由二甲胺与磺酰氯合成二甲胺基磺酰氯,产品计划用气相做检测,柱子是se-30的,结果不理想,大家给些意见,尤其是关于该产品的文献极少,在知网上没有几篇。也可能太简单了。
二甲胺基磺酰氯在合成过程中在主峰前出现一个1%的左右的杂质,请教高手帮我解疑是什么?
二甲胺基磺酰氯在[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱仪[/color][/url]中出峰吗?
二甲基氨基偶氮苯磺酰氯的分子式 有谁知道啊?敬请赐教!
OPA测定结果为70mg/片,丹磺酰氯测定结果为59mg/片。大家有用比较过用这2个物质衍生同一种氨基酸的结果差异么?
资料上看到用丹磺酰氯衍生氨基酸要加入一定量的2mol/L的碳酸氢钾,我想这里的碳酸氢钾应该是缓冲作用,因为现在没有这个试剂,可不可以用其他的代替?例如:碳酸氢钠?
想请教一下各位网友,我用苯胺衍生二甲氨基甲酰氯,但是随着衍生时间延长,以及苯胺用量,衍生物峰面积一直在增加,做不到反应彻底,有什么解决方法吗
想请教一下各位网友,我用苯胺衍生二甲氨基甲酰氯,但是随着衍生时间延长,以及苯胺用量,衍生物峰面积一直在增加,做不到反应彻底,有什么解决方法吗
磺胺药物对氨基苯磺酰胺的合成目的原理Ar-NHCOCH3 + 2HOSO2Cl → p-ClO2S-Ar-NHCOCH3+ HClp-ClO2S-Ar-NHCOCH3 + NH3 → p-CH3CONH-Ar-SO2NH2 + HClp-CH3CONH-Ar-SO2NH2 + H2O → p-H2N-Ar-SO2NH2 + CH2CO2H仪器药品乙酰苯胺(自制) 5g(0.037mol);氯磺酸(d=1.77) 22.5g(12.5ml,0.19mol);浓氨水(28%,d=0.9) 35ml 浓盐酸,碳酸钠。过程步骤(1)对乙酰氨基苯碘酰氯在100ml干燥的锥形瓶中,加入5g干燥的乙酰苯胺,在石棉网上用小火加热熔化。瓶壁上若有少量水气凝结,应用干净的滤纸吸去。冷却使熔化物凝结成块。将锥形瓶置于冰浴中冷却后,迅速倒入12.5ml氯磺酸,立即塞上带有氯化氢导气管的塞子。反应很快发生,若反应过于激烈,可用冰水浴冷却。待反应缓和后,旋摇锥形瓶使固体全溶,然后再在温水浴中加热10~15min使反应完全。将反应瓶在冷水中充分冷却后,于通风中在充分搅拌下,将反应液慢慢倒入盛75g碎冰的烧杯,用少量冷水洗涤反应瓶,洗涤液倒入烧杯中。搅拌数分钟,并尽量将大块固体粉碎,使成颗粒小而均匀的白色固体。抽滤收集,用少量冷水洗涤,压干,立即进行下一步反应。(2)对乙酰氨基苯磺酰胺将上述粗产物移入烧杯中,在不断搅拌中慢慢加入17.5ml浓氨水(在通风橱内),立即发生放热反应并产生白色糊状物。加完后,继续搅拌15min,使反应完全。然后加入19ml水,在石棉网上用小火加热10~15min,并不断搅拌,以除去多余的氨,得到的混合物可直接用于下一步合成。(3)对氨基苯磺酰胺(磺胺)将上述反应物放入圆底烧瓶中,加入3.5ml浓盐酸,在石棉网上用小火加热回流0.5h。冷却后,应得一几乎澄清的溶液,若有固体析出,应继续加热,使反应完全。如溶液呈黄色,并有极少量固体存在时,需加入少量活性炭煮沸10min,过滤。将滤液转入大烧杯中,在搅拌下小心加入粉状碳酸钠至恰呈碱性(约4g)。在冰水浴中冷却,抽滤收集固体,用少量冰水洗涤,压干。粗产物用水重结晶(每克产物约须12ml水),产量3~4g。熔点161~162℃。纯品对氨基苯磺酰胺为白色针状结晶,熔点163~164℃。注意事项1.氯磺酸对皮肤和衣服有强烈的腐蚀性,暴露在空气中会冒出大量氯化氢气体,遇水会发生猛烈的放热反应,甚至爆炸,故取用时需加小心。反应中所用仪器及药品皆需十分干燥,含有氯磺酸的废液不可倒入水槽,而应倒入废液缸中。工业氯磺酸常呈棕黑色,使用前宜用磨口仪器蒸馏纯化,收集148~150℃的馏分。2.酰磺酸于乙酰苯胺的反应非常剧烈,将乙酰苯胺凝结成快状,可使反应缓和进行,当反应过于激烈时,应适当冷却。3.在氯磺化过程中,将有大量氯化氢气体放出。为避免污染室内空气,装置应严密,导气管的末端要与接受器内的水面接近,但不能插入水中,否则可能倒吸而引严重事故!4.加入速度必须缓慢,必须充分搅拌,以免局部过热而使对乙酰胺基苯磺酰胺水解。这是实验成功的关键。5.尽量洗去固体所夹杂和吸附的盐酸,否则产物在酸性介质中放置过久,会很快水解,因此在洗涤后,应尽量压干,且在1~2h内将它转变为磺胺类化合物。6.粗制的对氨基苯磺酰氯久置容易分解,甚至干燥后也不可避免。若要得到纯品,可将粗产物溶于温热的氯仿中,然后迅速转移到事先温热的分液漏斗中,分出氯仿层,在冰水浴中冷却后即可析出晶体。纯品对氨基苯磺酰氯的熔点为149℃。7.为了节省时间,这一步的粗产物可不必分出。若要得到产品,可在冰水浴中冷却,抽滤,用冰水洗涤,干燥即可。粗品用水重结晶,纯品熔点为219~220℃。8.对乙酰胺基苯磺酰胺在稀酸中水解成磺胺,后者又与过量的盐酸形成水溶性的盐酸盐,所以水解完成后,反应液冷却时应无晶体析出。由于水解前溶液中氨的含量不同,加3.5ml盐酸有时不够,因此,在回流至固体全部消失前,应测一下溶液的酸碱性,若酸性不够,应补加盐酸回流一段时间。9.用碳酸钠中和滤液中的盐酸时,有二氧化碳产生,故应控制加热速度并不断搅拌使其逸出。磺胺是一两性化合物,在过量的碱溶液中也易变成盐类而溶解。故中和操作必须仔细进行,以免降低产量。分析思考 1.为什么在氯磺化反应完成以后处理反应混合物时,必须移到通风橱中,且在充分搅拌下缓缓倒入碎冰中?若在未倒完前冰就化完了,是否应补加冰块?为什么?2.为什么苯胺要乙酰化后在氯磺化?直接氯磺化行吗?3 .如何理解对氨基苯磺酰氨是两性物质?试用反应式表示磺胺与稀酸和稀碱的作用。
各位大侠,丹磺酰氯与25%氨水反应吗?丹磺酰氯的结构式我想上传个图片的,总是传不上,辛苦大家帮我分析下哈!
我做生物胺的柱前衍生,用的是10mg/ml的丹磺酰氯丙酮容液,但是配完之后丹磺酰氯很多都沉在底部并不溶解,用超声混匀后,用移液枪吸取能明显看到颗粒。然后将丹磺酰氯丙酮容液加进生物胺单标里混匀后会底部出现白色沉淀,是不是有发生什么化学反应?这个沉淀看了很多文献都没提到过。然后就是去跑液相,8个标品,只有精氨酸一直跑不出,时间感觉也很充足了,就是一直很平坦不出峰,是不是和上面的操作有关系啊?
朋友们谁有苯甲酸甲酯、对甲苯磺酸、TEBAC、甲醇钠、4-二甲胺基吡啶、苯甲酰氯、乙硫醇、巴豆醛、乙酰乙酸甲酯、丙酰氯、DCP的国标、行标或企业标准啊?帮忙找一下啊,谢谢!
求助: 对甲苯磺酰氯 CAS 98-59-9 国家标准
复方氨基酸注射液采用丁基胶塞密封,胶塞中的硫磺可能会浸出到注射液中,需要对药液中的硫磺含量进行测定。但是,方法学建立过程中,进行药液的加标回收率测定,发现其不合格,并且随氨基酸浓度升高,硫磺回收率降低。求助可能的原因,有哪位大神做过相关的实验吗?请不吝赐教,大谢
称取5克对氨基苯磺酰胺,于1比7盐酸溶液中,稀释至500M。这溶液时间用长了,瓶底有一层白色结晶,这是什么问题,刚配时是全部融化,看不到什么结晶,为什么时间长就会出现这个问题,结晶是什么?请各位高手指教,谢谢。
[size=2][font=宋体]2-氨基苯酚-4-磺酰胺的国标或行标[/font][/size][size=2][font=宋体][/font][/size]
专家你好!我再将我问的问题解释清楚点:我们生产的上原料药的中间体,我们用2,4-二氯-5-磺酰胺基苯甲酸做原料生产速尿,在分析中发现,此原料的熔点越高(正常为232-234度,如我们测出234-235度)生产的产率就比正常的原料产率要高。请问,这该怎么解释?
请问谁用过丹磺酰氯,我要求购。
请问氯磺酰异氰酸酯的理化性质,它遇水爆炸,那么废液如何处理,谢谢!
[color=#444444][url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]中,顶空进样,OV-1301色谱柱,平衡温度75摄氏度,二氯甲烷作溶剂,0.01mg/ml丁基磺酰氯为什么不重现??[/color]
NA-215磺酰氯的检测方法 由于我们公司新产品需要用到 NA-215磺酰氯做原料 需要用HPLC进行检测。在网上查到该物质的检测方法是94 年检测方法 且附带图形也不是很理想它的检测方法是:流动相 甲醇 流速1ML/MIN 波长 254柱子 YQG-5C18 4.6mmID*30CM浓度1MG/ML反相色谱 想问下有那位大侠知道有最新的检测方法吗 还有一个问题就是我 每次做完HPLC就把普图和检测结果直接粘贴在WOLD文档上但WOLD文档好像不能上传 有什么软件能将 WOLD文档转化为可以直接上传的文档 谢谢大家了先
我在做生物胺的实验 其中需要配10mg/mL的丹磺酰氯丙酮溶液 最后发现不溶 丹磺酰氯是新买新开封的 丙酮也开了瓶新的 超声效果也不理想 这是什么原因 有人能帮帮我吗
异喹啉-5-磺酰氯 用液相测定含量的方法?
0.1mol/L碳酸氢钠溶液复溶,加入等体积1g/L的丹磺酰氯丙酮溶液,60℃孵育5min,上机测试,无信号,有大佬做过这个衍生化么
怎么用液相测呀,吡啶三磺酰氯在水中易解离生成吡啶三磺酸,怎么用液相测呢
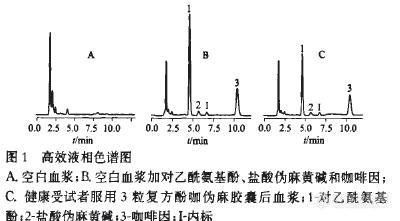
【作者】 胡晓; 张丽芳; 温金华; 蔡军;【机构】 南昌大学医学院临床药理研究所;【摘要】 目的:建立一种同时测定人血浆中对乙酰氨基酚、伪麻黄碱和咖啡因的高效液相色谱(HPLC)法,并用于含上述组分的复方制剂的人体药动学研究。方法:以茶碱为内标,血样经醋酸乙酯提取后,采用高效液相色谱紫外(HPLC-UV)法进行测定。色谱柱为Diamonsil C18柱(4.6mm×150mm,5μm);流动相为甲醇-0.05mol.L-1磷酸二氢钾(23∶77,pH2.4);流速1mL.min-1。检测波长210nm。结果:人血浆中对乙酰氨基酚、盐酸伪麻黄碱和咖啡因质量浓度测定的线性范围分别为0.12~11.52mg.L-1,0.008~0.432mg.L-1和0.03~2.16mg.L-1;最低可定量质量浓度分别为0.12,0.008,0.03mg.L-1;各组分日内、日间RSD均小于15%,方法回收率均大于88%。结论:该方法能快速可靠地同时测定人血浆中对乙酰氨基酚、伪麻黄碱和咖啡因的浓度,可用于含上述组分的复方制剂的人体药动学或生物等效性研究。【关键词】 http://ng1.17img.cn/bbsfiles/images/2012/08/201208142012_383851_1609970_3.jpg
请问有大神做过胺类用丹黄酰氯衍生化吗本人衍生化后直接离心进质谱 发现没有所需要的物质想请教一下
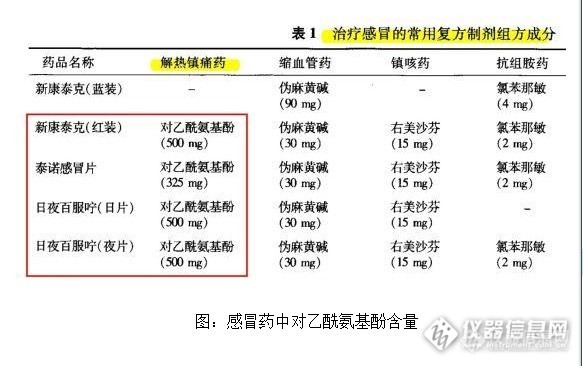
点击查看FDA声明原文 FDA警告,过量服用对乙酰氨基酚药物将会导致肝功能衰竭甚至死亡,高危人群包括在24小时内服用超过处方规定剂量药物的患者,同时服用超过一种含对乙酰氨基酚成分药物的患者,以及在服用含对乙酰氨基酚成分药物时饮用含酒精饮料的患者。 FDA自2011年起开始要求制药企业将对乙酰氨基酚的含量控制在每单位325毫克以内,但截止至2014年1月14日,市场上仍有部分单位对乙酰氨基酚含量超过325毫克的药物正在流通销售。 FDA表示,将在近期启动相关法律程序,禁止开具每单位对乙酰氨基酚含量超过325毫克的药物处方。 国内康泰克、百服宁等超标近54% 调查发现,国内多家知名品牌感冒药的对乙酰氨基酚含量高于每单位325毫克,包括新康泰克、百服宁等。 中美史克生产的新康泰克美扑伪麻片(红装),其每片对乙酰氨基酚含量同样是500毫克。 上海施贵宝制药有限公司生产的日夜百服咛、加合百服宁的每单位对乙酰氨基酚含量为500毫克,超出美国FDA规定含量上限54%。 此外,有多个品牌感冒药的单位对乙酰氨基酚含量达到美FDA规定的上限325毫克,它们分别是:白加黑、泰诺、银得菲。 其他品牌如快克、感康、康必得等单位对乙酰氨基酚含量在250毫克以内。 走访沪上多家药房后发现,最为常见的白加黑、新康泰克等感冒药均为处方药,需要处方及身份证才能购买。 大量非处方药含对乙酰氨基酚 不过,一药房工作人员提醒道,前述感冒药被转为处方药管理,并非缘于对乙酰氨基酚的过量,而是系其含有伪麻黄碱的缘故。 事实上,由于良好的镇痛祛热疗效,对乙酰氨基酚药物使用普及度很高,国内亦将其列为非处方药(OTC),即不需要持有医生处方即能买到,同时也能通过网络合法销售。 搜索淘宝等线上商城后发现,在网上不仅可以随意买到各种各样品牌的对乙酰氨基酚药品,而且购买数量亦完全没有限制。 沪上一医药销售人员表示,由于在各种药物中使用广泛,以及非处方药可以任意购买的缘故,患者容易同时购买多种含有对乙酰氨基酚的药物,导致患者在不知情的情况下过量服用。 根据FDA统计,在1998年至2003年间,对乙酰氨基酚过量服用是导致病人肝衰竭的主要原因。美国疾病控制中心在2007年也曾经发布报告称,全美每年有1600起急性肝功能衰竭,其中对乙酰氨基酚过量服用是最大缘由。 该销售人员解释道,美FDA长期以来都对药物中对乙酰氨基酚的含量作出规定,但因为非处方药的关系,仍有大量患者过量服用,所以FDA将对乙酰氨基酚的含量限制在325毫克以内,也是为了在不影响疗效的情况下尽可能的让患者减少服用,降低副作用。 FDA在此次声明中亦表示,将采取新的行动针对对乙酰氨基酚类的非处方药进行监管。 小知识:对乙酰氨基酚(扑热息痛) 对乙酰氨基酚是乙酰苯胺类解热镇痛药,别名有乙酰氨基酚、扑热息痛、醋氨酚、退热净等。该药具有解热镇痛作用,主要用于缓解轻、中度的疼痛,如关节痛、头痛、神经痛、牙痛及痛经等。 作为非处方药,虽然有质量稳定,疗效确切的优点,但该药的滥用,使用不当也会产生不良反应或严重的肝毒性和肾毒性。 此药对肝损害是最主要的不良反应,其次是引起肾衰,此外也可能导致血小板减少、哮喘发作等症状。 在无其他药物或无酒精干扰情况下,对乙酰氨基酚的安全剂量为:成人口服每次300-600毫克,最大日剂量不超过4000毫克,退热疗程不宜超过3天,镇痛疗程不宜超过5天。儿童12岁以下按体重每次10-15mg/kg,疗程不宜超过3天。 若患者饮酒或空腹或与其他药物有相互作用的药物合用时,应调低剂量或禁用。
最近我公司要分析对甲苯磺酰氯,现在化验室有台气象色谱仪,FID检测器,毛细管柱型号是:SE-30 不知道能不能分析对甲苯磺酰氯这个产品?有哪位朋友知道的帮助一下。
氯唑沙宗对照品和对乙酰氨基酚对照品的出峰时间分别是多久?







 我要推广仪器
我要推广仪器
 下载APP
下载APP