SVHC的5种蒽油及煤焦油,沥青标准品或者分析纯级别物质在哪里买啊?找了几家都没有找到。。。。。http://simg.instrument.com.cn/bbs/images/brow/em09501.gif
[size=3][/size][img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=58670]煤焦分析标准汇总[/url]上传点煤焦分析的最新技术标准,希望对同行有所帮助!有大型焦化厂的分析工作者希望大家共同交流,本人还有其他大量资料,相继会上传.需要焦炉煤气分析的作业指导书,当做参考.
[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]分析脂肪酸甲酯标准品,怎样处理标准品?用什么溶剂稀释?
求ISO国际标准对应的煤质分析国家标准号如下:煤中全水分的测定方法GB/T 211-1996煤的工业分析方法GB/T 212-2001煤的发热量测定方法GB/T 213-2003煤中全硫测定方法GB/T 214-1996煤的元素分析方法煤的可磨分析方法GB/T 2565-1998煤燃点分析方法煤的胶质层分析方法有以上标准的ISO国际英文版的请与本人tianhaibiwu@126.com联系。感激万分!
我们实验室是检测饲料和原料含量的,以前别人介绍用过中国农业科技院分析检测中心研制的氨基酸分析用校核标准品(参比物),名字我忘记了,只记得有代号,1#、2#、3#、4#、5# 这5种。有知道的老师们请帮帮忙!
连日来,反式脂肪酸都处于舆论的风头浪尖。继央视报道市面上许多用到植物奶油的食品都含有对人体有一系列副作用的反式脂肪酸之后,卫生部开始针对反式脂肪酸展开风险监测评估。这一现象再次引发了消费者对食品安全的担忧。请关注—— 前不久,关于反式脂肪酸对健康有害的报道引起了社会的广泛关注,有媒体将它与杀虫剂DDT相媲美,并声称其为“食品史上的又一灾难”。反式脂肪酸连日来受到了社会各界的口诛笔伐,食品安全问题也再次引发了人们的担忧。 反式脂肪酸存在健康风险 不久前,央视报道市面上所销售的很多用到植物奶油(又称氢化油)的食品都含有反式脂肪酸,而反式脂肪酸对人体有一系列副作用,极有可能导致冠心病、糖尿病、肥胖等疾病高发的危害。 笔者走访北京许多超市发现,在饼干、蛋黄派、巧克力、沙拉酱等商品的成分表中,随处可见“氢化植物油”、“植物奶油”等字样,也就是说,这些食品中都含有反式脂肪酸。除此之外,各种美味的生日蛋糕、冰激凌等也都含有反式脂肪酸。 那么,反式脂肪酸到底有哪些危害呢? 据了解,反式脂肪酸其实是一种具有反式结构的不饱和脂肪酸,它是在液态植物油氢化过程中产生的。上世纪80年代,由于担心存在于荤油中的饱和脂肪酸可能会对心脏带来威胁,植物油又有高温不稳定及无法长时间储存等问题,科学家就利用氢化的过程,将液态植物油改变成固态,反式脂肪酸由此开始被人们广泛使用。 中国农业大学食品营养与工程学院教授李里特介绍说:“当时的植物油由于成本低及胆固醇含量少,故而很受欢迎。然而,近年来,随着科学家们的深入研究,发现这种反式脂肪酸的摄入竟然比胆固醇对人体的危害还大,从而引起了广泛关注。” “研究表明反式脂肪酸摄入量多时可升高低密度脂蛋白(坏胆固醇),降低高密度脂蛋白(好胆固醇),增加患动脉硬化和冠心病的危险,还会影响儿童生长发育和神经系统健康以及可能使患肥胖病,糖尿病等慢性病的几率增加。”中国营养学会主任医师高慧英指出。 我国缺乏食用标准 自反式脂肪酸被曝可能存在健康风险后,关于该不该使用含有大量反式脂肪酸的植物奶油成为社会热议的焦点。食物中含多少反式脂肪酸才在安全范围以内?每人每天摄入反式脂肪酸的量需要控制在多少范围内? 李里特表示:“目前国家还没有权威的文件出台,也没有设定相关标准。它对于人体的危害不是一蹴而就的,而是慢慢发展的,就好比香烟对于人体的危害一样,虽然没有人试验过摄入多少反式脂肪酸才
乐伐替尼,作为现代医学的瑰宝,广泛应用于肿瘤治疗领域。然而,就像其他药物一样,乐伐替尼在生产过程中也可能会产生杂质。这些微小的杂质,虽然量少,却可能对药物的疗效和安全性产生不可忽视的影响。为了确保乐伐替尼的纯净与安全,科学家们引入了CATO标准品进行杂质分析。CATO标准品,就像一把精准的尺子,能够帮助研究人员准确地检测和衡量乐伐替尼中的杂质。通过对比和分析,我们可以清楚地了解杂质的种类、数量以及可能对药物产生的影响。这项应用研究不仅提升了乐伐替尼的生产质量,更为患者的安全用药提供了有力保障。借助CATO标准品,我们能够及时发现并控制杂质,确保每一颗乐伐替尼都是纯净、有效的。未来,随着科学技术的不断进步,我们期待看到更多关于乐伐替尼杂质分析的研究成果,为患者带来更加安全、可靠的治疗方案。同时,也期待CATO标准品在更多药物杂质分析中发挥重要作用,守护人类的健康与安全。[img=,603,515]https://ng1.17img.cn/bbsfiles/images/2024/02/202402021836237186_8744_6381568_3.png!w603x515.jpg[/img]广州佳途科技股份有限公司是一家专业的CATO标准品生产厂家,我们目前库存有全套乐伐替尼杂质,能够提供相应的系列图谱和产品COA证书,并支持溯源。我们公司已经通过了国内外双ISO 17034质量体系认证,欢迎广大客户选购。
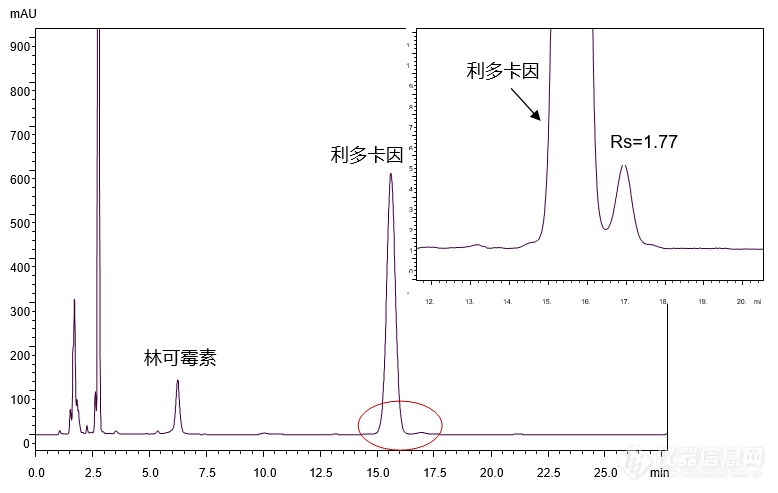
[align=center][b]【国家药品标准】林可霉素利多卡因凝胶的分析[/b][/align][align=center][b][/b][/align][align=right][b]——依据国家药品标准WS-10001-(HD-0140)-2002方法[/b][/align][b]林可霉素利多卡因凝胶[/b]为复方制剂,每克含林可霉素5毫克,利多卡因4毫克。适应症为用于轻度烧伤、创伤及蚊虫叮咬引起的各种皮肤感染。 [img=,193,127]http://ng1.17img.cn/bbsfiles/images/2018/07/201807260834522166_2994_2222981_3.gif!w193x127.jpg[/img] [img=,140,64]http://ng1.17img.cn/bbsfiles/images/2018/07/201807260834520028_3541_2222981_3.gif!w140x64.jpg[/img] 林可霉素 利多卡因 Lincomycin Lidocaine M.W.: 406.54 M.W.: 234.34客户提供林可霉素利多卡因凝胶样品,希望本实验室帮忙通过筛选色谱柱及调节分析条件,依据[color=#ff0000][b]国家药品标准WS-10001-(HD-0140)-2002[/b][/color]方法,实现林可霉素利多卡因凝胶样品的良好分析。首先,使用能在纯水条件下稳定使用的高极性色谱柱[color=#ff0000][b]CAPCELL PAK C[sub]18[/sub] AQ S5 4.6 mm i.d. × 150 mm[/b][/color],对林可霉素利多卡因凝胶样品进行分析,结果如图1所示,[color=#330099]利多卡因与其峰后杂质之间分离度为1.77[/color]。[align=center][img=,690,437]http://ng1.17img.cn/bbsfiles/images/2018/07/201807260858200006_8607_2222981_3.png!w690x437.jpg[/img][/align][align=center]图1 CAPCELL PAK C[sub]18 [/sub]AQ分析所得色谱图[/align]注:峰上标数字为分离度。[img=,528,205]http://ng1.17img.cn/bbsfiles/images/2018/07/201807260858202566_2695_2222981_3.png!w528x205.jpg[/img]为进一步提高利多卡因与其峰后杂质之间的分离度,在原条件基础上将柱温由30℃降低至25℃,并分别使用 CAPCELL PAK C[sub]18[/sub] AQ、CAPCELL PAK C[sub]18[/sub] MG及高含碳量ODS色谱柱SUPERIOREX ODS进行分析,结果如图2所示。[align=center][img=,690,490]http://ng1.17img.cn/bbsfiles/images/2018/07/201807260859201516_7229_2222981_3.png!w690x490.jpg[/img][/align][align=center]图2 25℃条件下不同色谱柱分析结果对比[/align]注:峰上标数字为分离度。[img=,637,223]http://ng1.17img.cn/bbsfiles/images/2018/07/201807260859204236_7198_2222981_3.png!w637x223.jpg[/img]如图2所示,在柱温25℃条件下使用三款色谱柱进行分析,其中,[color=#ff0000][b]CAPCELL PAK C[sub]18[/sub] AQ色谱柱分析结果最好,利多卡因与其峰后杂质分离得到最佳分离,分离度为4.23[/b][/color];[color=#330099][b]使用CAPCELL PAK C[sub]18[/sub] MG色谱柱进行分析时,利多卡因与其峰后杂质分离度为3.27[/b][/color];而使用SUPERIOREX ODS色谱柱分析时,利多卡因与其峰后杂质未得到有效分离。综上,在国家药品标准WS-10001-(HD-0140)-2002方法基础上,将色谱柱柱温由30℃降低至25℃,使用高极性色谱柱CAPCELL PAK C[sub]18[/sub] AQ及中等极性色谱柱CAPCELL PAK C[sub]18[/sub] MG进行分析,均可在25 min内完成林可霉素利多卡因凝胶样品的分析,并得到利多卡因与其峰后杂质之间的良好分离结果。[align=right][/align][align=right][/align][align=right] [/align][align=right]三耀精细化工品销售(中国)有限公司[/align][align=right]技术开发部[/align][align=right]地址:北京经济技术开发区宏达南路5号[/align][align=right]宏达利德工业园1栋418室[/align][align=right]邮编:100176[/align]
请问谁有以下标准的ISO国际标准煤中全水分的测定方法GB/T 211-1996煤的工业分析方法GB/T 212-2001煤的发热量测定方法GB/T 213-2003煤中全硫测定方法GB/T 214-1996煤的元素分析方法煤的可磨分析方法GB/T 2565-1998煤燃点分析方法煤的胶质层分析方法有以上标准的ISO国际英文版的请与本人tianhaibiwu@126.com联系。感激万分!
标定盐酸标准滴定溶液的不确定度分析 作者:吴文英 张春雨 唐惠兰 来源:中华医学研究杂志 在理化分析过程中,一切测量结果都不可避免地具有不确定度。盐酸标准溶液是常用化学定量参比物质,其标定值的准确性直接影响常规分析质量。笔者以GB/T601《滴定分析(容量分析)用标准液的制备》为依据配制并标定盐酸根据JJF1059-1999《测定不确定度评定与表示》分析其测量不确定度。简述由标定过程中得到的不确定度。 1 实验部分 1.1 测定方法[1,2] 准确称量270℃~300℃干燥至恒重的基准碳酸钠(99.95%~100.05%)约0.2g左右,电子分析天平(精度为0.1mg),置于三角瓶中,加入50ml水使之溶解,加指示剂,用盐酸标准液滴定至终点同时作试剂空白实验。 1.2 主要计量仪器与试剂 电了分析天平:AG204;酸式滴定管:50ml A级。 1.3 建立数学模型 C=m (V1-V2)×0.05300 式中 C:盐酸标准滴定溶液的浓度(mol/L);m:基准无水碳酸钠的质量(g);V1:盐酸标准滴定溶液用量(ml);V2:试剂空白实验中盐酸标准滴定溶液用量(ml);0.05300:与1.00ml盐酸标准溶液[C(HCl)=1.000mol/L]相当于以克表示的无水碳酸钠的质量。 1.4 盐酸标准滴定溶液的标定结果 为获得标准溶液重复测量的不确定度分量,对同一标准溶液进行8次独立的标定。测定数据见表1。 表1 盐酸标准滴定溶液的标定结果 略 2 测量不确定度来源 从检测过程和数学模型分析,标定盐酸标准溶液的不确定度主要来源,由四个方面所引起。(1)测量的重复性(A类不确定度);(2)基准无水碳酸钠的纯度;(3)测量使用的电子分析天平及量具;(4)其他相关常数。 3 测量不确定度分析 3.1 A类不确定度的分析 利用表1中的测量结果,按照A类评定测量重复性的标准不确定度。具体计算过程:重复测量的平均值计算式:=1 n∑8 i=1xi=0.09951mol/L 单次测量的标准差按贝塞尔公式计算s(x)为 s(x)=∑8 i=1(xi-)2 n-1=0.0001555mol/L 的标准差s()为 s()=s(x) n=0.000155 8=0.0000548mol/L=5.48×10-5mol/L 由测量重复性引起的相对标准不确定度为U(x):0.0000548/0.09951=0.055%。 3.2 B类不确定度分析 3.2.1 基准碳酸钠的纯度 基准碳酸钠的纯度为1.0000±0.0005,视为矩形分布0.00053=0.00029,则标准不确定度为:由基准碳酸钠的纯度引入的相对不确定度u(p)为:0.029%。 3.2.2 天平称量所引入的标准不确定度 干燥器与天平称量仓内均放置同质硅胶,视为相同湿度,称量时无吸潮。电子天平检定证书标出线性为上0.2mg;可视为矩形分布,则标准不确定度为:因为称量采用的是减量法,故称量的标准不确定度为0.2mg /3=0.12mg:因为称量采用的是减量法,故称量的标准不确定度为:2×0.122=0.17mg,则由称量引入的相对标准不确定度u(m)为:0.17mg/0.2018g=0.084%。 3.2.3 标定体积的不确定度 (1)滴定管的校准:滴定使用50ml酸式滴定管(A级),按照检定规程,其最大允许误差为±0.05ml,相对允许误差为±0.1%,按照矩形分布,则滴定体积的相对标准不确定度u(V)为:u(V)=0.1%/3=0.0577%。(2)环境温度:实验环境在空调条件下,室温近似20℃。温度在20℃左右,标准溶液的温度补正值非常小,对实验结果影响可忽略不计,所以在不确定度分析中不把一温度影响引起的不确定度列入考虑范围。(3)滴定终点的判断:终点时的误差±0.05ml(1滴的体积),两点分布,现由终点分布判断引入的标准不确定度为0.05ml:相对标准不确定度为0.05ml/38.32ml=0.13%标定体积的影响引入相对标准不确定度U(V)为0.0572+0.132=0.142%。 3.2.4 其他常数 基准无水碳酸钠摩尔质量引起的标准不确定度很小,可以忽略。 4 合成标准不确定度 测量重复性、基准无水碳酸钠的纯度、天平称量、标定体积等的不确定度相互独立,故将上述数据合成得盐酸的相对合成标准不确定度U(C)为0.0552+0.0292+0.0842+0.1422=0.176%。 5 扩展不确定度 实验测得盐酸标准溶液浓度为0.09951mol/L,则测量结果的合成标准不确定度U(C)=0.09951mol/L×0.176%=0.000175mol/L。若取包含因子K=2,得测量结果的扩展不确定度U=2U(C)=0.00035mol/L。 6 测量结果的表示 盐酸标准滴定溶液的浓度可表示为:(0.09951±0.00035mol/L,K=2)。 【参考文献】 1 姚正堂,将已峰.奶制品中蛋白质测定的不确定度分析.中华医学研究杂志,2005,5(6):6. 2 国家技术监督局.JJF1059-1999测量不确定度与表示.北京:中国计量出版社,1997,81. 作者单位: 214171 江苏无锡,无锡市惠山区疾病预防控制中心
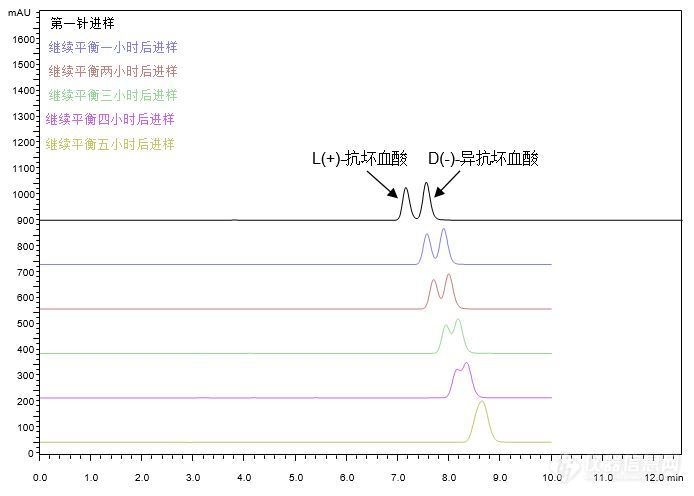
[align=center][b]GB 5009.86-2016 食品安全国家标准 食品中抗坏血酸的测定[/b][/align][align=center][b] ——附不添加离子对试剂方法[/b][/align][b] [/b]客户要求参考《GB 5009.86-2016 食品安全国家标准食品中抗坏血酸的测定》的测定方法,筛选合适的色谱柱,要求使L(+)-抗坏血酸、D(-)-异抗坏血酸的[color=#ff0000]分离度大于0.90,且保留稳定。[/color]首先,尝试使用CAPCELL PAK C[sub]18 [/sub]MGII S5 4.6 mm i.d. × 250 mm色谱柱,按照《GB 5009.86-2016 食品安全国家标准食品中抗坏血酸的测定》方法,对L(+)-抗坏血酸、D(-)-异抗坏血酸混合样品进行分析,实验中发现,[color=#3333ff]随着色谱柱平衡时间的延长[/color],L(+)-抗坏血酸、D(-)-异抗坏血酸峰[color=#3333ff]保留时间逐渐向后移动,并且两个峰逐渐合成一个峰[/color],如图1所示。[align=center][img=,690,493]http://ng1.17img.cn/bbsfiles/images/2017/12/201712210903_1412_2222981_3.jpg!w690x493.jpg[/img][/align][align=center]图1 混合样品分析色谱图(平衡时间延长)[/align][align=left][img=,690,212]http://ng1.17img.cn/bbsfiles/images/2017/12/201712210903_3096_2222981_3.jpg!w690x212.jpg[/img][/align][align=center][/align][align=left]由于标准方法中流动相含有离子对试剂(十六烷基三甲基溴化铵),因此我们需要对色谱柱进行充分的平衡,以得到相对稳定的保留时间。[color=#ff0000][b]连续对色谱柱平衡约48小时[/b][/color]后,待测物保留时间为10.33min,保留时间稳定,[color=#ff0000]但两个色谱峰重合[/color],三针重复性结果如图2所示。[/align][align=center][/align][align=center][img=,690,488]http://ng1.17img.cn/bbsfiles/images/2017/12/201712210903_7944_2222981_3.jpg!w690x488.jpg[/img][/align][align=center]图2 混合样品分析色谱图(三针重复性)[/align][align=center][/align][align=left]尝试在原条件基础上提高流动相中有机相比例,以期得到二者分离。如图3,当流动相中含有25%CH[sub]3[/sub]OH时,L(+)-抗坏血酸、D(-)-异抗坏血酸峰分离效果最好,分离度为0.95,但峰形略差。[/align][align=center][/align][align=center][img=,688,504]http://ng1.17img.cn/bbsfiles/images/2017/12/201712210910_4695_2222981_3.jpg!w688x504.jpg[/img][/align][align=center]图3 混合样品分析色谱图(调节有机相比例)[/align][align=left][img=,690,191]http://ng1.17img.cn/bbsfiles/images/2017/12/201712210910_2154_2222981_3.jpg!w690x191.jpg[/img][/align][align=left][/align][align=left]为改善峰形,尝试[color=#3333ff]将柱温由25°C提高到30°C[/color],结果如图4所示。L(+)-抗坏血酸、D(-)-异抗坏血酸[color=#3333ff]峰形得到明显改善,二者分离度为1.28,大于客户要求的0.90[/color]。我们也尝试进一步提高流动相中有机相比例、温度以及改变有机相种类,未得到更好的分析结果。[/align][align=left][/align][align=center][img=,690,506]http://ng1.17img.cn/bbsfiles/images/2017/12/201712210919_83_2222981_3.jpg!w690x506.jpg[/img][/align][align=center]图4 混合样品分析色谱图(25%CH3OH 30°C)[/align][align=left][img=,690,209]http://ng1.17img.cn/bbsfiles/images/2017/12/201712210920_2143_2222981_3.jpg!w690x209.jpg[/img][/align][align=center][/align][align=left]此外,我们还尝试使用保留能力较MGII稍弱的CAPCELL PAK C[sub]18[/sub] UG120 S5 4.6 mm i.d. × 250 mm色谱柱,经流动相充分平衡后,在同样条件下对L(+)-抗坏血酸、D(-)-异抗坏血酸混合溶液进行分析,也未得到更好的分离结果。[/align][align=left][/align][align=left]上述实验中,我们在原条件基础上将流动相中有机相比例及柱温调整为25%CH[sub]3[/sub]OH、30°C,使用MGII色谱柱进行分析,可得到L(+)-抗坏血酸、D(-)-异抗坏血酸分离度为1.28的最佳分离结果,但二者仍未达到基线分离。[b]且使用添加离子对试剂的流动相进行分析时,色谱柱需要2天左右的平衡时间,因此,我们也开发了不含离子对试剂的分析条件。[/b][/align][align=left][b][/b][/align][align=left]在流动相为[color=#3333ff]20mM NH[sub]4[/sub]H[sub]2[/sub]PO[sub]4[/sub](pH2.0) / CH[sub]3[/sub]OH = 98 / 2[/color]的酸性条件下,分别使用键合金刚烷基的高极性色谱柱CAPCELL PAK ADME S5、在纯水条件下也能稳定使用的高极性色谱柱CAPCELL PAK C[sub]18[/sub] AQ S3,以及之前实验中使用的CAPCELL PAK C[sub]18[/sub] MGII S5,对L(+)-抗坏血酸、D(-)-异抗坏血酸混合溶液进行分析。结果如图5所示,[b][color=#ff0000]三款色谱柱对L(+)-抗坏血酸、D(-)-异抗坏血酸均可得到基线分离的良好结果,且峰形良好,分离度分别为1.89、2.05、2.10。[/color][/b][/align][align=left][b][color=#ff0000][/color][/b][/align][align=center][b][color=#ff0000][img=,690,509]http://ng1.17img.cn/bbsfiles/images/2017/12/201712210923_4302_2222981_3.jpg!w690x509.jpg[/img] [/color][/b][/align][align=center]图5 混合样品分析色谱图(三款色谱柱对比)[/align][align=left][b][color=#ff0000][img=,538,176]http://ng1.17img.cn/bbsfiles/images/2017/12/201712210923_7418_2222981_3.jpg!w538x176.jpg[/img][/color][/b][/align][align=left][b][color=#ff0000][/color][/b][/align][align=left][b][color=#ff0000][/color][/b][/align][align=center][/align][align=left]在此条件下使用ADME色谱柱进行精密度及线性实验的考察。精密度实验结果如表1所示,连续进样六针,L(+)-抗坏血酸峰面积RSD为0.32%、D(-)-异抗坏血酸峰面积RSD为0.25%。线性实验结果如图6、7所示,[color=#ff0000][b]在0~50 μg/mL的浓度范围内,L(+)-抗坏血酸、D(-)-异抗坏血酸均呈现良好的线性关系。[/b][/color][/align][align=left][/align][align=center]表1 抗坏血酸精密度实验[/align][align=center][img=,631,295]http://ng1.17img.cn/bbsfiles/images/2017/12/201712210926_8294_2222981_3.jpg!w631x295.jpg[/img][/align][align=center][/align][align=center][img=,578,357]http://ng1.17img.cn/bbsfiles/images/2017/12/201712210926_2295_2222981_3.jpg!w578x357.jpg[/img][/align][align=center]图6 抗坏血酸线性试验(L(+)-抗坏血酸)[/align][align=center][/align][align=center][img=,575,358]http://ng1.17img.cn/bbsfiles/images/2017/12/201712210926_7247_2222981_3.jpg!w575x358.jpg[/img][/align][align=center]图7 抗坏血酸线性试验(D(-)-异抗坏血酸)[/align][align=left]综上,在不添加离子对试剂的酸性条件下,使用键合金刚烷基的ADME色谱柱及MGII、AQ S3两款C[sub]18[/sub]色谱柱进行分析,均可对L(+)-抗坏血酸、D(-)-异抗坏血酸得到基线分离的良好结果。[/align][align=left][/align][align=left]此外,在实验中我们发现使用流动相(20mM NH[sub]4[/sub]H[sub]2[/sub]PO[sub]4 [/sub](pH2.0) / CH[sub]3[/sub]OH= 98 / 2)作为[b][color=#ff0000]样品溶剂[/color][/b]时,随着分析时间的延长,L(+)-抗坏血酸、D(-)-异抗坏血酸的峰面积有逐渐减小趋势;而当样品溶剂为20 g/L的偏磷酸时,L(+)-抗坏血酸、D(-)-异抗坏血酸峰面积基本保持稳定,但死时间处的溶剂峰较大。[/align][align=left][/align]
《食品安全国家标准 食品中农药最大残留限量》2016版正式颁布实施,这一农药残留的新国标,在标准数量和覆盖率上都有了较大突破,规定了433种农药在13大类农产品中4140个残留限量,较2014版增加490项,基本涵盖了我国已批准使用的常用农药和居民日常消费的主要农产品。 食品伙伴网标法中心结合2016版标准前言部分内容与2014版进行相应的对比分析,供参考: 1、对原标准中氟唑磺隆、甲咪唑烟酸、氟吡菌胺、三唑酮和三唑醇等5种农药残留物定义,敌草快等5种农药每日允许摄入量等信息进行了核实,修订了敌草快、三环锡等5种农药的ADI值。 2、增加了2,4-滴异辛酯等46种农药;增加了490项农药最大残留限量标准 2014版规定了食品中2,4-滴等387种农药3650项最大残留限量,2016版规定了433种2,4-滴等农药4140项最大残留限量。增加了46种农药:2,4-滴异辛酯、2甲4氯异辛酯、苯嘧磺草胺、苯嗪草酮、吡唑草胺、丙硫多菌灵、除虫菊素、毒草胺、多抗霉素、呋虫胺、氟吡菌酰胺、复硝酚钠、甲磺草胺、井冈霉素、抗倒酯、苦参碱、醚苯磺隆、嘧啶肟草醚、扑草净、嗪草酸甲酯、氰氟虫腙、氰烯菌酯、炔苯酰草胺、噻虫胺、三苯基乙酸锡、三氯吡氧乙酸、杀螺胺乙醇胺盐、莎稗磷、虱螨脲、特丁津、调环酸钙、五氟磺草胺、烯丙苯噻唑、烯肟菌酯、烯效唑、辛菌胺、辛酰溴苯腈、溴氰虫酰胺、唑胺菌酯、唑啉草酯、啶菌噁唑、丁吡吗啉、噁唑酰草胺、甲哌鎓、丁酰肼、唑嘧菌胺。 3、增加 12 项检测方法标准,删除1项检测方法标准 增加了SN/T 0162、SN 0198、SN/T 0931、SN/T 1624、SN/T 1989、SN/T 2229、SN/T 2231、SN/T 2237、SN/T 2323、SN/T 2387、SN/T 2795、SN/T 2807,删除了SN/T 0711,其中SN 0198标准已于2015年12月31日被认监委废止,废止依据为《国家认监委办公室关于公布2015年检验检疫行业标准复审结论的通知》。 4、修改了丙环唑等8种农药的英文通用名 修改了丙环唑、六六六、烯肟菌胺、氯啶菌酯、杀虫双、四氯苯酞、氯氟吡氧乙酸和氯氟吡氧乙酸异辛酯 5、将苯噻酰草胺和灭锈胺的限量值由临时限量修改为正式限量;对资料性附录 A 进行了修订,增加了干制蔬菜等3种食品名称,修改1项作物名 食品伙伴网对附录A部分内容的对比发现如下变化: 1)水果(核果类)的类别说明增加了青梅,枣修改为枣(鲜)。 2)水果(浆果和其他小型水果)的类别说明中露莓增加了备注:包括波森莓和罗甘莓。 3)水果(热带和亚热带水果)的类别说明中将大型果的木瓜修改为番木瓜。 4)干制水果的类别说明中增加了枣(干)等。 5)食品类别名称修改:将饮料修改为饮料类。 6、食品伙伴网在对比过程中发现,2016版标准除了以上所列变化外,还修正了其他一些内容: 1)引用的标准名称的修正,如GB/T 19648、GB/T 19469等部分标准的名称中 “兽”字已删除。 2)引用的作废标准的修正,如2014版标准中引用的是2006版GB/T 20770的标准名称,2016版标准已经修正为2008版GB/T 20770的标准名称。 3)农药中文名称修改:2014版标准中的2甲4氯(钠)修改为2甲4氯钠。 4)附录A中动物源食品部分类别的测定部位描述进行了修正。
各类能源折算标准煤的参考系数能源名称平均低位发热量折标准煤系数原煤20934千焦/公斤0.7143公斤标煤/公斤洗精煤26377千焦/公斤0.9000公斤标煤/公斤其他洗煤8374 千焦/公斤0.2850公斤标煤/公斤焦炭28470千焦/公斤0.9714公斤标煤/公斤原油41868千焦/公斤1.4286公斤标煤/公斤燃料油41868千焦/公斤1.4286公斤标煤/公斤汽油43124千焦/公斤1.4714公斤标煤/公斤煤油43124千焦/公斤1.4714公斤标煤/公斤柴油42705千焦/公斤1.4571公斤标煤/公斤液化石油气47472千焦/公斤1.7143公斤标煤/公斤炼厂干气46055千焦/ 公斤1.5714公斤标煤/公斤天然气35588千焦/立方米12.143吨/万立方米焦炉煤气16746千焦/立方米5.714吨/万立方米其他煤气 3.5701吨/万立方米热力 0.03412吨/百万千焦电力 3.27吨/万千瓦时 1、热力 其计算方法是根据锅炉出口蒸汽和热水的温度压力在焓熵图(表)内查得每千克的热焓减去给水(或回水)热焓,乘上锅炉实际产出的蒸汽或热水数量(流量表读出)计算。如果有些企业没有配齐蒸汽或热水的流量表,如没有焓熵图(表),则可参下列方法估算: (1)报告期内锅炉的给水量减排污等损失量,作为蒸汽或热水的产量。 (2)热水在闭路循环供应的情况下,每千克热焓按20千卡计算,如在开路供应时,则每千克热焓按70千卡计算(均系考虑出口温度90℃,回水温度20℃)。 (3)饱和蒸汽,压力1-2.5千克/平方厘米,温度127℃以上的热焓按620千卡,压力3-7千克/平方厘米,温度135℃-165℃的热焓按630千卡。压力8千克/平方厘米,温度170℃以上每千克蒸汽按640千卡计算。 (4)过热蒸汽,压力150千克/平方厘米,每千克热焓:200℃以下按650千卡计算,220℃-260℃按680千卡计算,280℃-320℃按700千卡,350℃-500℃按700千卡计算。按4.1868焦耳折算成焦耳。 2.热力单位“千卡”与标准煤“吨”的折算 能源折算系数中“蒸汽”和“热水”的计算单位为“千卡”,但“基本情况表”中(能源消耗量中)“蒸汽”计算单位为“蒸吨”,在其它能源消耗量(折标煤)其中的“热水”计算单位为“吨”,因此需要进一步折算,才能适合“基本情况表”的填报要求,按国家标准每吨7000千卡折1千克标准煤计算: 3.电力的热值 一般有两种计算方法:一种是按理论热值计算,另一种是按火力发电煤耗计算。每种方法各有各的用途。理论热值是按每度电本身的热功当量860大卡即0.1229千克标准煤计算的。按火力发电煤耗计算,每年各不相同,为便于对比,以国家统计局每万度电折0.404千克标准煤,作为今后电力折算标准煤系数。
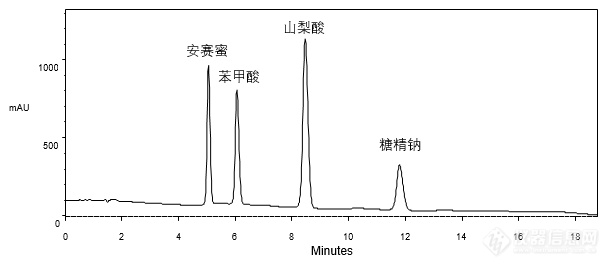
[align=center][b]GB 5009.28-2016食品安全国家标准 食品中苯甲酸、山梨酸和糖精钠的测定[/b][/align][align=center][b] ——标准品与乳品实际样品的分析[/b][/align][align=center][/align][align=left]本实验按照《GB5009.28-2016 食品安全国家标准食品中苯甲酸、山梨酸和糖精钠的测定》方法,分别对安赛蜜、苯甲酸、山梨酸、糖精钠的标准品混合溶液及加标乳品样品进行了分析。首先,使用CAPCELL PAK C[sub]18[/sub] MG S5 4.6 mm i.d. × 150mm色谱柱,对标准品混合溶液进行分析,如图1,安赛蜜、苯甲酸、山梨酸、糖精钠标准品均得到了良好的分析结果。[/align][align=left][/align][align=center][img=,611,268]http://ng1.17img.cn/bbsfiles/images/2018/03/201803221532276656_9890_2222981_3.png!w611x268.jpg[/img][/align][align=center]图1 标准品混合溶液分析色谱图[/align][img=,400,200]http://ng1.17img.cn/bbsfiles/images/2018/03/201803221532280132_6863_2222981_3.png!w400x200.jpg[/img][align=left][/align][align=left]其次,对乳品加标样品进行分析,如图2,糖精钠(Rt 12 min)与其后杂质峰之间未能取得基线分离,分离度仅为1.02。[/align][align=left][/align][align=center][img=,668,335]http://ng1.17img.cn/bbsfiles/images/2018/03/201803221533054905_2223_2222981_3.png!w668x335.jpg[/img][/align][align=center]图2 加标乳品样品分析色谱图[/align][align=left][img=,406,203]http://ng1.17img.cn/bbsfiles/images/2018/03/201803221533317202_2333_2222981_3.png!w406x203.jpg[/img][/align][align=left][/align][align=left]为改善糖精钠与杂质间的分离,在国标方法基础上,将流动相由[b]乙酸铵 / 甲醇 = 95 / 5[/b]调整为[b][b]乙酸铵 / 甲醇[/b][color=red]([/color][color=red]2 mmol/L [/color][color=red]甲酸)[/color]= 92 / 8[/b],再次对混合标准溶液和加标样品进行分析,结果如图3所示。[/align][align=left][/align][align=center][img=,690,545]http://ng1.17img.cn/bbsfiles/images/2018/03/201803221534141056_4073_2222981_3.png!w690x545.jpg[/img][/align][align=center]图3 混标与加标乳品样品分析色谱图[/align][align=left][img=,464,171]http://ng1.17img.cn/bbsfiles/images/2018/03/201803221535548985_7176_2222981_3.png!w464x171.jpg[/img][/align][align=left][/align][align=left]如图3,在酸性条件下,出峰顺序发生了变化,安赛蜜保留时间略有缩短,糖精钠保留时间明显缩短,由12 min缩短至8 min,苯甲酸和山梨酸保留时间分别延长至2 min和6 min;在分离度方面,糖精钠与苯甲酸之间分离度为2.79,苯甲酸与峰后杂质间分离度为2.04,所有色谱峰之间都达到了基线分离。[/align][align=left][/align][align=left]为使客户有更多选择,实验室又在国标原方法条件下继续筛选色谱柱,最终使用SUPERIOREX ODS S5 4.6 mm i.d. × 250 mm色谱柱时,仅微调有机相比例即可实现加标乳品样品的良好分析结果。如图4,杂质峰与糖精钠之间分离度达到2.48,达到基线分离要求。[/align][align=left][/align][align=center][img=,580,332]http://ng1.17img.cn/bbsfiles/images/2018/03/201803221537130173_1058_2222981_3.png!w580x332.jpg[/img][/align][align=center]图4 加标乳品样品分析色谱图[/align][align=left]*注:峰上标所示数字由下至上依次为分离度与不对称因子。[/align][align=left][img=,326,177]http://ng1.17img.cn/bbsfiles/images/2018/03/201803221537540634_9437_2222981_3.png!w326x177.jpg[/img][/align][align=left][/align][align=left]综上所述,按照国标《GB 5009.28-2016 食品安全国家标准食品中苯甲酸、山梨酸和糖精钠的测定》方法进行分析,使用CAPCELL PAK C[sub]18 [/sub]MG色谱柱对标准品混合溶液能得到良好分析结果,但在对加标乳品样品进行分析时,糖精钠与样品中的杂质未能实现基线分离,通过在流动相中添加甲酸可实现安赛蜜、糖精钠、苯甲酸、山梨酸及杂质的基线分离;另一方面,使用SUPERIOREX ODS色谱柱,在原条件基础上微调即可实现乳品中安赛蜜、苯甲酸、山梨酸、糖精钠及杂质间的良好分离。[/align]
β-Glucuronidase/aryl sulfatase β-葡萄糖醛酸酶\芳香基硫酸酯酶( 葡萄糖苷酸酶/硫酸芳酯酶 ) 标准品——检瘦肉精等用的除了北京希凯创新科技有限公司提供,还可以从哪里购买么?
用没食子酸标准曲线测茶多酚标准品为什么实际值比理论值还高,是我计算的问题吗,按照国标计算的
GB 26400-2011 食品安全国家标准 食品添加剂 二十二碳六烯酸油脂(发酵法)
论坛上煤炭分析的常用国家标准很多,也有很多重复了.本次上传到资料中心的85个煤炭分析相关国家标准为论坛没有的(资料中心和附件都搜索过,应该没有重复的,但不排除).认证会员免积分下载.本人上传的标准链接:http://www.instrument.com.cn/download/search.asp?keywords=dgtg105&sel=admin_name&SN=&Submit=%C1%A2%BC%B4%B2%E9%D1%AF以下所列标准目录为本论坛都有的.不知道怎么搜索的朋友学习一下发哥的【推荐】查找国家标准GB的便捷方法 http://www.instrument.com.cn/bbs/shtml/20060814/516721/论坛里煤炭分析相关国家标准目录如下:注:标准名称后面带"(2006)",表示最新标准为2006年,而论坛找不到,只有以前的老标准.GB 474-1996 煤样的制备方法 GB 475-1996 商品煤样采取方法 GB 481-1993 生产煤样采样方法GB 482-1995 煤层煤样采取方法GB 3812-1983褐煤蜡试样的采取和缩制方法GB 4632-1997 煤的最高内在水分测定方法GB 5751-1986 中国煤炭分类 GB 14181-1997 测定烟煤粘结指数专用无烟煤技术条件GB 20426-2006 煤炭工业污染物排放标准GBT 189-1997 煤炭粒度分级GBT 211-1996 煤中全水分的测定方法 GBT 212-2001 煤的工业分析方法GBT 213-2003 煤的发热量测定方法 GBT 214-1996 煤中全硫的测定方法GBT 215-2003 煤中各种形态硫的测定方法GBT 216-2003 煤中磷的测定方法 GBT 217-1996 煤的真相对密度测定方法 GBT 218-1996 煤中碳酸盐二氧化碳含量的测定方法GBT 219-1996 煤灰熔融性的测定方法GBT 220-2001 煤对二氧化碳化学反应性的测定方法GBT 397-1998 冶金焦用煤技术条件GBT 476-2001 煤的元素分析方法GBT 477-1998 煤炭筛分试验方法GBT 478-2001 煤炭浮沉试验方法GBT 479-2000 烟煤胶质层指数测定方法GBT 480-2000 煤的铝甑低温干馏试验方法 GBT 483-1998 煤炭分析试验方法一般规定 GBT 1341-2001 煤的格金低温干馏试验方法GBT 1572-2001 煤的结渣性测定方法 GBT 1573-2001 煤的热稳定性测定方法 GBT 1574-1995 煤灰成分分析方法 GBT 1575-2001 褐煤的苯萃取物产率测定方法 GBT 2559-2005 褐煤蜡测定方法GBT 2560-1981 褐煤蜡滴点测定方法GBT 2561-1981 褐煤蜡中溶于丙酮物质(树脂物质)测定方法GBT 2562-1981 褐煤蜡中苯不溶物测定方法GBT 2563-1981 褐煤蜡灰分测定方法GBT 2564-1981 褐煤蜡酸值和皂化值测定方法GBT 2565-1998 煤的可磨性指数测定方法(哈德格罗夫法) GBT 2566-1995 低煤阶煤的透光率测定方法 GBT 3058-1996 煤中砷的测定方法GBT 3558-1996 煤中氯的测定方法GBT 3715-1996 煤质及煤分析有关术语GBT 3813-1983 褐煤蜡密度测定方法GBT 3814-1983 褐煤蜡粘度测定方法GBT 3815-1983 褐煤蜡加热损失量测定方法GBT 3816-1983 褐煤蜡中地沥青含量测定方法GBT 4063-2001 蒸汽机车用煤技术条件GBT 4633-1997 煤中氟的测定方法GBT 4634-1996 煤灰中钾、钠、铁、钙、镁、锰的测定方法([url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]分光光度法) GBT 4757-2001 煤粉(泥)实验室单元浮选试验方法GBT 5447-1997 烟煤粘结指数测定方法GBT 5448-1997 烟煤坩埚膨胀序数的测定 电加热法GBT 5449-1997 烟煤罗加指数测定方法GBT 5450-1997 烟煤奥阿膨胀计试验GBT 6948-1998 煤的镜质体反射率显微镜测定方法GBT 6949-1998 煤的视相对密度测定方法GBT 7186-1998 煤矿科技术语 选煤GBT 7560-2001 煤中矿物质的测定方法GBT 7561-1998 合成氨用煤技术条件GBT 7562-1998 发电煤粉锅炉用煤技术条件GBT 7563-2000 水泥回转窑用煤技术条件GBT 8207-1987 煤中锗的测定方法GBT 8208-1987 煤中镓的测定方法GBT 8899-1998 煤的显微组分组和矿物测定方法GBT 9143-2001 常压固定床煤气发生炉用煤技术条件GBT 11957-2001 煤中腐植酸产率测定方法 GBT 12937-1995 煤岩术语GBT 15224.1-2004 煤炭质量分级 第1部分 灰分GBT 15224.2-2004 煤炭质量分级 第2部分 硫分GBT 15224.3-2004 煤炭质量分级 第3部分 发热量GBT 15334-1994 煤的水分测定方法 微波干燥法 GBT 15458-1995 煤的磨损指数测定方法(2006)GBT 15459-1995 煤的抗碎强度测定方法(2006)GBT 15460-2003 煤中碳和氢的测定方法 电量-重量法GBT 15588-2001 烟煤显微组分分类GBT 15589-1995 显微煤岩类型分类GBT 15590-1995 显微煤岩类型测定方法GBT 15591-1995 商品煤反射率分布图的判别方法GBT 15715-2005 煤用重选设备工艺性能评定方法GBT 15716-2005 煤用筛分设备工艺性能评定方法GBT 16415-1996 煤中硒的测定方法 氢化物发生[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]法GBT 16416-1996 褐煤中溶于稀盐酸的钠和钾测定用的萃取方法GBT 16417-1996 煤炭可选性评定方法GBT 16658-1996 煤中铬、镉、铅的测定方法GBT 16659-1996 煤中汞的测定方法GBT 16660-1996 选煤厂用图形符号GBT 16772-1997 中国煤炭编码系统GBT 16773-1997 煤岩分析样品制备方法GBT 17607-1998 中国煤层煤分类GBT 17608-2006 煤炭产品品种和等级划分GBT 17609-1998 铸造焦用煤技术条件GBT 17610-1998 水煤气两段炉用煤技术条件GBT 18023-2000 烟煤的宏观煤岩类型分类GBT 18510-2001 煤和焦炭试验可替代方法确认准则GBT 18511-2001 煤的着火温度测定方法GBT 18512-2001 高炉喷吹用无烟煤技术条件GBT 18666-2002 商品煤质量抽查和验收方法GBT 18702-2002 煤炭安息角测定方法GBT 18711-2002 选煤用磁铁矿粉试验方法GBT 18712-2002 选煤用絮凝剂性能试验方法GBT 18855-2002 水煤浆技术条件GBT 18856.1-2002 水煤浆质量试验方法 水煤浆采样方法GBT 18856.2-2002 水煤浆质量试验方法 水煤浆浓度测定方法 GBT 18856.3-2002 水煤浆质量试验方法 水煤浆筛分试验方法GBT 18856.4-2002 水煤浆质量试验方法 水煤浆表观粘度测定方法GBT 18856.5-2002 水煤浆质量试验方法 水煤浆稳定性测定方法GBT 18856.6-2002 水煤浆质量试验方法 水煤浆发热量测定方法GBT 18856.7-2002 水煤浆质量试验方法 水煤浆工业分析方法GBT 18856.8-2002 水煤浆质量试验方法 水煤浆全硫测定方法GBT 18856.9-2002 水煤浆质量试验方法 水煤浆密度测定方法GBT 18856.10-2002 水煤浆质量试验方法 水煤浆灰熔融性测定方法GBT 18856.11-2002 水煤浆质量试验方法 水煤浆碳氢测定方法GBT 18856.12-2002 水煤浆质量试验方法 水煤浆氮测定方法GBT 18856.13-2002 水煤浆质量试验方法 水煤浆灰成分测定方法GBT 18856.14-2002 水煤浆质量试验方法 水煤浆pH值测定方法GBT 19092-2003 煤粉浮沉试验方法GBT 19093-2003 煤粉筛分试验方法GBT 19094-2003 选煤厂 流程图原则和规定GBT 19222-2003 煤岩样品采取方法GBT 19224-2003 烟煤相对氧化度测定方法GBT 19225-2003 煤中铜、钴、镍、锌的测定方法GBT 19226-2003 煤中钒的测定方法GBT 19227-2003 煤和焦炭中氮的测定方法 半微量蒸汽法GBT 19494.1-2004 煤炭机械化采样 第1部分:采样方法GBT 19494.2-2004 煤炭机械化采样 第2部分:煤样的制备GBT 19494.3-2004 煤炭机械化采样 第3部分:精密度测定和偏倚试验GBT 19560-2004 煤的高压等温吸附试验方法 容量法GBT 19952-2005 煤炭在线分析仪测量性能评价方法GBT 20104-2006 煤自燃倾向性色谱吸氧鉴定法GBT 20475.1-2006 煤中有害元素含量分级 第1部分:磷欢迎大家补充上传.不对的地方跟指正.
GB 26401-2011 食品安全国家标准 食品添加剂 花生四烯酸油脂(发酵法)
核心提示:GB2760-2011与GB2760-2007标准对比分析之一——食品分类系统卫生部在2011年第12号卫生部公告中发布了4项食品安全国家标准,其中之一即为《GB 2760-2011 食品安全国家标准 食品添加剂使用标准》(以下简称新标准),该新标准将于2011年6月20日起正式实施,新标准的前言中明确指出对2007版的《GB 2760-2007 食品添加剂使用卫生标准》(以下简称旧标准)部分食品分类系统进行了调整,为使广大网友对该新旧标准的食品分类系统的调整有更具体的了解,食品伙伴网对食品论坛中网友发布的相关讨论帖进行了整理,仅供行业人士参考,对比分析具体结果: 1、对比过程中发现如下问题:和旧标准相比新标准的食品分类系统中删除了01.01.02.01调味乳,但搜索发现表A1里有涉及到该分类号。 2、新增了如下食品分类:01.01.02 灭菌乳,同时将旧标准中的01.01.02 调制乳调整为01.01.03 调制乳。01.02 发酵乳和风味发酵乳,同时将旧标准中的01.02 发酵乳调整为01.02.01。01.02.02 风味发酵乳。同时删除了旧标准中的01.02.01、01.02.02。04.04.03 其他豆制品。06.05.02.04 粉圆。13.02.01 婴幼儿谷类辅助食品、13.02.02 婴幼儿罐装辅助食品。14.03.01.01 发酵型含乳饮料、14.03.01.02 配制型含乳饮料、14.03.01.03 乳酸菌饮料(旧标准中的分类号为14.07)、14.03.03 复合蛋白饮料。 3、调整了如下食品分类:将旧标准中03.02雪糕类删除,在新标准中将其合并到03.01冰淇淋类。将旧标准中如下分类删除05.02.03、05.02.04、05.02.05、05.02.06、05.02.07、05.02.08、05.02.08.01、05.02.08.02、05.02.09,合并到05.02.02。旧标准中14.07乳酸菌饮料调整为新标准的14.03.01.03 乳酸菌饮料。乳品的分类调整比较复杂,在其他几点中都有所涉及。 4、删除了如下食品分类:04.02.02.03.01 酱渍的蔬菜、04.02.02.03.02 盐渍的蔬菜、04.02.02.03.03 糖醋渍的蔬菜、04.02.02.03.04 其他腌渍的蔬菜。04.03.02.03.01酱渍的食用菌和藻类、04.03.02.03.02盐渍的食用菌和藻类、04.03.02.03.03糖醋渍的食用菌和藻类、04.03.02.03.04其他腌渍的食用菌和藻类。06.03.01.03 蛋糕预拌粉、06.03.01.04 其他专用粉。08.03.05.01 高温蒸煮肠、08.03.05.02 低温蒸煮肠、08.03.05.03 其他肉肠。14.01.02 自然来源饮用水、14.01.04 饮用矿物质水。16.05 油炸食品、16.05.01 油炸小食品、16.05.02 其他油炸食品。01.02.01 原味发酵乳(全脂、部分脱脂、脱脂)、01.02.02 调味和果料发酵乳。13.05.01 孕产妇(乳母)配方食品、13.05.02 运动营养食品(运动饮料除外)。 5、修改了如下食品分类名称:01.0 乳及乳制品(13.0 特殊营养用食品涉及品种除外)修改为乳及乳制品(13.0 特殊膳食用食品涉及品种除外)。01.01 乳及调制乳修改为巴氏杀菌乳、灭菌乳和调制乳。01.01.01 纯乳(全脂、部分脱脂、脱脂),包括复原乳修改为巴氏杀菌乳。01.03.01 乳粉(全脂奶粉、脱脂奶粉和部分脱脂奶粉)和奶油粉修改为乳粉和奶油粉04.04.01.01 豆腐类(北豆腐、南豆腐、内酯豆腐、冻豆腐)修改为豆腐类。04.05.02.01 烘焙/炒制坚果与籽类修改为熟制坚果与籽类。04.05.02.01.01带壳烘焙/炒制坚果与籽类修改为带壳熟制坚果与籽类。04.05.02.01.02脱壳烘焙/炒制坚果与籽类修改为脱壳熟制坚果与籽类。04.05.02.05 其他方法(如腌渍的果仁)修改为其他加工的坚果与籽类(如腌渍的果仁)。
跪求百康Biochrom全自动氨基酸分析30+的操作视频,科研小白啥也不会[img=,690,920]https://ng1.17img.cn/bbsfiles/images/2023/12/202312012213077671_2508_6258352_3.png[/img]
我现在想测某提取物中的酚类含量。制作标准曲线要用到没食子酸,但手里只有焦性没食子酸。想请问高手,能用焦性没食子酸代替没食子酸使用吗?
食品分析中标准物的管理及其标准溶液的校正一、意义食品分析标准物质是分析方法质控的核心,是定性和定量的依据。标准物质以一定纯度和浓度配制的溶液称标准溶液,其稳定性受其自身降解、化学转化、溶质和溶剂挥发等内在因素的影响,又受其存放条件如温度、湿度、光线照射、存放时间、存放容器及其配制技巧等外界条件的影响。由于影响标准物及其标准液的因素较多,如果条件控制不当或管理不严密,标准物质浓度易发生变化,这是食品分析中难以进行质量控制的主要原因。以上原因也是实验室食品分析测定产生误差的最主要因素,要减小这些产生误差的主要因素,就要特别注重标准物管理,以及进行标准溶液的稳定性观察和校正工作,也是实验室质量控制关键工作之一。当标准溶液发生变化时就要重新配制,并找出变化原因,为分析工作积累经验,并可写入方法注释中。在食品分析质量控制中,准确度和精密度的提高,是以标准溶液稳定性和准确性为前提的,因此对于标准溶液稳定性和准确性的关键技术问题,是质量控制的核心问题。二、标准物质的管理 1、容量分析的基准物是标定其它标准溶液的基准,应购买基准试剂,它可以保证其纯度。另一个因素是水份的影响,用前应充分干燥和恒重,配制时量器要校正。溶剂要纯化,使用的容器要充分洗涤。最终目的都是防止基准物的化合损失。2、用于分光光度分析的标准物,分有机的和无机的标准物,无机标准稳定性好,但使用液浓度低时,极易被容器吸附,并与容器中离子进行交换,因此决定了其稀溶液使用时间短。玻璃容器在碱性介质中易溶出,塑料容器在成型时加入助剂时也含有不同金属杂质,容易溶出如Zn、Ca等金属离子。有机标准最好不放在塑料制容器中,因塑料在成型时加入的有成份比较复杂的助剂和增塑剂。标准使用液应现用现配为最佳。三、标准物存放使用1、无机的标准物要求在干燥并无化学干扰物的条件下存放,选择合适干燥剂如硅胶和分子筛。有机标准物最好分装封入安培瓶中低温避光保存。固体的多环芳烃可配制成溶液,再分装在安瓿瓶中保留溶剂封存,也可把溶剂挥掉后干燥封存,使用时再定容,后一种方法更为稳妥些。配制好一批标准溶液,再一支一支使用也是很方便的。如果液体的标准物特别是几种标准的混合物用于色谱分析同系物如醇类物质,可同时配制一批分装安瓿瓶低温存放,再一支一支使用,能避免溶剂挥发体积变化产生的误差。这种做法更适于实验室间的标准分发和校正工作。最难办的是气体,标准如氯乙烯、氟里昂,最好是钢瓶中存放,或配制钢瓶标准气。这些条件不具备时也可以选择高沸点溶剂,密封溶解这些气体,称量溶质重量,一次性使用。从这一事实出发,气体,测定误差可以稍加放宽,因为标准自身稳定性差。2、用于色谱分析的标准物要求色谱纯,其配剂溶液剂也要求色谱纯,准确配制前要在色谱上进行检查。特别是几种标准进行混合更应慎重,每种要严格检查否则给定性定量带来很多麻烦。如果纯度不够时可以纯化,再结晶或用制备色谱制备。勉强使用是无益的。四、标准溶液的校正1、从安瓿瓶分装标准溶液无论是有机的或是无机的用于校正是很方便的。如原子吸收测定金属,从安瓿瓶中取一定量配制浓度系列,再封存。每隔一段时间(1~2周)再用原溶液配制同样浓度系列,严格控制仪器条件来比较二次标准曲线的斜率,斜率下降时表明有损失。2、相同浓度同时配制的标准的几支安瓿瓶,先用打开的一支标准的测定值与间隔一段时间后打开的另一支标准的测定值进行比较,以此类推最后在一段时间内几支同时测定,其变异程度就是标准在这段时间稳定性变化程度。3、几个实验室用同一标准物分别配制相同浓度标准液,各自进行标准曲线的测定,再按规定交换该标准液再进行测定,比较测定结果差异来观察同一标准的时间和空间变异。如果标准液稳定,配制不准确的实验室很容易查出。配制都准确时,标准液若不稳定时,会使各实验室的测得值都偏低。4、同种标准物来源不同,也应采用分别配制交叉测定的办法来检查标准的纯度及配制是否准确。在食品分析中无论用何种手段分析样品,所使用的标准物应作统一的或确切的规定。例如:过硫酸铵测锰,用MnSO4·H2O作为标准使用,到底硫酸锰需要不需要烘烤呢?对于这个问题,在一部份的教科书中有规定烘烤的,也有不烘烤的。按照MnSO4·H2O的性质遇到空气可能吸潮或风化,如果直接称重计算Mn量,就有可能出现误差。用烘烤称重测得水分所含的量比理论值高1.6%,有同一硫酸锰配制锰标准溶液测一合成水样,使用烘烤后配制的锰标准溶液,测得的Mn含量为0.205mg/L,未烘烤过的则高达约2.3%,从中说明硫酸锰在配制标准溶液时应经过烘烤,使标准一致。
韩国拟定修改健康或功能食品标准和禁用成分相关法规来源:中国技术性贸易措施网 2009年10月12日,韩国发布通报:韩国食品药物管理局拟定修改健康/功能食品标准规范。本拟定法规制定和增加包括食物纤维在内的32种健康/功能食品吸收成分提示的相关规定。它还制定了卵磷酯、瓜尔胶/瓜尔胶水解物、葡甘露聚糖、阿拉伯树胶及聚葡萄糖内的铅含量规范,增加了瓜尔胶/瓜尔胶水解物及抗麦芽糖糊精的功效要求。此外,准许蜂胶提取物成品制成软胶囊。 韩国食品药物管理局拟定修改健康/功能食品禁用成分相关法规。 本拟定法规扩大健康/功能食品禁用植物成分的范围。本拟定措施还将减肥药、糖尿病药物及其它同类物质纳入健康/功能食品禁用植物成分清单。
前些日子,食品中常见的植物奶油因被曝光含有大量反式脂肪酸,一时间成为众矢之的。植物奶油危机生成后,卫生部随即表示:相关部门已对反式脂肪酸进行管理,并将开展对反式脂肪酸的风险监测评估。这一事件,不禁让人联想起曾经的苏丹红、三聚氰胺等“食品安全门”,同样先由媒体曝光披露危害,同样事后才由相关部门表态制定标准。关乎无数人群健康安全的食品标准,为何总是在媒体曝光后才姗姗来迟?食品安全监管该如何完善,才能让消费者吃得放心? 研究滞后令标准“迟到” 科技进步日新月异,食品品种也日渐繁多。作为工业时代的食品新品种,反式脂肪酸等物质的危害,近些年才被发现。中国疾控中心相关专家解释,上世纪七八十年代,当植物奶油普遍应用时,人们尚未意识到反式脂肪酸的问题所在。直到近些年,它可能导致糖尿病、肥胖、高血压等慢性疾病的危害,才逐渐为人们所熟知。至于苏丹红、三聚氰胺等食品添加剂,它们正式问世与发现危害之间,同样存在一定的“时差”。 复旦大学公共卫生学院营养与食品专家郭红卫教授认为,新食品问世后,相关的健康研究本身就需要一段时日,迄今为止,反式脂肪酸是否对人体有害在学界仍存在一定分歧,少数专家还不认同这类观点;将学界理论应用到实际监管之中,也需要一个时间段,这便导致标准制定总是晚一步。也有专家分析说,我国监管行政部门、学界专家沟通不足、交流不畅,令许多标准未能及时跟上,最终造成“有了结石宝宝才发现三聚氰胺,找到慢性病祸首才重视植物奶油”的尴尬境遇。 监管不到位标准难制定 据悉,我国专家在制定植物奶油标准前竟然发现:各种食品中到底含有多少植物奶油、多少反式脂肪酸,没有任何权威部门做过统一检测和调研。监管检测漏洞多多,成为标准制定难的最关键原因。 上海理工大学食品研究所所长徐斐分析,监管首先需要大量的调研工作,这方面我国做得较为欠缺,许多食品的成分调研并不细致,为之后的标准制定带来重重阻碍。其次,监管手段过于单一、陈旧,常会使监管出现纰漏。以含三聚氰胺的奶粉为例,传统检测只查蛋白质,找不到什么差错;如能查一查奶粉中的脂肪、乳糖等比例,三聚氰胺问题也就不难发现了。用“昨天”的标准来评判“今天”、“明天”的食品,待到发现问题再做补充,这样的被动跟进,只能使标准制定姗姗来迟。再者,监管后的惩罚力度不够,使标准制定的应有功效打了折扣。前不久,三聚氰胺“再出江湖”,令众多消费者咋舌之时,还折射出食品添加剂标准未能“掷地有声”,这也为之后制定标准带来阻力。 开展安全风险评估须置前 确保食品安全,标准制订该如何缩短“时差”?怎样才能让标准及时更新、不至于造成严重的健康威胁?权威专家认为,针对新食品开展安全风险评估至关重要。具体说来,学术研究专家、行政部门间应多沟通,让临床新发现及时应用在标准制订上,实现科学评价食品新添加剂,防患于未然。与之相匹配,监管部门的监管手段、监管范围也应与时俱进,做到监管更周密、到位。 令人欣慰的是,今年本市已在全国率先成立“食品安全风险评估专业委员会”,委员会将汲取过往经验,着手开展对大米中的镉成分、凉拌菜中的相关成分等进行安全风险评估,争取早日制订标准,保障消费者权益。 郭红卫教授补充,除了政府监管外,企业和消费者的知识普及、信息获取,也是加强食品安全的重要一环。比如,此次反式脂肪酸的利弊得到评估后,有关部门应即刻加强对企业、消费者的宣教。一方面,告诫食品企业不应完全以利润为考量,更当兼顾人体健康;一方面,消费者也应有意识选择少吃同类食品。消费者的科学选择,反过来又可左右企业行为,最终为食品质量织起安全网。
食品药品胶体金分析仪是一种先进的科学仪器,主要用于检测食品和药品中的胶体金含量。胶体金是一种微小颗粒的金纳米材料,其在食品和药品中的含量是一个关键的指标,对产品的质量和安全性具有决定性的影响。[img=,690,690]https://ng1.17img.cn/bbsfiles/images/2024/04/202404261526428439_4734_6238082_3.jpg!w690x690.jpg[/img] 该分析仪的工作原理基于胶体溶液中纳米颗粒的光学性质,通过测量光的散射强度来得到颗粒的浓度信息。当光与金纳米颗粒相互作用时,根据散射光的洛仑兹-米耳斯理论,可以得到散射强度与颗粒的浓度之间的关系。因此,胶体金分析仪可以精确、高效地测量食品和药品中的胶体金含量。 此外,食品药品胶体金分析仪还有便携式及台式两种款式,利用免疫层析胶体金原理及干式化学原理,可以对畜禽产品中瘦肉精、抗生素残留,水产品中非法化学品、抗生素药物残留、罂壳成分等进行快速检测。这种分析仪现已广泛应用于各级食药环侦快检实验室,成为实验室常规检测的有益补充。 使用食品药品胶体金分析仪时,用户需要按照以下步骤操作:首先,根据实际需求,准备好需要检测的样品和相应的试剂 其次,将准备好的样品和试剂分别装载到胶体金免疫层析分析仪的样品槽和试剂槽中 然后,启动仪器,仪器会自动进行样品混合和分析 最后,观察屏幕上显示的结果,并根据仪器提供的标准曲线,确定目标物质的浓度。 总的来说,食品药品胶体金分析仪为食品和药品行业的质量控制和安全监测提供了强有力的支持,是一种具有广泛应用前景的科学仪器。
请大家给我提供一些关于煤焦油成分的分析与研究资料,比如:煤焦油里面一些常见组分的测定方法之类的,主要是煤焦油中某种组分的具体测定方法,有步骤和实验数据更好,无论是企业的标准和国标都行,先谢谢大家了!
最近发现用ICP分析Sb元素,弱酸介质下GBW配置的标准曲线分析的其他品牌的标准溶液回收率偏低。实验室用的仪器是瓦里安 ICP-720,标准曲线是使用国家计量院的GBW(E)080545锑单元素溶液标准物质,浓度100ug/mL,基体是5%HCl。作为交叉验证的CK是使用国家钢铁材料测试中心钢铁研究总院的GSB G 62043-90锑标准溶液,浓度500ug/mL,基体是25%硫酸。或者是ACCU的ICP-02N-1,浓度1000ug/mL,基体是2~5%HNO3。我发现使用弱酸基体(若0.07mol/L HCl或者5%HNO3)配置的标准曲线(使用计量院的Sb标准溶液配置),分析相同基体的CK(由另外品牌的Sb标准溶液配置),测量第一个CK时,Sb回收率只有70-85%(酸度越低,回收率越差),继续测量,回收率会慢慢增大。可是即使连续测量(不拔出进样管)10次以上,Sb的回收率也只有93-94%。而测量由计量院的Sb标准溶液配置的同样基体的Sb溶液,回收率却没有问题;若是使用浓酸基体(35%HNO3),两个品牌的标准溶液的回收率却又没有问题。我知道Sb会有残留,可是分析每一只样品(包括标准曲线的点),我都会快泵进样十几秒再分析溶液的,就算有残留,没道理厉害到分析了10多样品还有残留。母溶液的基体可能有影响,可是同一支溶液的其他元素的回收率都OK啊最近有支PT样(5%硝酸基体,什么品牌的母溶液配出来的就不知道了),就是因为这个原因,Sb的读数偏低了。现在要整改,可是什么原因都不知道……现在只好到论坛来求助各位老大了,希望各位能给点意见。先谢谢了。
DL/T 465-1992 煤的冲刷磨损指数试验方法 查看 DL/T 498-1992 粉煤灰游离氧化钙测定方法 查看 DL/T 660-1998 煤灰高温黏度特性试验方法 查看 DZ 48-1987 岩石中有机碳分析方法 查看 GB 14181-1997 测定烟煤粘结指数专用无烟煤技术条件 查看 GB 189-1963 煤炭粒度分级 查看 GB 2566-1995 低煤阶煤透光率测定方法 查看 GB 4632-1997 煤的最高内在水分测定方法 查看 GB 474-1996 煤样的制备方法 查看 GB 475-1996 商品煤样采取方法 查看 GB 5751-1986 中国煤炭分类 查看 GB/T 11957-2001 煤中腐植酸产率测定方法 查看 GB/T 1341-2001 煤的格金低温干馏试验方法 查看 GB/T 14181-1993 测定烟煤粘结指数专用无烟煤技术条件 查看 GB/T 15334-1994 煤的水分测定方法 微波干燥法 查看 GB/T 15458-1995 煤的磨损指数测定方法 查看 GB/T 15459-1995 煤的抗碎强度测定方法 查看 GB/T 15460-1995 煤中碳和氢的测定方法 电量-重量法 查看 GB/T 1572-2001 煤的结渣性测定方法 查看 GB/T 1573-2001 煤的热稳定性测定方法 查看 GB/T 1574-1995 煤灰成分分析方法 查看 GB/T 1575-2001 褐煤的苯萃取物产率测定方法 查看 GB/T 16415-1996 煤中硒的测定方法 氢化物发生[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]法 查看 GB/T 16416-1996 褐煤中溶于稀盐酸的钠和钾测定用的萃取方法 查看 GB/T 16658-1996 煤中铬、镉、铅的测定方法 查看 GB/T 16659-1996 煤中汞的测定方法 查看 GB/T 18510-2001 煤和焦炭试验可替代方法确认准则 查看 GB/T 18511-2001 煤的着火温度测定方法 查看 GB/T 18666-2002 商品煤质量抽查和验收方法 查看 GB/T 18855-2002 水煤浆技术条件 查看 GB/T 18856.1-2002 水煤浆质量试验方法 第1部分:水煤浆采样方法 查看 GB/T 18856.10-2002 水煤浆质量试验方法 第10部分:水煤浆灰熔融性测定方法 查看 GB/T 18856.11-2002 水煤浆质量试验方法 第11部分:水煤浆碳氢测定方法 查看 GB/T 18856.12-2002 水煤浆质量试验方法 第12部分:水煤浆氮测定方法 查看 GB/T 18856.13-2002 水煤浆质量试验方法 第13部分:水煤浆灰成分测定方法 查看 GB/T 18856.14-2002 水煤浆质量试验方法 第14部分:水煤浆PH值测定方法 查看 GB/T 18856.2-2002 水煤浆质量试验方法 第2部分:水煤浆浓度测定方法 查看 GB/T 18856.3-2002 水煤浆质量试验方法 第3部分:水煤浆筛分试验方法 查看 GB/T 18856.4-2002 水煤浆质量试验方法 第42部分:水煤浆表观粘度测定方法 查看 GB/T 18856.5-2002 水煤浆质量试验方法 第5部分:水煤浆稳定性测定方法 查看 GB/T 18856.6-2002 水煤浆质量试验方法 第6部分:水煤浆发热量测定方法 查看 GB/T 18856.7-2002 水煤浆质量试验方法 第7部分:水煤浆工业分析方法 查看 GB/T 18856.8-2002 水煤浆质量试验方法 第8部分:水煤浆全硫测定方法 查看 GB/T 18856.9-2002 水煤浆质量试验方法 第9部分:水煤浆密度测定方法 查看 GB/T 211-1996 煤中全水分的测定方法 查看 GB/T 212-1991 煤的工业分析方法 查看 GB/T 212-2001 煤的工业分析方法 查看 GB/T 213-1996 煤的发热量测定方法 查看 GB/T 214-1996 煤中全硫的测定方法 查看 GB/T 215-1996 煤中各种形态硫的测定方法 GB/T 216-1996 煤中磷的测定方法 查看 GB/T 217-1996 煤的真相对密度测定方法 查看 GB/T 218-1996 煤中碳酸盐二氧化碳含量的测定方法 查看 GB/T 219-1996 煤灰熔融性的测定方法 查看 GB/T 220-2001 煤对二氧化碳化学反应性的测定方法 查看 GB/T 2565-1998 煤的可磨性指数测定方法(哈德格罗夫法) 查看 GB/T 2566-1995 低煤阶煤的透光率测定方法 查看 GB/T 3058-1996 煤中砷的测定方法 查看 GB/T 3558-1996 煤中氯的测定方法 查看 GB/T 4633-1997 煤中氟的测定方法 查看 GB/T 4634-1996 煤灰中钾、钠、铁、钙、镁、锰的测定方法([url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]分光光度法) 查看 GB/T 476-1991 煤的元素分析方法 查看 GB/T 476-2001 煤的元素分析方法 查看 GB/T 478-2001 煤炭浮沉试验方法 查看 GB/T 479-2000 烟煤胶质层指数测定方法 查看 GB/T 480-2000 煤的铝甑低温干馏试验方法 查看 GB/T 483-1998 煤炭分析试验方法一般规定 查看 GB/T 5447-1997 烟煤粘结指数测定方法 查看 GB/T 5448-1997 烟煤坩埚膨胀序数的测定 电加热法 查看 GB/T 5449-1997 烟煤罗加指数测定方法 查看 GB/T 5450-1997 烟煤奥阿膨胀计试验 查看 GB/T 6949-1998 煤的视相对密度测定方法 查看 GB/T 7560-2001 煤中矿物质的测定方法 查看 JB/T 7610-1994 锅炉煤粉燃烧特性试验规范 查看 JB/T 7611-1994 锅炉煤粉气流着火指数测定试验规范 JB/T 7612-1994 锅炉煤粉粒度分布测试规范 查看 MT 190-1988 选煤厂煤泥水沉降试验方法 查看 MT 263-1991 烟煤宏观类型的划分与描述 查看 MT 265-1991 商品煤随机反射率分布图的判别方法 查看 MT 422-1996 煤矿粉尘粒度分布测定方法(质量法) 查看 MT 56-1981 中国煤炭可选性测定标准 查看 MT 80-1984 煤中灰分快速测定方法 查看 MT/T 1-1996 商品煤含矸率和限下率的测定方法 查看 MT/T 109-1996 煤和矸石泥化试验方法 查看 MT/T 340.1-1994 冶金焦用北淮矿务局煤技术条件 查看 MT/T 340.2-1994 发电煤粉锅炉用淮北矿务局煤技术条件 查看 MT/T 340.3-1994 水泥回转窑用淮北矿务局煤技术条件 查看 MT/T 341.1-1994 冶金焦用大屯煤电公司煤技术条件 查看 MT/T 341.2-1994 发电煤粉锅炉用大屯煤电公司煤技术条件 查看 MT/T 341.3-1994 蒸汽机车用大屯煤电公司煤技术条件 查看 MT/T 342.1-1994 冶金焦用七台河矿务局煤技术条件 查看 MT/T 342.2-1994 常压固定床煤气发生炉用七台河矿务局煤技术条件 查看 MT/T 342.3-1994 蒸汽机车用七台河矿务局煤技术条件 查看 MT/T 342.4-1994 水泥回转窑用七台河矿务局煤技术条件 查看 MT/T 342.5-1994 发电煤粉锅炉用七台河矿务局煤技术条件 查看 MT/T 343.1-1994 冶金焦用西山矿务局煤技术条件 查看 MT/T 343.2-1994 发电煤粉锅炉用西山矿务局煤技术条件 查看 MT/T 344.1-1994 发电煤粉锅炉用龙口矿务局煤技术条件 查看 MT/T 344.2-1994 常压固定床煤气发生炉用龙口矿务局煤技术条件 查看 MT/T 345.1-1994 发电煤粉锅炉用霍州矿务局煤技术条件 MT/T 345.2-1994 冶金焦用霍州矿务局煤技术条件 查看 MT/T 346.1-1994 发电煤粉锅炉用大雁矿务局煤技术条件 查看 MT/T 347.1-1994 发电煤粉锅炉用扎赉诺尔矿务局煤技术条件 查看 MT/T 348.1-1994 冶金焦用萍乡矿务局煤技术条件 查看 MT/T 348.2-1994 发电煤粉锅炉用萍乡矿务局煤技术条件 查看 MT/T 348.3-1994 合成氨用萍乡矿务局煤技术条件 查看 MT/T 348.4-1994 水泥回转窑用萍乡矿务局煤技术条件 查看 MT/T 349.1-1994 冶金焦用潞安矿务局煤技术条件 查看 MT/T 349.2-1994 发电煤粉锅炉用潞安矿务局煤技术条件 查看 MT/T 349.3-1994 蒸汽机车用潞安矿务局煤技术条件 查看 MT/T 357-1994 煤的三氯甲烷萃取物测定方法 查看 MT/T 358-1994 煤的三氯甲烷萃取物族组分测定方法 查看 MT/T 384-1994 煤中铀的测定方法 查看 MT/T 385-1994 煤中钒的测定方法 查看 MT/T 560-1996 煤的热稳定性分级 查看 MT/T 561-1996 煤的固定碳分级 查看 MT/T 562-1996 煤中磷分分级 查看 MT/T 574-1996 煤矸石生物肥料技术条件 查看 MT/T 594-1996 煤显微组分荧光光谱测定方法 查看 MT/T 595-1996 煤显微组分荧光强度测定方法 查看 MT/T 596-1996 烟煤粘结指数分级 查看 MT/T 597-1996 煤中氯含量分级 查看 MT/T 736-1997 无烟煤电阻率测定方法 查看 MT/T 737-1997 量热仪氧弹安全性能检验规范 查看 MT/T 739-1997 煤炭堆密度小容器测定方法 MT/T 740-1997 煤炭堆密度大容器测定方法 查看 MT/T 741-1997 煤系高岭岩 三氧化二铝浸出率测定方法 查看 MT/T 748-1997 工业型煤冷压强度测定方法 查看 MT/T 749-1997 工业型煤浸水强度和浸水复干强度的测定方法 查看 MT/T 750-1997 工业型煤中的全硫测定方法 查看 MT/T 751-1997 工业型煤发热量测定方法 查看 MT/T 791-1998 水煤浆采样方法 查看 MT/T 792-1998 水煤浆浓度测定方法 查看 MT/T 799-1999 煤系高岭岩(土)及其煅烧土沉降体积测定方法 查看 MT/T 800-1999 煤系高岭岩(土)煅烧土白度测定方法 查看 MT/T 801-1999 煤系高岭岩(土)及其煅烧土悬浮性能测定方法 查看 MT/T 802.1-1999 煤系硫铁矿及硫精矿中有效硫的测定方法 查看 MT/T 802.2-1999 煤系硫铁矿及硫精矿中全硫、硫酸盐硫、硫化铁硫的测定方法 查看 MT/T 802.3-1999 煤系硫铁矿及硫精矿中总碳量的测定方法 查看 MT/T 802.4-1999 煤系硫铁矿及硫精矿中砷含量的测定方法 查看 MT/T 846-1999 煤体导水性分类 查看 SD 210-1987 火电厂动力煤标准煤样(第一批) 查看
跪求有没有人有“硫酸银、碳酸银”分析标准啊?







 我要推广仪器
我要推广仪器
 下载APP
下载APP