《食品安全国家标准 食品中农药最大残留限量》2016版正式颁布实施,这一农药残留的新国标,在标准数量和覆盖率上都有了较大突破,规定了433种农药在13大类农产品中4140个残留限量,较2014版增加490项,基本涵盖了我国已批准使用的常用农药和居民日常消费的主要农产品。 食品伙伴网标法中心结合2016版标准前言部分内容与2014版进行相应的对比分析,供参考: 1、对原标准中氟唑磺隆、甲咪唑烟酸、氟吡菌胺、三唑酮和三唑醇等5种农药残留物定义,敌草快等5种农药每日允许摄入量等信息进行了核实,修订了敌草快、三环锡等5种农药的ADI值。 2、增加了2,4-滴异辛酯等46种农药;增加了490项农药最大残留限量标准 2014版规定了食品中2,4-滴等387种农药3650项最大残留限量,2016版规定了433种2,4-滴等农药4140项最大残留限量。增加了46种农药:2,4-滴异辛酯、2甲4氯异辛酯、苯嘧磺草胺、苯嗪草酮、吡唑草胺、丙硫多菌灵、除虫菊素、毒草胺、多抗霉素、呋虫胺、氟吡菌酰胺、复硝酚钠、甲磺草胺、井冈霉素、抗倒酯、苦参碱、醚苯磺隆、嘧啶肟草醚、扑草净、嗪草酸甲酯、氰氟虫腙、氰烯菌酯、炔苯酰草胺、噻虫胺、三苯基乙酸锡、三氯吡氧乙酸、杀螺胺乙醇胺盐、莎稗磷、虱螨脲、特丁津、调环酸钙、五氟磺草胺、烯丙苯噻唑、烯肟菌酯、烯效唑、辛菌胺、辛酰溴苯腈、溴氰虫酰胺、唑胺菌酯、唑啉草酯、啶菌噁唑、丁吡吗啉、噁唑酰草胺、甲哌鎓、丁酰肼、唑嘧菌胺。 3、增加 12 项检测方法标准,删除1项检测方法标准 增加了SN/T 0162、SN 0198、SN/T 0931、SN/T 1624、SN/T 1989、SN/T 2229、SN/T 2231、SN/T 2237、SN/T 2323、SN/T 2387、SN/T 2795、SN/T 2807,删除了SN/T 0711,其中SN 0198标准已于2015年12月31日被认监委废止,废止依据为《国家认监委办公室关于公布2015年检验检疫行业标准复审结论的通知》。 4、修改了丙环唑等8种农药的英文通用名 修改了丙环唑、六六六、烯肟菌胺、氯啶菌酯、杀虫双、四氯苯酞、氯氟吡氧乙酸和氯氟吡氧乙酸异辛酯 5、将苯噻酰草胺和灭锈胺的限量值由临时限量修改为正式限量;对资料性附录 A 进行了修订,增加了干制蔬菜等3种食品名称,修改1项作物名 食品伙伴网对附录A部分内容的对比发现如下变化: 1)水果(核果类)的类别说明增加了青梅,枣修改为枣(鲜)。 2)水果(浆果和其他小型水果)的类别说明中露莓增加了备注:包括波森莓和罗甘莓。 3)水果(热带和亚热带水果)的类别说明中将大型果的木瓜修改为番木瓜。 4)干制水果的类别说明中增加了枣(干)等。 5)食品类别名称修改:将饮料修改为饮料类。 6、食品伙伴网在对比过程中发现,2016版标准除了以上所列变化外,还修正了其他一些内容: 1)引用的标准名称的修正,如GB/T 19648、GB/T 19469等部分标准的名称中 “兽”字已删除。 2)引用的作废标准的修正,如2014版标准中引用的是2006版GB/T 20770的标准名称,2016版标准已经修正为2008版GB/T 20770的标准名称。 3)农药中文名称修改:2014版标准中的2甲4氯(钠)修改为2甲4氯钠。 4)附录A中动物源食品部分类别的测定部位描述进行了修正。
中华人民共和国进出口商品检验行业标准 中华人民共和国进出口商品检验行业标准 出口水果中氯硝胺残留量检验方法 SN 0280-93 Method for the determination of dicloran residues in fruits for export━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━1 主题内容与适用范围 本标准规定了出口水果中氯硝胺残留量检验的抽样、制样和[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]测定方法。 本标准适用于出口柑桔中氯硝胺残留量的检验。2 抽样和制样2.1 检验批 以不超过1500件为一检验批。 同一检验批的商品应具有相同的特征,如包装、标记、产地、规格、等级等。2.2 抽样数量 批量,件 最低抽样数,件 1~25 1 26~100 5 101~250 10 251~1500 152.3 抽样方法 按2.2规定的抽样件数随机抽取,逐件开启。每件至少取500 g作为原始样品,原始样品的总量不得少于2kg。加封后,标明标记,及时送实验室。2.4 试样制备 将所取原始样品缩分出1kg,取可食部分,经组织捣碎机捣碎,混匀后,均分成两份,装入洁净容器内,作为试样,密封,并标明标记。2.5 试样保存 将试样于—18℃以下冷冻保存。 注:在抽样和制样的操作过程中,必须防止样品受到污染或发生任何残留物的变化。3 测定方法3.1 方法提要 试样与丙酮一起匀浆,用石油醚和二氯甲烷提取,经弗罗里硅土柱净化后,用配有电子俘获检测器的[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url]测定,外标法定量。3.2 试剂和材料 除另有规定外,试剂均为分析纯,水为蒸馏水。3.2.1 丙酮:重蒸馏,收集56℃馏分。3.2.2 石油醚:重蒸馏,收集30~60℃馏分。3.2.3 二氯甲烷:重蒸馏,收集40℃馏分。3.2.4 无水乙醚-石油醚(15+85)。3.2.5 弗罗里硅土:60~100目, 于650℃灼烧4h,冷却后立即转入具玻璃塞的干燥玻璃容器内。使用前于130℃加热至少5h,趁热转入具玻璃塞的干燥玻璃容器内备用。3.2.6 无水硫酸钠:于650℃灼烧4h,冷却后,贮存于具玻璃塞的干燥玻璃容器内备用。3.2.7 氯化钠。3.2.8 氯硝胺标准品:含量≥99.5%。3.2.9 氯硝胺标准溶液:用丙酮将氯硝胺标准品配成0.100mg/mL的储备溶液,根据需要用无水乙醚-石油醚(15+85)配成适当浓度的标准工作溶液。3.3 仪器和设备3.3.1 [url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url]:配有电子俘获检测器。3.3.2 组织捣碎机。3.3.3 移液管:5mL。3.3.4 多功能微量化样品处理仪及配件(或相当装置)。3.3.5 微量注射器:10μL。3.3.6 具塞刻度离心管:5.00mL。3.3.7 微型层析柱:150mm×7mm(id)玻璃柱,带有10mL储液斗。在柱底塞入少量玻璃棉,然后依次加入1.0g弗罗里硅土、0.5g无水硫酸钠,轻轻敲打装实。用前经10mL石油醚淋洗。3.3.8 快速混匀器。3.3.9 离心机: 4000r/min。3.4 测定步骤3.4.1 提取 称取约1g(精确至0.01g)经捣碎、混匀的试样于8 mL离心管中,加入2mL丙酮,快速混匀1min, 3000 r/min离心2min,将上清液转入20 mL离心管中。残渣再用2mL丙酮同上提取一次,合并提取液。于丙酮提取液中加入2mL石油醚和2mL二氯甲烷,快速混匀提取。3000r /min离心2 min,用尖嘴吸管将上层有机相转入另一20mL离心管中。在下层水相中加入0.2g氯化钠并快速混匀至大部分氯化钠溶解后,再用2×2mL二氯甲烷提取两次,每次提取不得少于1min,合并提取液于20mL离心管中。加入1g无水硫酸钠至提取液中,混匀(1min),离心(3000 r/min,2min),脱水后转入另一20 mL磨口离心管中,再用2mL二氯甲烷洗涤硫酸钠,洗涤液并入磨口离心管中。在多功能微量化样品处理仪或相当装置上,于小于等于40℃温度下减压浓缩至干。3.4.2 净化 上述残渣以1.0mL无水乙醚-石油醚(15+85)溶解并移入层析柱(3.3.7)上,待液面降至无水硫酸钠表面时,弃去上述流出液。然后用5mL无水乙醚-石油醚(15+85)洗涤离心管,洗涤液倾入层析柱(3.3.7)上进行洗涤,用具塞刻度离心管收集洗脱液,准确收集5.00mL。混匀后供测定。3.4.3 测定3.4.3.1 色谱条件 a. 毛细管色谱柱:0.5μmSE-30, 20m×0.53mm(id)熔融石英柱; b. 色谱柱温度:180℃; c. 进样口温度:250℃; d. 检测器温度:300℃; e. 载气:氮气,纯度≥99.99%;柱流量:25mL/min;尾吹气流量:40mL/min; f. 进样方式:直接进样,不分流。3.4.3.2 色谱测定 根据样液中氯硝胺残留浓度,选定峰高相近的氯硝胺标准工作溶液。标准工作溶液和样液中氯硝胺的响应值均应在仪器检测线性范围内。标准工作溶液和样液等体积参插进样测定。在上述色谱条件下,氯硝胺的保留时间约为3.5min。3.4.4 空白试验 除不加试样外,按上述测定步骤测定。3.5 结果计算与表述 用色谱数据处理机或按下式计算试样中氯硝胺残留含量:hcVX= ─────hsm式中:X——试样中氯硝胺含量,mg/kg; h——样液中氯硝胺的峰高,mm; hs——标准工作溶液中氯硝胺的峰高,mm; c——标准工作溶液中氯硝胺的浓度,μg/mL; V——样液最终定容的体积,mL; m——称取的试样量,g。 注:计算结果需扣除空白值。4 测定低限和回收率4.1 测定低限 本方法测定低限为0.025mg/kg。4.2 回收率 回收率实验数据:氯硝胺添加浓度在0.025~0.25mg/kg范围内,回收率为81.0%~1094%。━━━━━━━━━━━附加说明:本标准由中华人民共和国国家进出口商品检验局提出。本标准由中华人民共和国湖南进出口商品检验局负责起草。本标准主要起草人袁智能、黄志强、戴华。━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━中华人民共和国国家进出口商品检验局1993-12-28批准 1994-05-01实施
[color=#444444]请问[/color][color=#444444]GB/T5009.84-2003 [/color][color=#444444]食品中硫胺素(维生素[/color][color=#444444]B1[/color][color=#444444])的测定中所用的硫胺素标准品指的是硝酸硫胺素还是盐酸硫胺素?这两种标准品都有的卖。[/color][color=#444444]但就是找不到[/color][color=#444444]“[/color][color=#444444]硫胺素[/color][color=#444444]”[/color]
PONY谱尼测试最新了解到,厚生劳动省医药食品局发布食安发0928第2号:部分修改食品、添加剂等的规格标准(2009年厚生劳动省告示第422号),设定农药氯虫酰胺、氰氟虫腙以及甲基碘在食品中的残留标准。根据此通知,厚生劳动省将如下记修改部分食品、添加剂等的规格标准(昭和34年厚生省告示第370号)。第1 修改的摘要根据食品卫生法(昭和22年法律第233号。以下简称“法”。)第11条第1项的规定,设定农药氯虫酰胺、氰氟虫腙以及甲基碘等在食品中的残留标准。第2 实施• 适用日期由公布之日起开始实施第3 应用须知1、此次,在设定了氯虫酰胺标准值的食品中,桃、西瓜以及香瓜是包括果皮的。2、此次,设定了标准值的氰氟虫腙是指:将氰氟虫腙(E-同分异构体)、氰氟虫腙(Z-同分异构体)以及作为氰氟虫腙代谢物的p-[m-(三氟甲基) 苯甲酰甲基] 苯甲腈换算为氰氟虫腙之后的和。第4 其它以残留标准值(根据“法”设定)以及农药取缔法(昭和23年法律第82号)为依据,在农林水产省将氯虫酰胺、氰氟虫腙以及甲基碘注册为农药。关于氯虫酰胺、氰氟虫腙以及甲基碘的检验法将在日后通知。PONY谱尼测试在农药残留方面具有丰富的经验,可以依据日本肯定列表进行检测。对于这三种农药,企业应该引起重视,PONY谱尼测试将鼎力帮助企业进行检测,顺利出口。
厚生劳动省医药食品局发布食安发0928第2号:部分修改食品、添加剂等的规格标准(2009年厚生劳动省告示第422号),设定农药氯虫酰胺、氰氟虫腙以及甲基碘在食品中的残留标准。根据此通知,厚生劳动省将如下记修改部分食品、添加剂等的规格标准(昭和34年厚生省告示第370号)。第1 修改的摘要根据食品卫生法(昭和22年法律第233号。以下简称“法”。)第11条第1项的规定,设定农药氯虫酰胺、氰氟虫腙以及甲基碘等在食品中的残留标准。第2 实施• 适用日期由公布之日起开始实施第3 应用须知1、此次,在设定了氯虫酰胺标准值的食品中,桃、西瓜以及香瓜是包括果皮的。2、此次,设定了标准值的氰氟虫腙是指:将氰氟虫腙(E-同分异构体)、氰氟虫腙(Z-同分异构体)以及作为氰氟虫腙代谢物的p-[m-(三氟甲基) 苯甲酰甲基] 苯甲腈换算为氰氟虫腙之后的和。第4 其它以残留标准值(根据“法”设定)以及农药取缔法(昭和23年法律第82号)为依据,在农林水产省将氯虫酰胺、氰氟虫腙以及甲基碘注册为农药。关于氯虫酰胺、氰氟虫腙以及甲基碘的检验法将在日后通知。
哪位版友知道N-亚硝胺标准品哪里有卖的?
[b]有机氯农药残留分析方法标准化的研究[/b] 摘要:概述了农药残留分析方法国家标准制订中在方法验证、分析步骤、条件对比试验、干扰及分离度研究、多个实验室质量控制等内容,为方法标准化提供可资借鉴的实例。 关锐词:BHC DDT 农药残留分析方法 国家标准 制订 研究[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=38242]有机氯农药残留分析方法标准化的研究[/url]文献为caj格式。下面是PDF格式的:[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=60733]有机氯农药残留分析方法标准化的研究.pdf[/url]
[color=#333333]食品中磷化铝的标准分析方法[/color]
2010年11月10日,日本厚生劳动省发布食安发1110第1号通知,设定部分农药和添加剂的标准:(1)根据食品卫生法第11条第1项的规定设定农药亚胺唑、氟哇唑以及异丙甲草胺在食品中的残留标准。(2)根据食品卫生法第11条第1项的规定设定苯乙胺、丁胺的使用标准以及成分规格。详细内容见附件。
本人系一名在校研究生,现需要采购多种农药标准品,用于科研。标准品浓度需在1000PPM以上或者固体,有意者请把标准品价格、浓度、含量,发到myou@xmu.edu.cn 或者mh_you@126.com氯氰菊酯、醚菌酯腈菌唑杀螟腈二甲戊乐灵氟虫腈乙草胺异菌脲氟丙菊酯丙溴磷异丙甲草胺丙环唑三氯杀螨醇ddv乙拌磷四氯间二甲苯氟乐灵甲拌磷乐果二嗪哝百菌清甲基毒死蜱七氯杀螟松马拉硫磷环氧七氯硫丹1丁草胺稻瘟灵异狄氏剂环氟菌胺硫丹2乙硫磷联苯菊酯甲氰菊酯三氯杀螨砜三氟氯氰菊酯氯菊酯氟氯氰菊酯氰戊菊酯溴氰菊酯三唑磷丙线磷敌敌畏甲胺磷异吸硫磷甲拌磷治螟磷内吸磷二嗪哝乙拌磷稻瘟净久效磷乐果甲基毒死蜱甲基对硫磷倍硫磷马拉硫磷杀螟松对硫磷甲基异柳磷喹硫磷稻丰散丙溴磷乙硫磷苯硫磷
[align=center][b][/b][/align][align=left][b][color=#ff0000]您好,如果您是代表国联质检团队发帖,建议您跟其他成员做好沟通。已给您留了站内短信,请确认。[/color][/b][/align][align=left][b][color=#ff0000]-----------------[/color][/b][/align][b][color=#ff0000][/color][/b][align=center][b]猪肉制品中9种亚硝胺的检测条件[/b][/align][align=center][b]国联质检食品事业部——任阳阳[/b][/align]基于国标GB5009.26-2016中食品中N亚硝胺类化合物的测定中第一法[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]-质谱法仅能测定出两种N亚硝胺NDMA和NDEA,本文对分析方法做出相应的优化,能够有效的分离出肉制品中9种亚硝胺。1.标准溶液的制备将9种N亚硝胺的标准储备液(1000μg/mL)用二氯甲烷稀释成0.01μg/mL、0.02μg/mL、0.05μg/mL、0.1μg/mL、0.2μg/mL、0.5μg/mL进行上机分析。2..测定条件仪器型号:岛津GCMS-QP2020GC条件:色谱柱:WAX柱,30m×0.25μm×0.25μm,柱温:40℃,进样口温度:230℃,不分流,柱流量:1.5mL/min程序升温条件:40℃,3min;10℃/min,升至110℃;15℃/min,升至200℃;40℃/min,升至240℃,保持10minMS条件:EI离子源温度:230℃,接口温度:240℃,溶剂延迟4min,扫描范围:30m/z ~ 450m/z ,采用SCAN模式确定目标组分,之后采用SIM模式。3. 样品前处理称取20.0g样品于锥形瓶中,加入5.0g无水硫酸钠,摇匀后加入80.0mL二氯甲烷溶液,超声萃取15min,收集上清液,再重复提取2次,合并3次提取液,用无水硫酸钠进行脱水,之后用35℃进行旋转蒸发至近干,用二氯甲烷溶解并定容至3.0mL,经0.22μm滤头过滤后进行上机分析。4.实验结果 9种N亚硝胺得到有效的分离,见图1,9种N亚硝胺的相关系数能够达到R=0.991~0.999之间,且能达到很好的分离,分离度R>1.5。[table][tr][td][align=center]化合物名称[/align][/td][td][align=center]目标离子[/align][/td][td][align=center]保留时间[/align][/td][td][align=center]参考离子[/align][/td][/tr][tr][td][align=center]N-二甲基亚硝胺[/align][/td][td][align=center]74[/align][/td][td][align=center]8.41[/align][/td][td][align=center]42.00-43.00[/align][/td][/tr][tr][td][align=center]N-亚硝基甲乙胺[/align][/td][td][align=center]88[/align][/td][td][align=center]9.15[/align][/td][td][align=center]42.00-43.00[/align][/td][/tr][tr][td][align=center]N-亚硝基二乙胺[/align][/td][td][align=center]102[/align][/td][td][align=center]9.59[/align][/td][td][align=center]44.00-42.00[/align][/td][/tr][tr][td][align=center]N-亚硝基二丙胺[/align][/td][td][align=center]70[/align][/td][td][align=center]11.48[/align][/td][td][align=center]43.00-42.00[/align][/td][/tr][tr][td][align=center]N-亚硝基二正丁胺[/align][/td][td][align=center]84[/align][/td][td][align=center]13.43[/align][/td][td][align=center]57.00-41.00[/align][/td][/tr][tr][td][align=center] N-亚硝基哌啶[/align][/td][td][align=center]42[/align][/td][td][align=center]13.59[/align][/td][td][align=center]114.00-55.00[/align][/td][/tr][tr][td][align=center] N-亚硝基吡咯烷[/align][/td][td][align=center]100[/align][/td][td][align=center]13.87[/align][/td][td][align=center]41.00-42.00[/align][/td][/tr][tr][td][align=center]N-亚硝基吗啉[/align][/td][td][align=center]56[/align][/td][td][align=center]14.38[/align][/td][td][align=center]86.00-116.00[/align][/td][/tr][tr][td][align=center]二苯胺[/align][/td][td][align=center]169[/align][/td][td][align=center]19.36[/align][/td][td][align=center]168.00-167.00[/align][/td][/tr][/table]0.5μg/ml标准溶液图谱5.总结经条件优化后,此方法能够很好的分离出肉制品中的9种亚硝胺类物质。[align=right][b]国联质检食品事业部——任阳阳[/b][/align]

[size=16px] 食品安全综合分析仪主要标准是什么 食品安全综合分析仪的主要标准包括以下几个方面: 检测项目:食品安全综合分析仪的检测项目涵盖了多个方面,主要包括营养成分、添加剂、农药残留、重金属和微生物等。这些项目的检测是确保食品安全的重要手段,可以全面了解食品的质量和安全状况。 检测精度和效率:食品安全综合分析仪需要具备高检测精度和效率,以确保测试结果的准确性和可靠性。为实现这一目标,需要选择可实现自校的仪器,并设置合理的自校时间间隔,按期校对仪器。此外,在仪器操作前也需进行校对,以及定期对仪器进行检修与保养,降低检测出现误差的可能性。 农残检测标准:食品安全综合分析仪的农残检测标准通常是由国家食品药品监管总局制定的。对于农药残留限量,一般采用“最大残留限量(MRL)”作为标准。这个限度是由卫生、环境和经济等多方面考虑制定的。在农残检测过程中,除了检测仪器的准确性外,样品的采样和制备也是非常重要的。 国家和行业标准:食品安全综合分析仪的检测方法应符合国家和行业标准。例如,对于营养成分、添加剂、重金属和微生物等的检测,应采用国家规定的检测方法,确保数据的准确性和可靠性。 仪器性能:食品安全综合分析仪应具备良好的稳定性、重复性和灵敏度等性能指标,以确保在不同环境和条件下都能获得准确的检测结果。 综上所述,食品安全综合分析仪的主要标准包括检测项目、检测精度和效率、农残检测标准、国家和行业标准以及仪器性能等方面。这些标准共同构成了保障食品安全的重要技术支持。[img=,690,690]https://ng1.17img.cn/bbsfiles/images/2024/02/202402200937557726_9841_6098850_3.jpg!w690x690.jpg[/img][/size]
N-二甲基亚硝胺是一种潜在致癌物,对其在食品中的检测和研究,以保障食品安全非常重要,本文建立了液相色谱串联质谱水蒸气蒸馏法测定食品中N-二甲基亚硝胺,样品通过水蒸气蒸馏装置,把分析物蒸馏出来,在通过和二氯甲烷液液萃取,减压浓缩后,采用[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url]质检测。该方法的定量限1.0μg/kg,仪器在进样体积5μL、1.0~100.0μg/L范围内,线性相关系数大于0.995,在1倍、3倍和10倍定量限添加水平,添加回收率在80%-92%,相对标准偏差(RSD)小于15%,满足检测标准的要求。
大家好 请教个问题 我做液相色谱分析标准曲线,用邻氯苯胺盐酸盐做标准曲线,但是样品是邻氯苯胺硫酸盐,怎么计算邻氯苯胺的含量?谢谢
食品安全国家标准GB2762-2012《食品中污染物限量》,将于2013年6月1日实施,本标准与GB 2762—2005相比,主要变化如下: ——修改了标准名称; ——增加了可食用部分的定义; ——增加了应用原则; ——取消了硒、铝、氟的限量规定; ——增加了锡、镍、3-氯-1,2-丙二醇及硝酸盐的限量规定; ——将N-亚硝胺限量指标由N-二甲基亚硝胺和N-二甲基乙硝胺调整为N-二甲基亚硝胺,并将N-亚硝胺限量指标名称修改为N-二甲基亚硝胺; ——增加了附录 A; ——稀土限量指标按原GB 2762—2005执行。与我们相关的就是,其中明确规定,铅的限量:酒类(蒸馏酒、黄酒除外) 0.2mg/kg ;蒸馏酒、黄酒 0.5mg/kg取消了酒类中无机砷的限量大家来讨论下在自己的领域中的变化!
卤代酚卤代酚是含酚的卤代化合物,对革兰氏阳性菌有强杀菌作用,用在化妆品中的卤代酚有六氯酚 等多种化合物。这类化合物通常是光敏物质。我国化妆品卫生标准规定为限用物质,限用量见表2-3-17。表 2-3-17 化妆品卫生标准中卤代酚的限用量品名 序号 最大使用量(%) 溴氯双酚 4-4 0.1 双氯酚 4-7 0.2 2,4-二氯二甲苯酚 4-8 0.1 三氯生 4-21 0.3 六氯酚 4-24 0.1 4-溴邻甲苯酚 4-31 0.3 苄氯酚 4-42 0.2 4-氯2-甲苯酚 4-55 0.2 4-氯3,5-二甲苯酚 4-56 0.2 * 指化妆品卫生标准(GB7916-87)中的序号,4-42即表4的序号42(一)薄层色谱法(TLC)1 适用范围本方法适用于化妆品中六氯酚,双二氯酚硫醚、二氯酚和三溴水杨酞替苯胺的定性。2 原理样品经预处理后,样液中的卤代酚用的薄层色谱法进行分离、呈色,然后与标准斑点比较,进行定性。3 试剂3.1 乙醇:分析纯。3.2 己烷:分析纯。 3.3 丙酮:分析纯。3.4 无水硫酸钠:分析纯。3. 5硫酸(lmol/L)。3.6 六氯酚标准溶液(1):准确称取用苯重结晶的六氯酚50.0mg,加丙酮溶解后移入50ml容量瓶中并定容至刻度,避光保存。此溶液1ml含1.0mg六氯酚。3.7 双二氯酚硫醚(2):准确称取用苯重结晶的双二氯酚硫醚50.omg,用丙酮溶解,移入50ml容量瓶中并定容至刻度,避光保存。此溶液lml含1.0mg二氯酚硫醚。3.8双氯酚标准溶液(3):准确称取用甲苯重结晶的双氯酚50.0mg,用丙酮溶解,移入50ml容量瓶中,定容至刻度。此溶液1.0ml含1.0mg二氯酚,避光保存。3.9三溴水杨酞替苯胺(4):准确称取用丙酮重结晶的三溴水杨酞替苯胺50.0mg,用丙酮溶解,移入50ml容量瓶中并定容至刻度。此溶液1.0ml含1.0mg三溴水杨酞替苯胺,避光保存。3.10 乙醇一己烷(1 9)。3.ll离子交换纤维素(5):将DEAE(二乙基氨基乙醇)纤维素,(交换量约0.9meg/g),浸泡于50倍量的0.lmol/L的盐酸中,用玻璃漏斗过滤,用20倍量的丙酮,30倍量的0.lmo1/L氢氧化钠溶液淋洗至OH-型后,用水洗成中性,再用20倍量的丙酮淋洗,弃去丙酮。空气中干燥。保存在乙醇十己烷(l+9)溶液中。3.12 硅胶:薄层用硅胶中加有荧光剂。3.13碱性氧化铝。3.14展开剂:石油醚 冰乙酸(89 12)3.15显色剂。3.15.1 浓氨水。3.15.2 2%4-氨基安替比林溶液:称取2g4-氨基安替比林用乙醇溶解稀释至100ml。3.15.3 8%铁氰化钾溶液(K3[Fe(CN)6])。3.15.4 2%三氯化铁溶液(FeCl36H20):称取2g三氯化铁用乙醇溶解稀释至100ml。3.15.5 2%铁氰化钾溶液。3.16 盐酸 丙酮溶液:9.5ml盐酸加丙酮至100ml(临用前配制)。4 仪器 4.1 层析柱:、内径10mm、高200mm的具塞玻璃管的下端熔接玻璃过滤器或塞有玻璃棉,4.2 紫外灯,具有8W功率,254nm波长。4.3离子交换柱(6):将离子交换纤维素用乙醇 已烷(3.11)配成混悬液,:用湿式填充法缓慢倾入层析柱中,以防止产生气泡,填充高度80mm。5 分析步骤5.1样品预处理(7)(8)称取含卤化酚0.5mg的样品(扑粉,除臭砂芯、香波约10g,膏霜约0.5g),置于100ml玻璃瓶中,连接好回流冷凝器:加50ml乙醇 已烷(1 9)溶液,2ml 1mol/L硫酸,于水浴上加热3min,冷却后用3号玻璃砂芯漏斗过滤,用乙醇 己烷(1 9)溶液5ml洗沉淀,滤液移入分液漏斗中静置分层。取己烷层用10ml水洗涤,无水硫酸钠脱水后以0.5ml/min的流速注入离子交换柱(10)。用50ml己烷洗涤。去除油脂等干扰物质,弃去淋洗液,依次用10ml丙酮、2ml丙酮 盐酸溶液(3.16),20ml丙酮洗脱(11)。溶出液在水溶上加热蒸去有机溶媒,加5ml乙醇,加热使盐酸挥发,重复此操作2次。残渣加2.0ml丙酮溶解,作为样品待测溶液。5.2 制备薄层板5.2.1硅胶薄层板:硅胶30g,加水约65ml,搅拌均匀,涂布成厚度0.25~0.3mm的薄层板,105~l10℃干燥30min,置干燥器中保存。 5.2.2含硝酸银的氧化铝薄层板:0.12g AgNO3,加少量水溶解,加30ml乙醇、20g氧化铝,调成浆状物,涂布厚度为0.25~O.3mm的薄层板,空气中干燥、于干燥器中避光保存。5.3 点样距薄层板底边2cm处将5~20μl待测溶液从左到右点样(12),两点间隔约1cm,薄员板.的右边点2μl标准溶液,空气中干燥。5.4 展开取适量展开剂(3.14)倾人展开槽中,将薄层板放入展开剂中,待溶剂上升约10cm,取出薄层板,空气中干燥。5.5显色(13)在薄层板上顺序喷雾显色剂3.15.1~3.15.3或3.15.4~3.15.5,六氯酚在显色剂3.15.1~3.15.3中为红色,在3.15.4~3.15.5中为蓝色斑点。二氯酚硫醚和三溴水杨酞替苯胺在3.15.1~3.15.5中为紫色,在3.15.4~3.15.5中呈现蓝色斑点。用加荧光剂的硅胶薄层板测定时,各种卤代酚在紫外线照射下,在各自的Rf 值位置上以荧光为背景呈现出暗黑色的斑点。
[color=#444444]用高效液相色谱分析生物胺标准品,梯度,温度,衍生方法等条件都用的一样。流动相是乙腈和超纯水。怎么跑出来的图差别这么大。求助大家,谢谢。两张图不一个时间[/color][color=#444444][img=,690,517]https://ng1.17img.cn/bbsfiles/images/2019/05/201905201035452605_6441_1676638_3.png!w690x517.jpg[/img][img=,690,517]https://ng1.17img.cn/bbsfiles/images/2019/05/201905201035471785_2554_1676638_3.png!w690x517.jpg[/img][/color]
SH409石油产品硫氯测定仪应用微库仑分析技术,采用氧化法将样品通过裂解炉氧化为可滴定离子,在滴定池中滴定,根据电解滴定过程中所消耗的电量,依据法拉第定律,计算出样品中硫或氯的含量。广泛应用于检测液体、固体或气体样品中的硫氯含量。微机硫氯分析仪符合SH/T 0253,GB/T 11061,SH/T 0222,SH/T 0254标准。 仪器具有性能稳定可靠,操作简便,分析精度高,重复性好等特点。[b]仪器执行标准:[/b]SH/T 0253-92 轻质石油产品中总硫含量测定法(电量法)标准 GB/T 11061-1997 天然气中总硫的测定-氧化微库仑法 标准 SH/T 0222-1992 液化石油气中总硫含量测定法(电量法)标准SH/T 0254、 ASTM D3120、ASTM D3246等[b]技术参数:[/b]1.样品种类:液体、气体和固体2.测量范围:S:0.1 ~10000 ng/μl—百分含量,标称下限:0.1ppm CL:0.1 ~10000 ng/μl—百分含量,标称下限:0.1ppm3、仪器准确度:浓度为0.1 ~1 ng/μl,样品绝对误差:≤0.1浓度为1.0 ~1 0ng/μl,样品相对误差:≤10%浓度为为1 0ng/μl以上的样品,样品相对误差:≤5%4、仪器重复性误差:A、浓度为0.1 ~1 ng/μl,样品绝对误差:≤50%B、浓度为1.0 ~1 0ng/μl,样品相对误差:≤10%C、浓度为为1 0ng/μl以上的样品,样品相对误差:≤5%5.测量精度:
1 绿色食品添加剂使用准则中华人民共和国农业行业标准NY/T 392--2000绿色食品添加剂使用准则,4 .2 .7规定:“在任何情况下,绿色食品中不得使用下列食品添加剂”。其中包括生产绿色食品粉条不使用食品漂白剂…硫磺。(硫磺对人体的不良作用……)不得使用硫磺,但在生产控制与检测监督工作中,怎样才能检查出所检粉条中是否含有并使用过硫磺,对申报产品是否进行绿色食品认可的重要基础之一。但恰恰在实际工作中因缺乏相应的检测方法,而使这一工作陷人困境。2 生产控制与检测监督工作存在的问题例如,有一甘薯淀粉制品…甘薯粉条产品,申报绿色食品产品认可,绿色食品授权检测中心对其进行质量品质检测与评价工作,依据NY/T 392-200绿色食品添加剂使用准则规定,硫磺不得使用于此类产品,但该准则并未规定禁止使用亚硫酸盐等添加剂 而且,目前可以采用的国家检测方法标准主要是GB/T5009 .34一 1996食品中亚硫酸盐的测定方法 其他产品中亚硫酸盐的测定方法还有多种,但这些方法都共同存在着一个问题,即最终结果对硫磺、亚硫酸盐等添加剂无法予以区别,都是在最后结果时通过计算以二氧化硫或亚硫酸盐计。如此以来,当依据现行国家标准检测方法进行粉条产品含硫磺检测时,会出现这样一种无法结论的情况,如该样品未检测出二氧化硫,则该样品可下结论未使用硫磺 但如检测出二氧化硫时,该样品却无法下结论是否是硫磺使用所致,即可能是使用硫磺所致,也可能不是硫磺而是其他亚硫酸盐所致。在这种情况下检测中心检测后无法明确进行被检样品是否使用硫磺的判定。至此,绿色食品申报程序与检测工作过程因检测结果无法下结论而中断。也就是绿色食品添加剂禁用的漂白剂硫磺,因无明确专一的检测方法讲行监督控制.形成限量标准禁用与检测方法标准无法明确检测之间的矛盾。没有监督的禁用本质上是无效的,在实际工作中会出现2个极端情况:(l)严格要求时,实际是只要检出硫元素存在,结果就可以换算为二氧化硫,此时,因不能判明是否是硫磺所致,粉条产品就不能判定未使用硫磺,即该产品不能成为绿色食品 (2)第一种检验结论方式对许多未使用硫磺的粉条类产品显失公平,但如果不这样判定,当检出样品中硫时,是不是加有硫碘检测中心和认证管理机构只有相信申报受检者自己的承诺保证了,如此,禁止使用的规定本质上成为一种无效的要求。同样的问题在绿色食品添加剂标准中仍然存在,如明矾禁止使用,但现行的标准方法亦不能检测粉条中的明矾,而是检测铝离子 检铝离子就无法判断铝是粉条中本身具有的,还是工艺过程中加人的明矾,如此,又同硫磺的检测一样进人一个不能下结论状态,从而使检测方法和限量标准之间存在矛盾。无法落实检查的要求等于没做要求,这种要求只会给实际工作造成麻烦,使检测中心无所适从,导致检测与判定工作无法作出科学明确的结论。3 建议标准制定单位对标准进行进一步完善,如果提出的禁用指标在现条件下还无法从检测技术上予以解决,到不如将此类指标给出最低限量指标要求(如铝),指标高于背景值即可,如此,似更为科学实用。象粉条中使用硫磺的问题,现阶段禁止使用一切含硫化合物进入绿色食品粉条加工过程,才能实现绿色食品二氧化硫不得检出的要求,绿色食品申报检测工作中部分粉条生产企业的产品证明是可以做到的.
我们做二氯四氟乙氧基苯胺,是用的液相色谱法,可惜没有合适的标准品,
问:请问乙酰氯的GC分析方法极其标准?我们是是化工企业,生产高纯乙酰氯,分析量很大,我们目前用GC-FID毛细管柱子,没有标准品,含量大约99。5%,请问这样分析的缺陷,用HPLC行不行?假如有其他微量杂质,是否还要用质谱仪?
[b]我国香蕉农药残留标准与国外标准的比较分析[/b][i]李玉萍,方佳,梁伟红,董定超[/i] 1 我国香蕉农药残留国家标准现状 在我国,水果农药残留国家标准开始于20世纪70年代末,起步较晚。经过20余年的努力,取得了可喜的成绩,截至到1999年9月底,我国已发布18个与水果有关的农药最大残留限量强制性国家标准,涉及50种农药。2005年1月我国又颁布新的国家标准《食品中农药最大残留限量》(GB2763-2005),将原先的18个标准代替。新标准规定了水果中70种农药的最大残留限量,其中仅有腈苯唑、咪鲜胺、丙环唑、戊唑醇、噻菌灵5种农药对香蕉的残留限量值作了专门规定;溴氰菊酯、乙烯利、代森锰锌3种农药对皮不可食的热带与亚热带水果作了专门规定;乙酰甲胺磷等13种农药对所有水果规定了统一的最大残留限量;多菌灵除对梨果类水果、葡萄作出专门规定外,对其他水果也都规定了相同的残留限量值。此外,在我国现行的《农产品安全质量无公害水果安全要求》(GB18406.2-2001)强制性国家标准中,也规定了无公害水果中22 种农药的最大残留限量值。综合上述国家标准,直接或间接涉及香蕉农药残留最大限量指标共33项,涉及农药33种。其中杀虫剂23种,杀菌剂8种,除草剂1种,植物生长调节剂1种。 2 国际及国外先进国家香蕉农药残留最大限量标准现状 食品法典委员会(Codex Alimentarius Commission,简称CAC)是世界上唯一的政府间协调国际食品标准法规的国际组织,成立于1962年,至今已拥有163个成员国。到目前为止,CAC 已规定了香蕉、葡萄、苹果等63种(类)水果及其产出品的最大农药残留限量,涉及农药100余种,其中有24种农药对香蕉作了专门规定(其中除草剂2种,杀虫剂9种,杀菌剂12种,杀螨剂1种)。 欧盟制定的香蕉农药最大残留限量标准涵盖180种农药。其中除草剂39种,杀虫剂73种,杀菌剂49种,杀螨剂9种,熏蒸剂2种,植物生长调节剂8种。 美国在美国联邦法规(CFR)第40篇第180部分对香蕉规定了详细的农药最大残留限量,涵盖农药47种,有些农药还分别对香蕉全果、果肉进行了规定。在47种农药中,包括除草剂10种,杀虫剂11种,杀菌剂23种,杀螨剂1种,熏蒸剂1种,植物生长调节剂1 种。 日本有91种农药对香蕉的农药最大残留限量进行了规定。其中除草剂16种,杀虫剂40种,杀菌剂27种,植物生长调节剂2种,薰蒸剂2种,杀螨剂4种。 3 我国与国外香蕉农药残留指标比对分 析3.1 我国香蕉农药残留指标与 CAC 比对分析 目前我国香蕉农药残留指标33项,涉及33种农药,CAC 香蕉农药残留指标24项,涉及24种农药。我国香蕉农药残留指标与CAC 相比,有6种农药与 CAC都有限量要求,其中指标相同的有腈苯唑、丙环唑、戊唑醇、噻菌灵4种,占我国香蕉指标的12.1%,占 CAC指标的16.7%;比 CAC 严的有克百威1种;比 CAC 宽的有百菌清1种。有27种农药我国有限量要求而CAC 却没有,有18种农药 CAC 有限量要求而我国却没有。
自“齐二药”事件以来,药监部门对药品原辅料把关日趋严格,要求原辅料全检。三乙醇胺(分析纯)是一种常用的乳化剂,质量标准一直查不到,后来才知道是企业自定标准,问企业索取,说是内部机密文件,不愿意提供。想我们为保证人民用药安全多此一举,做原来根本不做的检验项目,原料生产厂家竟然不愿意配合,真是可气!也许我们医院制剂是小客户,不值得一提吧。不得已只好到网上求助各位专业人士了!多谢大家支持!
请教各位老师: 在用气相色谱做有机溶剂残留时,质量标准中标准溶液配制方法如下:取无水乙醇约50mg,乙酸乙酯10mg,二氯甲烷60mg,均精密称定,分别置于500ml的容量瓶中,加水溶解并稀释至刻度,摇匀,即可。这里面我有以下几个疑问:1 在试验操作时,称取无水乙醇50mg,置于500ml容量瓶中,由于无水乙醇极易挥发,如果直接称到500ml的容量瓶中的话,已称好的无水乙醇不是会容易挥发吗,有可能实际由50mg挥发到40mg?这样标准品浓度不是降低了吗?误差不是很大?有没有必要先在容量瓶中加入适量的溶剂,再加入挥发性有机溶剂标准品?平时大家做有机溶剂残留,有机溶剂大多易挥发,如何保证已称好的标准品不挥发损失呢?2 气相有机溶剂残留有机溶剂标准品用分析纯的可以吗?如果分析纯纯度是大于99%,在计算标准品浓度时是按99%还是100%纯度计算?另外分析纯含有的杂质产生的杂质峰怎么办?3 气相配制标准品或样品溶液时,一般用什么溶剂来配制,是只要对标准品与样品溶解性好的溶剂吗?为什么经常有人用DMF(NN-二甲基甲酰胺),说是万能溶剂,也有用DMSO或水的,一般如何选择?
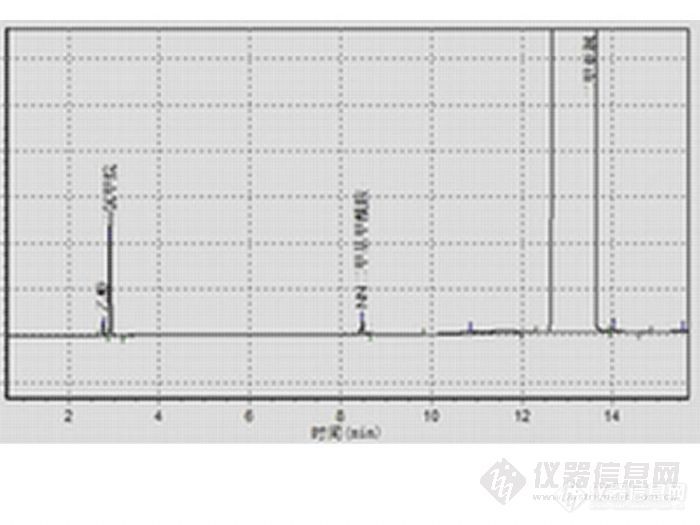
顶空毛细管柱气相色谱法分析测定药品中残留溶剂http://ng1.17img.cn/bbsfiles/images/2011/05/201105211013_295254_2242538_3.jpg 摘要 目前国家对药品中残留溶剂的检测还没有一个统一的国家标准,在关于有机溶剂残留量的指导原则中将二氯甲烷、DMF列为第二类必须控制的毒性试剂,将乙醇、丙酮列为第三类低毒性的溶剂。建立一种快速测定药品中残留溶剂的分析方法很有必要。为此南京科捷采用DK-300A顶空进样器取样,气相色谱毛细管柱分离,分析测定药品中常见的残留溶剂(乙醇、二氯甲烷、NN二甲基甲酰胺、二甲亚枫)。结合相关指标,本实验将二氯甲烷的限量规定为 600ppm,DMF的限量规定为 880ppm,乙醇及丙酮的限量规定为 5000ppm。实验结果表明:顶空毛细管柱气相色谱法快速、简便、准确是测定药品中残留溶剂较为理想的方法。关键词 乙醇 二氯甲烷 NN二甲基甲酰胺 顶空进样 毛细管气相色谱 药品 有机溶剂残留1.药品中乙醇、二氯甲烷、NN二甲基甲酰胺色谱图峰序1. 乙醇2.二氯甲烷3.NN二甲基甲酰胺4.二甲亚枫(溶剂)2.本方法应用范围在合成原料药,辅料或制剂生产的过程中使用或产生的挥发性有机化学物质,它们在实际的生产中未能被完全地清除。本方法可应用在药品中残留溶剂的检测中。近年来,药品中残留有机溶剂的毒性和致癌作用日益引起各方面的重视。药品中残留有机溶剂于1997年被美国FDA列为药品监控项目。我国药品中残留有机溶剂检测也越来越受到有关方面的重视。本文初步研究探索采用顶空毛细管柱气相色谱法分析药品中残留挥发性有机溶剂。顶空气相色谱法只将挥发和半挥发的组份引入柱子,可避免非挥发性的物质对系统的污染,样品前处理简便,分析效率高。结果表明,本方法快速、准确、重现性好。3.仪器及试剂配置色谱仪器配置色谱柱及试剂GC5890(FID检测器)毛细管专用柱30*0.32.*0.5乙醇、二氯甲烷各一瓶顶空进样器:DK-300ANN二甲基甲酰胺1瓶N2000色谱工作站(电脑自备1台)二甲亚枫1瓶氢氮氧一体发生器或钢瓶气各一瓶顶空压盖机1台顶空瓶20ml (带塞) 50只
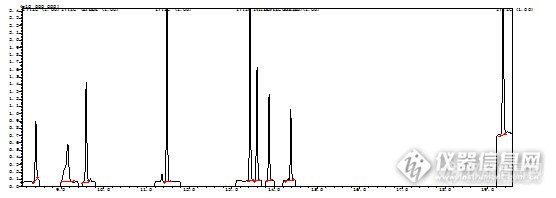
[align=center][b][b]猪肉制品中9种亚硝胺的检测条件[/b][/b][/align][align=center][b][b]作者:任阳阳[/b][/b][/align] 基于国标GB5009.26-2016中食品中N亚硝胺类化合物的测定中第一法[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]-质谱法仅能测定出两种N亚硝胺NDMA和NDEA,本文对分析方法做出相应的优化,能够有效的分离出肉制品中9种亚硝胺。1.标准溶液的制备将9种N亚硝胺的标准储备液(1000μg/mL)用二氯甲烷稀释成0.01μg/mL、0.02μg/mL、0.05μg/mL、0.1μg/mL、0.2μg/mL、0.5μg/mL进行上机分析。2.测定条件仪器型号:岛津GCMS-QP2020GC条件:色谱柱:WAX柱,30m×0.25μm×0.25μm,柱温:40℃,进样口温度:230℃,不分流,柱流量:1.5mL/min程序升温条件:40℃,3min;10℃/min,升至110℃;15℃/min,升至200℃;40℃/min,升至240℃,保持10minMS条件:EI离子源温度:230℃,接口温度:240℃,溶剂延迟4min,扫描范围:30m/z ~ 450m/z ,采用SCAN模式确定目标组分,之后采用SIM模式。3.样品前处理称取20.0g样品于锥形瓶中,加入5.0g无水硫酸钠,摇匀后加入80.0mL二氯甲烷溶液,超声萃取15min,收集上清液,再重复提取2次,合并3次提取液,用无水硫酸钠进行脱水,之后用35℃进行旋转蒸发至近干,用二氯甲烷溶解并定容至3.0mL,经0.22μm滤头过滤后进行上机分析。4.实验结果 9种N亚硝胺得到有效的分离,见图1,9种N亚硝胺的相关系数能够达到R=0.991~0.999之间,且能达到很好的分离,分离度R>1.5。[align=left]化工物理检测框[/align][align=center][table][tr][td][align=center][b]化合物名称[/b][/align][/td][td][align=center][b]目标离子[/b][/align][/td][td][align=center][b]保留时间[/b][/align][/td][td][align=center][b]参考离子[/b][/align][/td][/tr][tr][td][align=center]N-二甲基亚硝胺[/align][/td][td][align=center]74[/align][/td][td][align=center]8.41[/align][/td][td][align=center]42.00-43.00[/align][/td][/tr][tr][td][align=center]N-亚硝基甲乙胺[/align][/td][td][align=center]88[/align][/td][td][align=center]9.15[/align][/td][td][align=center]42.00-43.00[/align][/td][/tr][tr][td][align=center]N-亚硝基二乙胺[/align][/td][td][align=center]102[/align][/td][td][align=center]9.59[/align][/td][td][align=center]44.00-42.00[/align][/td][/tr][tr][td][align=center]N-亚硝基二丙胺[/align][/td][td][align=center]70[/align][/td][td][align=center]11.48[/align][/td][td][align=center]43.00-42.00[/align][/td][/tr][tr][td][align=center]N-亚硝基二正丁胺[/align][/td][td][align=center]84[/align][/td][td][align=center]13.43[/align][/td][td][align=center]57.00-41.00[/align][/td][/tr][tr][td][align=center] N-亚硝基哌啶[/align][/td][td][align=center]42[/align][/td][td][align=center]13.59[/align][/td][td][align=center]114.00-55.00[/align][/td][/tr][tr][td][align=center] N-亚硝基吡咯烷[/align][/td][td][align=center]100[/align][/td][td][align=center]13.87[/align][/td][td][align=center]41.00-42.00[/align][/td][/tr][tr][td][align=center]N-亚硝基吗啉[/align][/td][td][align=center]56[/align][/td][td][align=center]14.38[/align][/td][td][align=center]86.00-116.00[/align][/td][/tr][tr][td][align=center]二苯胺[/align][/td][td][align=center]169[/align][/td][td][align=center]19.36[/align][/td][td][align=center]168.00-167.00[/align][/td][/tr][/table][/align][align=left]0.5μg/ml标准溶液图谱[/align][align=left][img=,556,198]http://ng1.17img.cn/bbsfiles/images/2017/07/201707221434_01_2904018_3.jpg[/img][/align]5.总结 经条件优化后,此方法能够很好的分离出肉制品中的9种亚硝胺类物质。
化验分析硅石标准品,怎样测铝的含量,标准品如何处理?
[b]SRM 方法建立[/b]我们使用了Thermo Scientific TSQTM 8000 [url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]GC-MS[/color][/url]/MS 软件套件中的AutoSRM 软件进行了三重四极杆质谱方法的建立,且并未对AutoSRM 生成的方法进行任何手动修改。一个装有待分析亚硝胺化合物标准品溶液的自动进样器样品瓶专供AutoSRM 程序使用。AutoSRM 程序自动进行以下三个步骤:1. 首先对标准品溶液进行全扫描分析(图1.)。从全扫中得到的信号最强的离子将被作为一级离子。2. 对上一步确定的一级离子(母离子)进行二级离子(子离子)谱图获取(可以根据分析需求设定一级离子的个数)。找出每个一级离子产生的信号最强的二级离子(可以手动选择最感兴趣的一级离子进行进一步优化)。[img=,1009,623]https://i5.antpedia.com/attachments/att/image/20200518/1589800505412622.jpg[/img]表1. AutoSRM 生成的SRM 方法设置3. 对所有化合物的选定的母离子/ 子离子对进行碰撞能的优化,以获得最大化合物响应及最佳方法灵敏度(图2)。AutoSRM 程序能够根据需要从一个标准品样品瓶启动,完成所需的进样次数。表1 就是由AutoSRM 自动生成的SRM 离子对表格。该表同时还显示了TSQ8000 [url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]GC-MS[/color][/url]/MS 在Timed-SRM 模式下、在化合物洗脱时间左右用一个60 秒的短采集窗口进行采集的SRM 采集方法。无需对扫描时段进行任何其他的设置,或者说如果需要在某化合物的洗脱时间之外对其进行监测,则需要手动添加该化合物。[b]样品测定[/b]在大量各种可能的亚硝胺化合物之中,本方法涵盖了那些被报道与发芽麦芽干燥的过程相关的亚硝胺化合物。被分析的样品包括未添加标样的麦芽啤酒样品,以及作为空白样的4%乙醇。如需对其他食物基质进行分析,其它化合物可以随时参照前述AutoSRM 方法建立的步骤添加至本方法中。[b]实验结果[/b]本方法中包含的亚硝胺类化合物的色谱呈现了较快的流出,从7.87 的NDMA 到12.47 分,能够实现较短的循环时间并提高样品通量。图3 显示了用校准曲线中的最低浓度--1 ppb 的样品得到的峰强度。从图中可见NDMA 检测的信噪比依然很好。[img]https://i5.antpedia.com/attachments/att/image/20200518/1589800506229376.jpg[/img]图1. AutoSRM 对NDMA 从EI 全扫谱图中进行一级离子选择[img]https://i5.antpedia.com/attachments/att/image/20200518/1589800506329379.jpg[/img]图2. AutoSRM 对所有亚硝胺一级离子进行碰撞能优化[img=,611,468]https://i5.antpedia.com/attachments/att/image/20200518/1589800507403482.jpg[/img]图 3. 浓度为1ppb 的标准品混合物的色谱图
本人需要TS-Amine-011A标准的具体内容如果能够提供乙二胺纯度的其它气相色谱分析方法也可以,谢谢最后顺便求一下乙二胺水分的分析方法,什么方法都可以,不一定非得是气相的,拜谢!~~~http://simg.instrument.com.cn/bbs/images/brow/em09506.gif
食品中甲胺磷和乙酰甲胺磷农药残留量的测定方法1.适用范围本方法适用于谷物、蔬菜和植物油中甲胺磷和乙酰甲胺磷的残留量分析,其最小检出限分别为7.79×10-12g和1.79×10-11g。2.原理概要含有机磷的样品在富氢焰上燃烧,以HPO碎片的形式,放射出波长526nm的特征光,这种特征光通过滤光片选择后,由光电倍增管接收,转换成电信号,经微电流放大器放大后,被记录下来,样品的峰高与标准品的峰高相比,计算出样品相当的含量。3.主要试剂和仪器3.1.主要试剂丙酮;二氯甲烷:重蒸;无水硫酸钠;活性炭:用3mol/L盐酸浸泡过夜,抽滤,用水洗至中性,在120℃下烘干备用;甲胺磷(methamidophos):≥99%;乙酰甲胺磷(acephate):≥99%;甲胺磷和乙酰甲胺磷标准溶液的配制:分别准确称取甲胺磷和乙酰甲胺磷的标准品,用丙酮分别制成0.1mg/mL的标准储备液。使用时用丙酮稀释配制成单一品种的标准使用液(1mg/mL)和混合标准工作液(每个品种浓度为1mg/mL)。贮藏于冰箱中。3.2.仪器气相色谱仪:具有火焰光度检测器;电动振荡器;K-D浓缩器或旋转蒸发器;离心机。4.试样的制备取谷物实验样品经粉碎机粉碎,过20目筛后,制成谷物试样。取蔬菜实验样品洗净,晾干,去掉非食部分后剁碎或经组织捣碎机捣碎,制成蔬菜试样。5.过程简述5.1.提取和净化蔬菜:称取蔬菜试样10g,精确至0.001g,用无水硫酸钠(因蔬菜含水量不同而加入量不同,约50~80g)研磨呈干粉状,倒入具塞锥形瓶中,加入0.2~0.4g活性炭(根据蔬菜色素含量)及80mL丙酮,振摇0.5h,抽滤,滤液浓缩定容至5mL,待气相色谱分析。谷物:称取谷物试样10g,精确至0.001g,置于具塞锥形瓶中,加入40mL丙酮,振摇1h,抽滤,浓缩,定容至5mL,待气相色谱分析。小麦:称取小麦试样10g,精确至0.001g,置于具塞锥形瓶中,加入0.2g活性炭及40mL丙酮,振摇1h,抽滤,浓缩,定容至5mL,待气相色谱分析。植物油:称取植物油试样5g,用45mL丙酮分次洗入50mL的离心管内,加入5mL水,混匀,在3 000r/min下离心5min,吸取上清液,下面油层再加10mL水和10mL丙酮,离心5min,吸取上清液,合并两次上清液,用K-D浓缩器浓缩近干,残渣和水加入40g无水硫酸钠,研磨呈干粉状,倒入具塞锥形瓶中,加入0.3g活性炭、60mL二氯甲烷,振荡0.5h,抽滤,定容至5mL,待气相色谱分析。5.2.色谱条件色谱柱:玻璃柱,内径3mm,长0.5m,内装2%dEGS/Chromosorb W AWdMCS,80~100mesh。气流:载气,氮气70mL/min,空气0.7kg/cm2,氢气1.2kg/cm2。温度:进样口200℃,柱温180℃。5.3.测定定性:以甲胺磷和乙酰甲胺磷农药标样的保留时间定性。定量:用外标法定量,以甲胺磷和乙酰甲胺磷农药已知浓度的标准样品溶液作外标物,按峰高定量。6.结果计算Xi=hi•Esi•V1hsi•V2•m式中:Xi——样品中i组分有机磷含量,mg/kg;Esi——注入标样中i组分有机磷的含量,ng;hi——样品的峰高,mm;hsi——标样中i组分的峰高,mm;V1——浓缩定容体积,mL;V2——注入色谱样品的体积,μL;m——样品的质量,g。7.方法的精密度添加回收试验中甲胺磷和乙酰甲胺磷的变异系数分别为2.36%和3.95%。8.甲胺磷和乙酰甲胺磷的保留时间在5.2的气相色谱条件下,甲胺磷的保留时间为0.9min,乙酰甲胺磷的保留时间为1.9min。9.来源:GB 14876—94







 我要推广仪器
我要推广仪器
 下载APP
下载APP