸铵测锰,用MnSO4·H2O作为标准使用,到底硫酸锰需要不需要烘烤呢?对于这个问题,在一部份的教科书中有规定烘烤的,也有不烘烤的。按照MnSO4·H2O的性质遇到空气可能吸潮或风化,如果直接称重计算Mn量,就有可能出现误差。用烘烤称重测得水分所含的量比理论值高1.6%,有同一硫酸锰配制锰标准溶液测一合成水样,使用烘烤后配制的锰标准溶液,测得的Mn含量为0.205mg/L,未烘烤过的则高达约2.3%,从中说明硫酸锰在配制标准溶液时应经过烘烤,使标准一致。
我们实验室是检测饲料和原料含量的,以前别人介绍用过中国农业科技院分析检测中心研制的氨基酸分析用校核标准品(参比物),名字我忘记了,只记得有代号,1#、2#、3#、4#、5# 这5种。有知道的老师们请帮帮忙!
求草酸钴产品标准
做定量分析时,为了保证称量的准确性,配制的标准品母液浓度较高。而分析浓度较低,能连续稀释两次或三次吗?
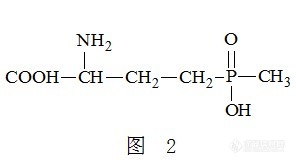
关于”新版GB 2763 食品安全国家标准 规定茶叶中限量农残草甘膦和草铵膦项目“的检测研究 一、研究意义及现状 随着新版GB 2763 食品安全国家标准的不断更新及发布实施,草甘膦和草铵膦已被明确列为茶叶中农药残留强检(必检)项目,草甘膦在茶叶中的限量为1mg/kg,草铵膦在茶叶中的限量为0.5mg/kg。同时,草甘膦和草铵膦也成为中国茶叶出口国外的检测项目(来源于中华人民共和国商务部),且已成为越来越严的限量指标。 文献(2013年农药行业预测和草甘膦市场机遇分析,杨益军,农药市场信息,2013.03)报道,除草剂草甘膦因其高效、广谱、低毒等特性使其被广泛应用,未来需求量也将大幅增加。但草甘膦的使用容易使植物产生抗性(IARC国际研究机构发布报告称草甘膦很可能对人类致癌),而草铵膦可克服该缺陷,现已有学者(草铵膦、百草枯、草甘膦对非耕地杂草的防效比较,凌进,农药,2014年第53卷第8期,613-615)对草铵膦和草甘膦的除草性能进行了研究,确证了草铵膦代替草甘膦的可行性。 因草甘膦和草铵膦为广谱除草剂,被广泛应用于农业、林业及园艺的栽培。我国作为农业大国,其茶叶产量世界第一、出口量世界第二,草甘膦和草铵膦的生产和使用量都位居世界前列(草甘膦 草铵膦及其代谢产物的检测方法,李小娟、周信康、孟品佳,公共安全中的化学问题研究进展)。同时,我单位对西南茶叶原料主产区进行了初步调研,进一步确认茶农使用草甘膦和草铵膦农药的现状。 随着草甘膦和草铵膦除草剂使用量的日益增大,使其常被发现存在于环境水样、土壤及植物中,这样长期积累会引起环境污染,从而对人类健康造成严重威胁。草甘膦和草铵膦结构类似,且均含有膦酸基、羟基、氨基,是极强的两性化合物,易溶于水,难挥发。鉴于草甘膦和草铵膦特殊的物化性质和茶叶基质自身的复杂性,无论国内外,茶叶中草甘膦和草铵膦同时检测的标准还未见发布。 目前,可用于检测草甘膦和草铵膦农药残留量的主要方法有液相色谱法,柱前衍生后气相色谱法、气相色谱-质谱法及液相色谱-质谱/质谱法。 快速发展起来的超高效液相色谱-质谱联用技术,具有检测灵敏度高、适用范围广、分析速度快和能有效排除复杂基质产生的干扰等优点,当今已成为检测型实验室检测农残的首选。然而,若采用液质质直接测定草甘膦和草铵膦,则仪器响应较低,无法满足茶叶中草甘膦和草铵膦农药残留量检测的要求。 近两年来,已有研究文献陆续发表,用柱前衍生-液相色谱串联质谱法检测。笔者结合其文献研究结果,对茶叶中草甘膦和草铵膦农药残留量的检测方法系统地进行研究,采用9-芴甲氧羰酰氯(FMOC-Cl)作为常用衍生剂,在硼酸盐缓冲盐溶液的条件下,能与草甘膦和草铵膦的提取液发生衍生反应,形成衍生产物,衍生产物注入UPLC进行色谱洗脱分离,采用串联质谱探测响应信号,外标法直接快速定量茶叶中的草甘膦和草铵膦的含量。二、液质质检测分析原理 质谱原理是先将物质离子化,按离子的质荷比分离,然后测量各种离子谱峰的强度而实现分析目的的一种分析方法。液质联用是将色谱的分离能力与质谱强大的定性功能结合起来,实现对复杂混合物更准确的定量和定性分析,简化样品的前处理流程,使样品分析更简便。主要针对不挥发性、极性、热不稳定、大分子量等化合物的分析测定。液质联用检测技术灵敏度高,且串联质谱(三重四级杆)定性准确,可有效杜绝微量甚至痕量物质分析时的假阳性现象,常用于目标物质的痕量分析。 采用柱前衍生-液相色谱串联质谱法检测茶叶中的草甘膦和草铵膦有以下优势: 1)、灵敏度高、线性好、检出限低(可达ng/mL级及其以下); 2)、定量结果准确、稳定、重复性好; 3)、实验操作简单、步骤少、耗时短、分析速度快、检测效率高; 4)、实验试剂无污染、无毒、安全; 5)、有效减弱基质对目标物检测的影响。三、茶叶中草甘膦和草铵膦农药残留量检测的前处理试验 茶叶经GB/T8303磨碎、过筛制得待测茶样(发酵茶应先低温去除水分,使样品易于磨碎); 准确称取已磨碎处理过的茶样1g(精确至0.001g)置于80mL具盖离心管中,加入10mL水,涡旋混匀静置,加入2mL二氯甲烷,混匀,超声提取或回旋振荡10min,低速离心机4500r/min离心5min,取上清液,制得提取上清液; 注意:若茶样为新采摘的鲜叶,则称取约5g鲜叶于研钵中,加入30mL水,研磨约10min,将其转入离心管中,用10mL水洗涤研钵后转移至离心管,重复洗涤一次,再次加入10mL二氯甲烷于离心管,均质至混匀,4500r/min离心10min,取上清液,制得提取上清液; 将提取上清液用净化柱CAX、C18,以及活性炭小柱等进行比对试验,确定以C18小柱净化提取液,制得提取净化液; 通过缓冲液浓度、衍生液浓度、衍生液用量、缓冲液用量、净化液用量,衍生时间等条件试验,得出最优衍生试验参数为缓冲液浓度为50g/L,衍生液浓度20g/L,衍生液用量:缓冲液用量:净化液用量的体积比为1:1:1,衍生时间为约3h,衍生液过0.22μm的有机滤膜后进样。四、茶叶中草甘膦和草铵膦农药残留量检测的衍生机理 茶叶中草甘膦和草铵膦农药残留量的衍生机理为:在硼酸钠缓冲盐溶液条件下,草甘膦(分子结构如图1所示)和草铵膦(分子结构如图2所示)中R-NH-R’的-H被FMOC-Cl(分子结构如图3所示)中的FMOC-取代,生成 http://ng1.17img.cn/bbsfiles/images/2017/10/2015070414494663_01_0_3.png,得到衍生目标产物草甘膦衍生物和草铵膦衍生物。 其中,草甘膦分子结构图,见图1;草铵膦分子结构图,见图2;9-芴甲氧羰酰氯(FMOC-Cl)分子结构图,见图3;草甘膦和草铵膦与9-芴甲氧羰酰氯(FMOC-Cl)的衍生机理图,见图4。http://ng1.17img.cn/bbsfiles/images/2017/10/2015070414424276_01_0_3.pnghttp://ng1.17img.cn/bbsfiles/images/2017/10/2015070414430242_01_0_3.pnghttp://ng1.17img.cn/bbsfiles/images/2017/10/2015070414432007_01_2275853_3.pnghttp://ng1.17img.cn/bbsfiles/images/2015/07/201507061123_553629_2275853_3.png五、茶叶中草甘膦、草铵膦农残衍生物在质谱中的裂解机理1、茶叶中草甘膦农残衍生物在质谱中的裂解机理 通过对草甘膦衍生物在串联质谱中的裂解机理进行系统的分析研究,可探索出草甘膦衍生物的裂解机理为:首先草甘膦衍生物裂解为离
草酸是一个具有酸性、络合性和还原性的化合物,广泛应用于分析测试中?
[align=center]吸附搅拌磁子SBSE-热脱附[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]GCMS[/color][/url]分析活菌型乳酸菌饮品风味成分[/align] 活菌型乳酸菌饮品?[font='simsun']是一种以乳或乳制品为原料,经过乳酸菌发酵后制成的饮料,其中含有大量的活性乳酸菌。这类饮品旨在通过摄入活性乳酸菌,帮助调节肠道菌群平衡,改善肠道健康。此外,对于肠道敏感或易受肠道问题困扰的人群,选择活菌型乳酸菌饮品可以帮助改善肠道环境,缓解肠道不适[/font][font='simsun'](AI)[/font][font='simsun']。[/font] 本文采用吸附搅拌磁子(SBSE)提取活菌型乳酸菌饮品的香气香味风味成分,大体积冷却进样口PTV热脱附TDU[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]质谱法分析鉴定活菌型乳酸菌饮品成分;利用安捷伦未知物分析质谱解卷积软件识别拆分共流出色谱峰或基线附近的小峰,得到更纯净的质谱图,更利于下一步质谱检索的工作;并结合保留指数校正使质谱检索结果更为准确。 1试验部分 1.1 仪器与装置 美国安捷伦7890/5975C[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]-质谱联用仪,带有德国Gerstel的MPS Robotic Pro多功能自动进样系统,德国Gerstel的CIS4大体积分流/不分流进样口和TDU2热脱附。 吸附搅拌子(PDMS, 0.10mmX10mm,Gerstel)。 1.2 样品和标样,试剂 样品:某活菌型乳酸菌饮品,市场样品,来自某上海某超市。 氯化钠(优级纯) 香气香味化合物标准品均来自Sigma-Aldrich等主要试剂公司,少数为实验室内部精制标样。C6-C33正构烷混合标准物来自安谱公司。 1.3 GC/MS条件 1.3.1 色谱条件: 色谱柱:安捷伦HP-Innowax (60m×0. 25 mm ( i.d.)×0.25μm)毛细管柱; 升温程序: 40℃保持2 min,以5 ℃/min升至250℃,保持20 min; 载气(He, 纯度99.999%以上)流速1.8mL/min 进样口:PTV大体积冷进样口,温度-20℃-250℃, 15℃/S;分流比:11:1 (MSD和ODP分流比为1:1) TDU:25-230℃, 100℃/min, 不分流,传输线温度:260℃ 1.3.2质谱条件: 电子轰击(EI)离子源;电子能量70eV;传输线温度280℃;离子源温度230℃;四级 杆温度150℃。SCAN扫描范围:29-400。EMV: 1253。 1.4样品的提取处理及分析方法 [font='simsun'][color=#000000]样品的提取[/color][/font][font='simsun'][color=#000000]处理[/color][/font][font='simsun'][color=#000000]:[/color][/font] [font='simsun'][color=#000000]样品[/color][/font][font='simsun'][color=#000000]主要成分配料表[/color][/font][font='simsun'][color=#000000]有[/color][/font][font='simsun'][color=#000000]水、[/color][/font][font='simsun'][color=#000000]聚葡萄糖、[/color][/font][font='simsun'][color=#000000]脱脂乳粉、乳酸钙、[/color][/font][font='simsun'][color=#000000]维生素[/color][/font][font='simsun'][color=#000000]E[/color][/font][font='simsun'][color=#000000]、维生素[/color][/font][font='simsun'][color=#000000]D[/color][/font][font='simsun'][color=#000000]、[/color][/font][font='simsun'][color=#000000]麦芽糖醇液、[/color][/font][font='simsun'][color=#000000]白砂糖、[/color][/font][font='simsun'][color=#000000]葡萄糖、[/color][/font][font='simsun'][color=#000000]可溶性大豆多糖、[/color][/font][font='simsun'][color=#000000]三氯蔗糖、[/color][/font][font='simsun'][color=#000000]食品用香精、[/color][/font][font='simsun'][color=#000000]干酪乳酪杆菌[/color][/font][font='simsun'][color=#000000]等原料[/color][/font][font='simsun'][color=#000000]。[/color][/font][font='simsun'][color=#000000]样品里面[/color][/font][font='simsun'][color=#000000]含有[/color][/font][font='simsun'][color=#000000]乳脂,蛋白[/color][/font][font='simsun'][color=#000000],[/color][/font][font='simsun'][color=#000000]干扰很大[/color][/font][font='simsun'][color=#000000],如果使用一般的有机溶剂提取是不可以的。故选用[/color][/font][font='simsun'][color=#000000]吸附搅拌磁子(SBSE)提取[/color][/font][font='simsun'][color=#000000]。[/color][/font][font='simsun'][color=#000000]取[/color][/font][font='simsun'][color=#000000]8[/color][/font][font='simsun'][color=#000000]g样品[/color][/font][font='simsun'][color=#000000],[/color][/font][font='simsun'][color=#000000]加入2[/color][/font][font='simsun'][color=#000000].4g[/color][/font][font='simsun'][color=#000000]氯化钠[/color][/font][font='simsun'][color=#000000],[/color][/font][font='simsun'][color=#000000]放入吸附磁力搅拌子[/color][/font][font='simsun'][color=#000000]。[/color][/font][font='simsun'][color=#000000]在磁力搅拌器以[/color][/font][font='simsun'][color=#000000]900[/color][/font][font='simsun'][color=#000000]rpm转速下,[/color][/font][font='simsun'][color=#000000]提取1小时。[/color][/font][font='simsun'][color=#000000]取出[/color][/font][font='simsun'][color=#000000]吸附磁力搅拌子[/color][/font][font='simsun'][color=#000000],[/color][/font][font='simsun'][color=#000000]用[/color][/font][font='simsun'][color=#000000]超纯[/color][/font][font='simsun'][color=#000000]水冲洗干净,用[/color][/font][font='simsun'][color=#000000]干净的[/color][/font][font='simsun'][color=#000000]餐巾纸吸干,放入热脱附的小管,运行序列。[/color][/font] 在分析样品前,和样品分析完全相同的条件下,用0.02%的C6-C33的正构烷标样注射到[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]GCMS[/color][/url],获得正构烷的保留时间,用于计算保留指数。分析样品后,用软件计算样品各个组分的保留指数,并和标样的保留指数对比来,结合质谱来定性。事先也用同样方法测定标样的保留指数备用。 1.5 数据处理软件 安捷伦未知物分析软件10.1 建立好带极性保留指数的质谱数据库 2 数据处理 基本流程:建立新分析,调用[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质[/color][/url]数据,设置合理的数据分析方法参数(包括解卷积),分析检索结果,参照检索匹配度和保留指数,以及相关基本风味化合物知识,仔细核对检索结果,选择合理的检索结果,删去不必要的峰,导出分析报告。 下面为利用未知物分析处理活菌型乳酸菌饮品样品的示例图: [img]https://ng1.17img.cn/bbsfiles/images/2024/09/202409032139059895_1247_1615838_3.png[/img] [align=center]图1 未知物分析处理活菌型乳酸菌饮品样品的图例[/align][align=center] [/align] 2.1 删掉不需要的非风味物质 例如28.28min吸附搅拌子的流失的硅氧烷峰和12.83的乙基苯,样品基质带来的某些化合物,烷烃化合物,邻苯化合物等。 [img]https://ng1.17img.cn/bbsfiles/images/2024/09/202409032139063962_5752_1615838_3.png[/img] [align=center]图2 删掉不需要的非风味物质峰[/align][align=center] [/align] 2.2核对结果 观察化合物匹配度,对比计算保留指数和数据库保留指数是否一致。如果两者不吻合,可以选择Show Alternate hits...来选择合适的候选化合物来确认。 [img]https://ng1.17img.cn/bbsfiles/images/2024/09/202409032139066619_6593_1615838_3.png[/img] [align=center]图3 19.25min检索结果和替换[/align][align=center] [/align] 例如19.25min的检索匹配度94虽然很好,但保留指数1341(实际计算)和1218(质谱数据库)相差较大,显然不正确。需要替换。替换为正己醇后是正确的而结果。 [font=SimSun][img=,554,311]https://ng1.17img.cn/bbsfiles/images/2024/09/202409032142564039_8020_1615838_3.jpg!w554x311.jpg[/img][/font][font='Times New Roman',serif][/font][font=SimSun][/font][font='Times New Roman',serif][/font][font=SimSun][/font] [align=center]图4 替换为正己醇[/align][align=center] [/align] 同理28.28min的Isoborneol的保留指数1704(实际计算)和1673(质谱数据库检索)相差大一些,用borneol替换就更为合理了。Borneol的保留指数1704(实际计算)和1706(质谱数据库检索)。 [img]https://ng1.17img.cn/bbsfiles/images/2024/09/202409032139069404_6138_1615838_3.png[/img] [align=center]图5 替换Isoborneol为Borneol[/align] [img]https://ng1.17img.cn/bbsfiles/images/2024/09/202409032139071204_7262_1615838_3.png[/img] [align=center]图6 替换Isoborneol为Borneol[/align] 同样核对其它保留指数不一致的峰,进行替换。 核对检索结果后,保存结果。导出分析报告如:表 活菌型乳酸菌饮品样品风味成分表 3 结果与讨论 3.1实验结果 下面为活菌型乳酸菌饮品样品风味成分表 [align=center]表 活菌型乳酸菌饮品样品风味成分表[/align] [img]https://ng1.17img.cn/bbsfiles/images/2024/09/202409032139072513_5809_1615838_3.png[/img] [img]https://ng1.17img.cn/bbsfiles/images/2024/09/202409032139073520_6632_1615838_3.png[/img] 2.4讨论 2.4.1 从上表看出,鉴定出91种香气香味风味化合物。包括各种醛类,醇类,酯类,酸类,呋喃类,硫化物,含氮化合物,萜烯化合物,烃类化合物等。(注:一些烷烃化合物,邻苯化合物,基质带来的双酯化合物已经删掉)。风味化合物种类繁多,还是比较丰富的。 含量较大(占比10%以上)的化合物有芳樟醇,甲位松油醇,辛酸,香兰素。 乙酸,己酸,辛酸等酸类化合物是乳酸菌饮品的酸味体现。 酯类化合物提供香气。 2-酮类化合物,内酯化合物,香兰素有酸酸乳的特征气味。 甲硫醇,二硫醚,三硫醚等硫化物提供特征气味。 吡嗪化合物,甲基邻氨基苯甲酸甲酯等含氮化合物更使香气丰富。 大量的萜烯化合物有植物等气味。 2.4.2此活菌型乳酸菌饮品样品含有水、[font='simsun']聚葡萄糖、[/font][font='simsun']脱脂乳粉、乳酸钙、[/font][font='simsun']维生素[/font]、[font='simsun']麦芽糖醇液、[/font][font='simsun']白砂糖、[/font][font='simsun']葡萄糖、[/font][font='simsun']大豆多糖、[/font][font='simsun']三氯蔗糖、[/font][font='simsun']食品用香精、[/font][font='simsun']干酪乳酪[/font]杆菌等原料。如果用一般的液液萃取, 里面的脂肪会进入有机相,严重干扰[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]GCMS[/color][/url]分析,同时一些成分也很容易造成乳化现象,不易提取;如果用固相微萃取(SPME),也需要一定的加热温度,对不同挥发性的化合物的提取比例可能不一致,高沸点化合物提取效果差,难以得到较准确的定量,另外灵敏度比SBSE低,不能更好的反映活菌型乳酸菌饮品样品的风味化合物的全貌;如果用同时蒸馏萃取(SDE),有加热过程,可能在提取过程会带来热分解或反应,产生新物质,基质的影响较大。这些提取方法的不足之处,提取效果差,费事耗时,样品量大,使用大量有机溶剂,灵敏度低等。用磁力吸附搅拌子(SBSE)提取活菌型乳酸菌饮品样品,灵敏度高,需要样品量很少,无溶剂,操作简单方便,可以在不加热情况下提取,减少热分解。非常有利于香气香味风味化合物的提取。 样品中适当加入氯化钠有助于更多极性化合物的提取。
微量金标准物质定值均匀性分析检验 利用王水溶样,活性炭吸附-灰化-王水溶解-硫脲提取-原子吸收光谱法测定微量金标准物质“GAu-9b”的30个子样的含量,每个子样重复测定3次。考察了天平称量精度、玻璃器皿刻度精度和活性炭吸附率造成的测量不确定度,并估算出合成不确定度范围为1.6%(A级品)~2.2%(A级品),论证了方法的可行性;对单个样品重复分析结果的可靠性及样品均匀性进行评估,计算出样品均匀性质量参数为0.77,样品测定结果的RSD均符合地质实验室质量管理规范要求,依此判定该样品的均匀性质量等级为合格。均匀性是标准物质必须具备的特性之一,均匀性检验得到的数据实际上包含了两种不确定度,一是样品不均匀引起的不确定度,另一是测量不确定度,所以在均匀性检验中应该使用高精度的方法,本文采用吸附效果好的活性炭吸附-灰化-王水溶解-石墨炉原子吸收光谱法测定微量金标准物质,以此方法为基础,结合石墨炉原子吸收对测定介质的要求,制定了微量金标准物质的分析步骤。通过查阅不确定度有关文献,计算了样品分析的合成不确定度,将合成的相对不确定度与重复分析相对偏差允许限进行比较,确定了拟定的分析方法的可靠性;均匀性检验方法很多,有方差法、极差法区间法和三分之一法等等 ,其中方差法和摄差法经多年的实践证明是可靠的,本文采用方差法(F)检验,得到样品均匀性良好;标准物质中各成分经均匀性检验合格后可进入定值,本文采用哥拉布斯 (Grubbs)法对离群值进行剔除,.最后得到标准物质的标准参考值和不确定度。1 实验部分1.1 仪器和主要试剂日立5000石墨炉原子吸收分光光度计,其测金条件略标准溶液:(ρ)=0.1μg/ml,工作液根据需要稀释,介质为Ψ=10%(体积分数,下同)的王水;盐酸、硝酸、硫脲试剂均为分析纯,水为蒸馏水;活性炭:粒径0.074mm,化学纯(北京光华木材厂[
《食品安全国家标准 食品中农药最大残留限量》2016版正式颁布实施,这一农药残留的新国标,在标准数量和覆盖率上都有了较大突破,规定了433种农药在13大类农产品中4140个残留限量,较2014版增加490项,基本涵盖了我国已批准使用的常用农药和居民日常消费的主要农产品。 食品伙伴网标法中心结合2016版标准前言部分内容与2014版进行相应的对比分析,供参考: 1、对原标准中氟唑磺隆、甲咪唑烟酸、氟吡菌胺、三唑酮和三唑醇等5种农药残留物定义,敌草快等5种农药每日允许摄入量等信息进行了核实,修订了敌草快、三环锡等5种农药的ADI值。 2、增加了2,4-滴异辛酯等46种农药;增加了490项农药最大残留限量标准 2014版规定了食品中2,4-滴等387种农药3650项最大残留限量,2016版规定了433种2,4-滴等农药4140项最大残留限量。增加了46种农药:2,4-滴异辛酯、2甲4氯异辛酯、苯嘧磺草胺、苯嗪草酮、吡唑草胺、丙硫多菌灵、除虫菊素、毒草胺、多抗霉素、呋虫胺、氟吡菌酰胺、复硝酚钠、甲磺草胺、井冈霉素、抗倒酯、苦参碱、醚苯磺隆、嘧啶肟草醚、扑草净、嗪草酸甲酯、氰氟虫腙、氰烯菌酯、炔苯酰草胺、噻虫胺、三苯基乙酸锡、三氯吡氧乙酸、杀螺胺乙醇胺盐、莎稗磷、虱螨脲、特丁津、调环酸钙、五氟磺草胺、烯丙苯噻唑、烯肟菌酯、烯效唑、辛菌胺、辛酰溴苯腈、溴氰虫酰胺、唑胺菌酯、唑啉草酯、啶菌噁唑、丁吡吗啉、噁唑酰草胺、甲哌鎓、丁酰肼、唑嘧菌胺。 3、增加 12 项检测方法标准,删除1项检测方法标准 增加了SN/T 0162、SN 0198、SN/T 0931、SN/T 1624、SN/T 1989、SN/T 2229、SN/T 2231、SN/T 2237、SN/T 2323、SN/T 2387、SN/T 2795、SN/T 2807,删除了SN/T 0711,其中SN 0198标准已于2015年12月31日被认监委废止,废止依据为《国家认监委办公室关于公布2015年检验检疫行业标准复审结论的通知》。 4、修改了丙环唑等8种农药的英文通用名 修改了丙环唑、六六六、烯肟菌胺、氯啶菌酯、杀虫双、四氯苯酞、氯氟吡氧乙酸和氯氟吡氧乙酸异辛酯 5、将苯噻酰草胺和灭锈胺的限量值由临时限量修改为正式限量;对资料性附录 A 进行了修订,增加了干制蔬菜等3种食品名称,修改1项作物名 食品伙伴网对附录A部分内容的对比发现如下变化: 1)水果(核果类)的类别说明增加了青梅,枣修改为枣(鲜)。 2)水果(浆果和其他小型水果)的类别说明中露莓增加了备注:包括波森莓和罗甘莓。 3)水果(热带和亚热带水果)的类别说明中将大型果的木瓜修改为番木瓜。 4)干制水果的类别说明中增加了枣(干)等。 5)食品类别名称修改:将饮料修改为饮料类。 6、食品伙伴网在对比过程中发现,2016版标准除了以上所列变化外,还修正了其他一些内容: 1)引用的标准名称的修正,如GB/T 19648、GB/T 19469等部分标准的名称中 “兽”字已删除。 2)引用的作废标准的修正,如2014版标准中引用的是2006版GB/T 20770的标准名称,2016版标准已经修正为2008版GB/T 20770的标准名称。 3)农药中文名称修改:2014版标准中的2甲4氯(钠)修改为2甲4氯钠。 4)附录A中动物源食品部分类别的测定部位描述进行了修正。
请问草酸亚铁的国家标准或行业标准那里有?
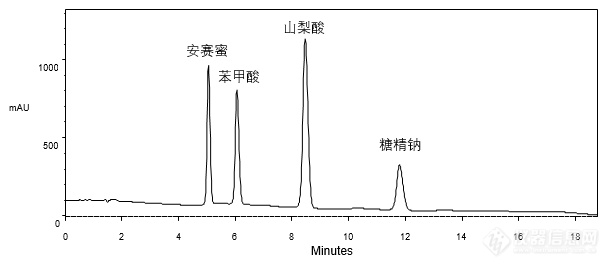
[align=center][b]GB 5009.28-2016食品安全国家标准 食品中苯甲酸、山梨酸和糖精钠的测定[/b][/align][align=center][b] ——标准品与乳品实际样品的分析[/b][/align][align=center][/align][align=left]本实验按照《GB5009.28-2016 食品安全国家标准食品中苯甲酸、山梨酸和糖精钠的测定》方法,分别对安赛蜜、苯甲酸、山梨酸、糖精钠的标准品混合溶液及加标乳品样品进行了分析。首先,使用CAPCELL PAK C[sub]18[/sub] MG S5 4.6 mm i.d. × 150mm色谱柱,对标准品混合溶液进行分析,如图1,安赛蜜、苯甲酸、山梨酸、糖精钠标准品均得到了良好的分析结果。[/align][align=left][/align][align=center][img=,611,268]http://ng1.17img.cn/bbsfiles/images/2018/03/201803221532276656_9890_2222981_3.png!w611x268.jpg[/img][/align][align=center]图1 标准品混合溶液分析色谱图[/align][img=,400,200]http://ng1.17img.cn/bbsfiles/images/2018/03/201803221532280132_6863_2222981_3.png!w400x200.jpg[/img][align=left][/align][align=left]其次,对乳品加标样品进行分析,如图2,糖精钠(Rt 12 min)与其后杂质峰之间未能取得基线分离,分离度仅为1.02。[/align][align=left][/align][align=center][img=,668,335]http://ng1.17img.cn/bbsfiles/images/2018/03/201803221533054905_2223_2222981_3.png!w668x335.jpg[/img][/align][align=center]图2 加标乳品样品分析色谱图[/align][align=left][img=,406,203]http://ng1.17img.cn/bbsfiles/images/2018/03/201803221533317202_2333_2222981_3.png!w406x203.jpg[/img][/align][align=left][/align][align=left]为改善糖精钠与杂质间的分离,在国标方法基础上,将流动相由[b]乙酸铵 / 甲醇 = 95 / 5[/b]调整为[b][b]乙酸铵 / 甲醇[/b][color=red]([/color][color=red]2 mmol/L [/color][color=red]甲酸)[/color]= 92 / 8[/b],再次对混合标准溶液和加标样品进行分析,结果如图3所示。[/align][align=left][/align][align=center][img=,690,545]http://ng1.17img.cn/bbsfiles/images/2018/03/201803221534141056_4073_2222981_3.png!w690x545.jpg[/img][/align][align=center]图3 混标与加标乳品样品分析色谱图[/align][align=left][img=,464,171]http://ng1.17img.cn/bbsfiles/images/2018/03/201803221535548985_7176_2222981_3.png!w464x171.jpg[/img][/align][align=left][/align][align=left]如图3,在酸性条件下,出峰顺序发生了变化,安赛蜜保留时间略有缩短,糖精钠保留时间明显缩短,由12 min缩短至8 min,苯甲酸和山梨酸保留时间分别延长至2 min和6 min;在分离度方面,糖精钠与苯甲酸之间分离度为2.79,苯甲酸与峰后杂质间分离度为2.04,所有色谱峰之间都达到了基线分离。[/align][align=left][/align][align=left]为使客户有更多选择,实验室又在国标原方法条件下继续筛选色谱柱,最终使用SUPERIOREX ODS S5 4.6 mm i.d. × 250 mm色谱柱时,仅微调有机相比例即可实现加标乳品样品的良好分析结果。如图4,杂质峰与糖精钠之间分离度达到2.48,达到基线分离要求。[/align][align=left][/align][align=center][img=,580,332]http://ng1.17img.cn/bbsfiles/images/2018/03/201803221537130173_1058_2222981_3.png!w580x332.jpg[/img][/align][align=center]图4 加标乳品样品分析色谱图[/align][align=left]*注:峰上标所示数字由下至上依次为分离度与不对称因子。[/align][align=left][img=,326,177]http://ng1.17img.cn/bbsfiles/images/2018/03/201803221537540634_9437_2222981_3.png!w326x177.jpg[/img][/align][align=left][/align][align=left]综上所述,按照国标《GB 5009.28-2016 食品安全国家标准食品中苯甲酸、山梨酸和糖精钠的测定》方法进行分析,使用CAPCELL PAK C[sub]18 [/sub]MG色谱柱对标准品混合溶液能得到良好分析结果,但在对加标乳品样品进行分析时,糖精钠与样品中的杂质未能实现基线分离,通过在流动相中添加甲酸可实现安赛蜜、糖精钠、苯甲酸、山梨酸及杂质的基线分离;另一方面,使用SUPERIOREX ODS色谱柱,在原条件基础上微调即可实现乳品中安赛蜜、苯甲酸、山梨酸、糖精钠及杂质间的良好分离。[/align]
用LC—MS做定量分析时,为了保证称量的专业性,标准品配制的较浓,请问能连续稀释2—3次得到想要的浓度吧,会不会导致误差偏大呢?
目前我们分析草酸钴的杂质先将草酸钴在400度氧化,再加盐酸硝酸溶解试样,程序比较麻烦,而且易污染,有谁知道更简洁的处理样品的方法?
哪位有非发酵性豆制品中吊白块(次硫酸氢钠甲醛)的分析方法啊,麻烦提供下,最好是现行有效版本的标准。
根据检测项目,建议选择两个品牌或两个批次的标准品进行对比验证,确保标准曲线的准确性。注意商品化标准品和标准储备溶液的有效期,如果条件允许,建议能力验证实验使用新的未开瓶的标准品制备标准曲线。纯品型标准品需要使用分析天平进行准确称量,并记录数值,同时需要关注标准品的纯度,计算目标物浓度时需要带入标准品纯度。标准品的选择还需关注目标物的形式,例如四环素的标准品是以四环素盐酸盐的形式存在的,需要确认最终要求以何种形式进行报告,在进行浓度计算时需要注意换算。
中华人民共和国国家标准 GB\T 5009.48--1996蒸馏酒及配制酒卫生标准的分析方法 代替 GB 5009.48--85Method for analysis of hygienic standardof distilled wines and mixed wines____________________________________________________________________________1 主题内容与适用范围 本标准规定了以含糖或淀粉的物质为原料,经糖化发酵蒸馏而制得的白酒及以发酵酒或蒸馏酒作酒基,经添加可食用的辅料制成的配制酒中各项卫生指标的分析方法。 本标准适用于蒸馏酒和配制酒中各项卫生指标的分析。2 引用标准 GB 2757 蒸馏洒和配制洒卫生标准 GB 5009.2 食品中相对密度的测定方法。 GB 5009.11 食品中总砷的测定方法。 GB 5009.12 食品中铅的测定方法。 GB 5009.36 粮食卫生标准的分析方法。 GB 5009.35 食品中着色剂的测定方法。 GB 12396 食品中铁、锰的[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]测定方法3 感官检查3.1 量取 30mL 样品,倒入 50mL 清洁干燥无色玻璃烧杯中,观察其颜色,应透明,无沉淀或杂质。3.2 尝其味应有该种酒特有的芳香味和滋味,不应有霉味、酸味、异味。应符合GB 2757的规定。4 理化检验4.1 乙醇浓度(比重计法)4.1.1 原理 同GB 5009.2的原理。4.1.2 仪器 酒精比重计。4.1.3 分析步骤 吸取 100mL样品于 250mL或 500mL全玻璃蒸馏器中,加 50mL 水,再加入玻璃珠数粒,蒸馏,用100mL 容量瓶收集馏出液 100mL。 将蒸馏后的样品倒入量筒中,将洗净擦干的酒精计缓缓沉入量筒中,静止后再轻轻按下少许,待其上升静止后,从水平位置观察其与液面相交处的刻度,为乙醇浓度, 同时测定温度,按测定的温度与浓度,查《酒精计温度浓度换算表》,换算成温度为20℃时的乙醇浓度(%)。4.2 甲醇4.2.1 原理 甲醇经氧化成甲醛后,与品红亚硫酸作用生成蓝紫色化合物,与标准系列比较定量。最低检出量为 0.02g/100mL。4.2.2 试剂4.2.2.1 高锰酸钾-磷酸溶液:称取 3g 高锰酸钾,加入15mL磷酸 (85%)与 70mL水的混合液中,溶解后加水至 100mL。贮于棕色瓶内,防止氧化力下降,保存时间不宜过长。4.2.2.2 草酸-硫酸溶液:称取 5g 无水草酸(H2C2O4) 或7g含 2分子结晶水草酸(H2C2O4.2H2O ), 溶于硫酸 (1+1)中至 100mL. 4.2.2.3 品红- 亚硫酸溶液 :称取 0.1g碱性品红研细后,分次加入共60mL 80℃的水,边加入水边研磨使其溶解,用滴管吸取上层溶液滤于100mL 溶量瓶中,冷却后加10mL亚硫酸钠溶液(100g/L), 1mL盐酸 ,再加水至刻度, 充分混匀,放置过夜,如溶液有颜色,可加少量活性炭搅拌后过滤,贮于棕色瓶中,置暗处保存,溶液呈红色时应弃去重新配制。4.2.2.4 甲醇标准溶液:称取 1.000g 甲醇,置于 100mL 容量瓶中, 加水稀释至刻度。此溶液每毫升相当于 10mg 甲醇。置低温保存。4.2.2.5 甲醇标准使用液:吸取 10.0mL 甲醇标准溶液,置于 100mL容量瓶中,加水稀释至刻度。再取 10.0mL 稀释液置于 50mL 容量瓶中,加水至刻度,该溶液每毫升相当0.50mg甲醇。4.2.2.6 无甲醇的乙醇溶液:取 0.3mL按操作方法检查,不应显色。如显色需进行处理。取 300mL乙醇 (95%),加高锰酸钾少许,蒸馏,收集馏出液。在馏出液中加入硝酸银溶液(取1g硝酸银溶于少量水中)和氢氧化钠溶液(取 1.5g 氢氧化钠溶于少量水中),摇匀,取上清液蒸馏,弃去最初50mL馏出液,收集中间馏出液约200mL,用酒精比重计测其浓度,然后加水配成无甲醇的乙醇(60%)。4.2.2.7 亚硫酸钠溶液(100g/L)。4.2.3 仪器 分光光度计。







 我要推广仪器
我要推广仪器
 下载APP
下载APP