有谁检测化妆品中黄樟素这种成份的,本人需要黄樟素标物,不知如何购买
请问各位老师们香精原料标准品都在哪买的比较靠谱,香豆素和黄樟素标准品采购链接有推荐的吗~~
如题,测定香精中的黄樟素和香豆素,用什么内标呢?
谁有或者哪里有最新的香豆素和黄樟素的[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]-质谱检测方法?不胜感激。
[color=#444444]用二氯甲烷提取后,进行机械振荡。利用[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质联用[/color][/url]进行进行加标回收率计算,黄樟素能达到90%,而香豆素只有60%,从结构上可看出,香豆素不是很稳定。色谱柱:HP-5ms(30,0.25,0.25)[/color][color=#444444]进样量:1微升,分流比为10:1,[/color][color=#444444]进样口温度:280[/color][color=#444444]He:1ml/min[/color][color=#444444]用SIM定量。[/color][color=#444444]感觉色谱方法没问题,两种组分分离较好,峰形也很好,关键就是香豆素稳定性的问题。有没有好的建议?[/color]
有老师按照中国兽药典2020年一部,丙二醇的乙二醇含量的项目吗?按照药典的方法设置,对照品溶液中乙二胺的峰没有出!不知道怎么回事?
甘油乙二醇二甘醇图为3针对照品,完全不平行,且二甘醇峰(14.5左右)越来越小……做了很多遍,都是这样,先后用db_624,0.53*3,ov_1301,0.25*1.4柱子试过,二甘醇不是和甘油包一起,就是这样不怎么出峰,而且面积也并不平行,求各位大神看看,指点一下??
领导让做聚丙烯酰胺的离子度,标准17514-2017里面提到甲基乙二醇甲壳素(MGC)标准溶液0.005mol/L,请问我要怎么采购,供应商那里说没有这东西,只有甲基乙二醇壳聚素
各位大侠,有谁有乙二醇中氮元素的测定方法啊,谢谢
如题,有朋友用的是ZB624柱(固定液:6%氰丙基苯基-94%二甲基聚硅氧烷),柱长30m,内径530um,膜厚0.3um。载气流速4.5ml/min。其他条件同10版药典。结果发现,对照品溶液图谱情况良好,各峰分离度很好,峰面积之比(该法为内标法测定)的RSD5%,出峰顺利乙二醇-正己醇-二甘醇。但系统适应性图谱就不行了,溶剂峰(甲醇)拖尾严重,干扰乙二醇,更纳闷的是二甘醇峰会消失,在二甘醇应该出现的保留时间前后也无峰出现,加大二甘醇的浓度也没有峰出现,真是怪了。做了好多次了,都是如此。可以确定的是,做供试分析会严重污染柱子,对照品溶液(无甘油)进样分析正常(多次进样也正常),供试品溶液和系统适应性样品溶液(均含甘油)分析溶剂峰就会拖尾,影响乙二醇峰,然后再进对照品溶液也会如此。但柱子老化后,对照品溶液进样分析就会正常了。请教做过这个品种的同行,有没有遇到这问题?这种情况可能是那里出现了问题?
乙二醇、二甘醇和三甘醇取本品4g,精密称定。置 100ml量瓶中,取1,3-丁二醇0.004g,精密称定,置同一量瓶中,加丙酮使溶解,相同溶剂稀释至刻度,作为供试品溶液。取乙二醇0.0025g, 二甘醇0.004g,三甘醇0.004g,精密称定,置同一100ml量瓶中,取1,3-丁二醇0.004g,置 该量瓶中,加丙酮使溶解,相同溶剂稀释至刻度,作为对照品溶液。照[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法(通则0521)试验。以50%苯基-50% 甲基聚硅氧烷为固定液(液膜厚度1.0μm)的毛细管柱,起始 温度为40℃,以每分钟10℃的速率升温至60℃,维持5分钟,再以每分钟10℃的速率升温至170℃,维持0分钟,再以每分钟15℃的速率升温至280℃。维持60分钟(可根据具体情况调整)。检测器为氢火焰离子化检测器。检测器温度 290℃,进样口温度为270℃。取对照品溶液作为系统适用性 试验溶液,载气为氦气,流速5.0ml/min,分流比2:1,进 样体积1.0μl。乙二醇,二甘醇和三甘醇与内标1,3-丁二醇的分离度均不得小于2.0,各峰间的拖尾因子应符合规定,[color=#ff0000]乙二醇,二甘醇和三甘醇峰面积相对于内标1,3-丁二醇的峰面积相对标准偏差不得过5.0%[/color]。以1,3-丁二醇峰面积计算乙二醇,二甘醇和三甘醇的峰面积,以下式计算:[color=#ff0000]结果=(Ru / Rs) X (Cs X Cu)X F X100 [/color] 式中Ru为供试品溶液中各待测物质与内标的峰面积比率;Rs为对照品溶液中各对照物质(乙二醇,二甘醇和三 甘醇)与内标的峰面积比率; Cs为对照品溶液中各对照物质(乙二醇,二甘醇和三 甘醇)的浓度,ug/ml;Cu为供试品溶液中待测物质的浓度,mg/ml F为转换因子,103mg/g。(10的三次方 )依法检测,乙二醇,二甘醇和三甘醇均不得过0.01%。以上标红文字不能理解其含义,望解答,谢谢 !

检测甘油,有对照乙二醇、二甘醇,内标正己醇,条件:DB-624(30m*0.53mm 3.um),进样量1ul,分流比10:1,进样口200°,FID250°,程序:起始100(维持4′),以50°/min升120°(维持10′),再以50°/min升220°(维持6′).后加设降温和平衡时间。样品处理:系统适用性性乙二醇、二甘醇,内标正己醇各100mg稀释至100ml(系统储备液),精取1ml+4g甘油样品至100容量瓶, 所 有 溶剂都是色谱甲醇。 对照液:乙二醇、二甘醇,内标正己醇各50mg至100ml(标储液),取5ml稀释至25ml。问题:6月份同样方法检测,一切正常(当时柱子新买来活化后检1批乙醇,) 这两天同一根柱子检测(中间检测了3批乙醇),结果系统适用性乙二醇出不来峰了。正己醇和二甘醇峰面积无论是储备液还是系统适用性都没什么差异。储备液中的乙二醇有峰面积,但与之前浓度相当情况下峰面积小1/3,系统适用性就出不来了,对照液要算校正因子f,之前差不多2-3左右,现在超过10了。乙二醇的安剖瓶色标5ml有之前开启后密封冷藏的,也有新开的,两种情况都差不多,批号都是081226。http://ng1.17img.cn/bbsfiles/images/2012/07/201207251613_379663_2481522_3.jpg6月份的对照液,峰依次是:乙二醇--正己醇--二甘醇。http://ng1.17img.cn/bbsfiles/images/2012/07/201207251616_379664_2481522_3.jpg6月的系统适用性,7.8′乙二醇峰还是不错的,但这次此峰消失了。后面的正己醇、二甘醇相当浓度峰面积也相当。请问问题可能在哪里呢?
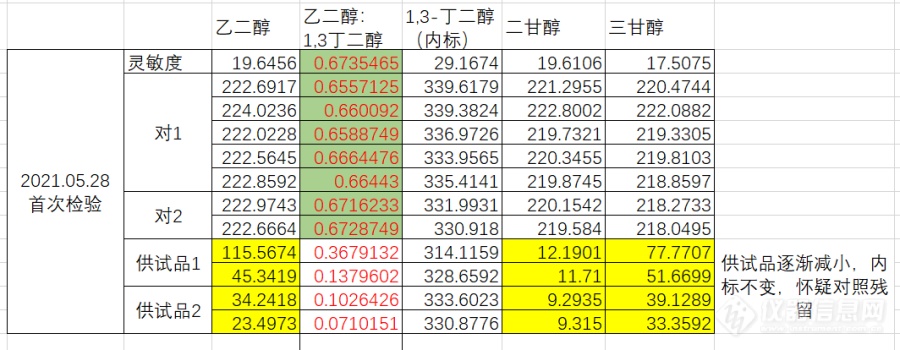
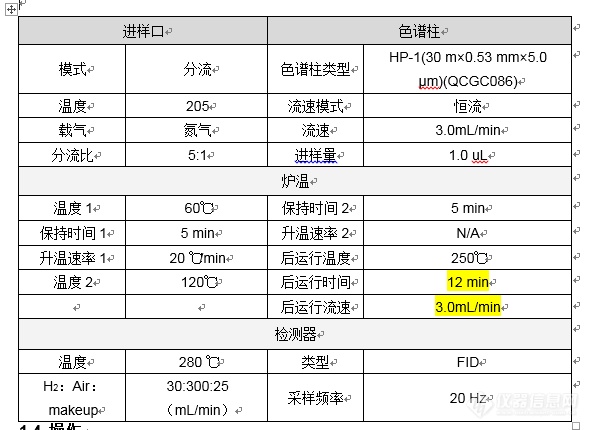
在进行聚山梨酯80Ⅱ 乙二醇、二甘醇和三甘醇检测过程中,发现对照品溶液重复性相当不错,但是供试品中目标峰逐渐减小,见如下数据:[img=第一次实验时实验数据,690,268]https://ng1.17img.cn/bbsfiles/images/2021/07/202107031131180784_3076_3356999_3.png!w690x268.jpg[/img]当时出现这种情况,便怀疑是对照品残留到了供试品中,然后减少了进样针数,实验数据如下:[img=减少对照进样次数,690,147]https://ng1.17img.cn/bbsfiles/images/2021/07/202107031133386103_879_3356999_3.png!w690x147.jpg[/img]供试品确实小了很多,而且也没有逐渐减小的情况,当时基本确定是对照品残留,因此后续实验调整了进样顺序,首先进供试品,但是又出现跟首次检验一样的情况,供试品目标峰面积逐渐减小,而后面的对照重复性也是相当不错做到此时怀疑是色谱柱问题,因此新采购了色谱柱,但供试品逐渐减小的问题仍然存在,后续又按照空白2针,供试品2针,空白2针,供试品2针的顺序进样,数据如下:[img=只进供试品和空白,690,267]https://ng1.17img.cn/bbsfiles/images/2021/07/202107031139383447_5262_3356999_3.png!w690x267.jpg[/img]令本人百思不得其解,请各位分析解答,谢谢~最后附上检验方法(仪器为安捷伦7890B,色谱柱为安捷伦VF-17ms,砂芯衬管)[img=,690,596]https://ng1.17img.cn/bbsfiles/images/2021/07/202107031143388912_5805_3356999_3.png!w690x596.jpg[/img][img=,690,368]https://ng1.17img.cn/bbsfiles/images/2021/07/202107031143564333_3950_3356999_3.png!w690x368.jpg[/img]
片碱+氯化钙+尿素+木钙+乙二醇为什么生成沉淀怎么解决
易制毒化学品的分类和品种目录第一类1.1-苯基-2-丙酮2.3,4-亚甲基二氧苯基-2-丙酮3.胡椒醛4.黄樟素5.黄樟油6.异黄樟素7. N-乙酰邻氨基苯酸8.邻氨基苯甲酸9.麦角酸*10.麦角胺*11.麦角新碱*12.麻黄素、伪麻黄素、消旋麻黄素、去甲麻黄素、甲基麻黄素、麻黄浸膏、麻黄浸膏粉等麻黄素类物质*第二类1.苯乙酸2.醋酸酐3.三氯甲烷4.乙醚5.哌啶第三类1.甲苯2.丙酮3.甲基乙基酮4.高锰酸钾5.硫酸6.盐酸说明:一、第一类、第二类所列物质可能存在的盐类,也纳入管制。二、带有*标记的品种为第一类中的药品类易制毒化学品,第一类中的药品类易制毒化学品包括原老药及其单方制剂。
实验室常会用到一些有毒有害的试剂,了解这些易制毒化学试剂,注意安全,至关重要。易制毒化学品分为三类:第一类是可以用于制毒的主要原料;第二类、第三类是可以用于制毒的化学配剂。易制毒化学品的分类和品种目录 第一类1. 1-苯基-2-丙酮;2. 3,4-亚甲基二氧苯基-2-丙酮;3. 胡椒醛;4. 黄樟素;5. 黄樟油;6. 异黄樟素;7. N-乙酰邻氨基苯酸;8. 邻氨基苯甲酸;9. 麦角酸*;10. 麦角胺*;11. 麦角新碱*;12. 麻黄素、伪麻黄素、消旋麻黄素、去甲麻黄素、甲基麻黄素、麻黄浸膏、麻黄浸膏粉等麻黄素类物质*;第二类1.苯乙酸;2.醋酸酐;3.三氯甲烷;4.乙醚;5.哌啶;第三类1.甲苯;2.丙酮;3.甲基乙基酮;4.高锰酸钾;5.硫酸;6.盐酸;第一类、第二类所列物质可能存在的盐类,也纳入管制。带有*标记的品种为第一类中的药品类易制毒化学品,第一类中的药品类易制毒化学品包括原料药及其单方制剂。
最近在开发一个原料药的乙二醇溶剂残留方法时,遇到一个问题,乙二醇在[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]中保留时间不一致,容易峰飘,时间相差0.1min中,我们产品是盐酸舒托必利,[img=,679,260]https://ng1.17img.cn/bbsfiles/images/2020/06/202006121037129934_4992_3176442_3.png!w679x260.jpg[/img]是一个盐酸盐,自动进样基质效应很强,所以样品前处理,需要加入氢氧化钠溶液,空白,对照,加标均同法处理,产品在水,甲醇,DMF,DMSO中溶解,开发方法选择用DMF做稀释剂,具体方法如下:见图,后续尝试过,把稀释剂换成水,甲醇,DMSO,依然对照中的乙二醇和加标中的乙二醇峰叠加图中,保留时间还是不能完全重叠,相差0.1min,也尝试过换色谱柱,DB-WAX、DB-624,情况还是没有改变,依然峰飘,一直解决不了这个问题,方法无法进行转移至QC,目前已经在做液相柱前衍生检测,各位老师能否帮忙分析一下,求指导。[img=,591,430]https://ng1.17img.cn/bbsfiles/images/2020/06/202006121036532031_7719_3176442_3.png!w591x430.jpg[/img]
各位师傅,小弟想知道乙二醇不同温度下的密度对照表。望知道的告知一下,谢谢
[color=#333333]乙二醇,丙二醇和二甲基是食品添加剂吗[/color]
供试品溶液制备:取本品1g,精密称定,置顶空瓶中,精密加入超纯水1.0ml, 密封,摇匀,作为供试品溶液。环氧乙烷对照贮备液制备:量取环氧乙烷300μl(相当于0.25g环氧乙烷),置含50ml经过滤处理的聚乙二醇400(以60℃,1.5~2.5kPa旋转蒸发6小时,除去挥发性成分)的100ml量瓶中,加入相同溶剂稀释至刻度,摇匀,作为环氧乙烷对照品贮备液。环氧乙烷对照品溶液制备:精密称取1g冷的环氧乙烷对照品贮备液,置含40ml经过处理的聚乙二醇400的50ml量瓶中,加相同溶剂稀释至刻度。精密称取10g,置含30ml水的50ml量瓶中,加水稀释至刻度。精密量取10ml,置50ml量瓶中,加水稀释至刻度,摇匀,作为环氧乙烷对照品溶液。二氧六环对照品溶液制备:取二氧六环适量,精密称定,用水制成每1ml中含0.1mg的溶液,作为二氧六环对照品溶液。混合对照品溶液制备:精密称取本品1g,置顶空瓶中,精密加入0.5ml环氧乙烷对照品溶液及0.5ml二氧六环对照品溶液,密封,摇匀,作为对照品溶液。系统适用性试验溶液制备:量取0.5ml环氧乙烷对照品溶液置顶空瓶中,加入新鲜配制的0.001%乙醛溶液0.1ml及二氧六环对照品溶液0.1ml,密封,摇匀,作为系统适用性试验溶液。试验条件:照气相色谱法试验,以聚二甲基硅氧烷为固定液,起始温度为35℃,维持5分钟,以每分钟5℃的速率升温至180℃,然后以每分钟30℃的速率升温至230℃,维持5分钟(可根据具体情况调整)。进样口温度150℃,检测器温度250℃,顶空瓶温度70℃,平衡时间为45分钟。取系统适用性试验溶液顶空进样,调节检测器灵敏度使环氧乙烷峰和乙醛峰的峰高约为满量程的15%,乙醛峰和环氧乙烷峰之间的分离度不小于2.0,二氧六环峰高应为基线噪音的5倍以上,分别取供试品溶液及对照品溶液顶空进样,重复进样至少3次。环氧乙烷峰面积的相对标准偏差应不得过15%,二氧六环峰面积的相对标准偏差应不得过10%,按标准加入法计算,环氧乙烷不得过0.0001%,二氧六环不得过0.001%。问题1:“相同溶剂稀释至刻度“是指水吗?问题2:求组计算过程或者指点一下,多谢。实在搞不清楚标准加入法的计算过程,一些人说要扣什么,已经糊涂了,灰常感谢。好人好报
题目:聚乙二醇化重组人生长激素的性质及活性
环氧乙烷和二氧六环 取本品1g,精密称定,置顶空瓶中,精密加入超纯水1.0ml,密封,摇匀,作为供试品溶 液。量取环氧乙烷300uL(相当于0.25g环氧乙烷),置含 50ml经过处理的聚乙二醇400(以60℃,1.5~2. 5kPa旋转蒸发6小时,除去挥发性成分)的100ml量瓶中,加入相同溶剂稀释至刻度,摇匀,作为环氧乙烷对照品贮备液,精密称取lg冷的环氧乙烷对照品贮备液,置含40ml经过处理的聚乙二醇400的50ml量瓶中,加相同溶剂稀释至刻度。精密称取10g,置含30ml水的50ml量瓶中,加水稀释至刻 度。精密量取10ml,置50ml量瓶中,加水稀释至刻度,摇匀,作为环氧乙烷对照品溶液。取二氧六环适量,精密称定,用水制成每1ml中含0.1mg的溶液,作为二氧六环对照品溶液。精密称取本品lg,置顶空瓶中,精密加入0.5ml 环氧乙烷对照品溶液及0.5ml 二氧六环对照品溶液,密封, 摇匀,作为对照品溶液。量取0.5ml环氧乙烷对照品溶液置 顶空瓶中,加入新鲜配制的0.001%乙醛溶液0.1ml及二氧六环对照品溶液0.1ml,密封,摇匀,作为系统适用性试验溶液,照[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url](通则0521)试验,以聚二甲基硅氧烷为固定液,起始温度为35℃,维持5钟,以每分钟5℃的 速率升温至180℃,然后以每分钟30℃的速率升温至 230℃,维持5分钟(可根据具体情况调整)。进样口温度为 150℃,检测器温度为250℃,顶空平衡温度为70℃,平衡时间为45分钟。取系统适用性试验溶液顶空进样,调节检测器灵敏度使环氧乙烷峰和乙醛峰的峰高约为满量程的 15%,乙醛峰和环氧乙烷峰之间的分离度不小于2.0,二氧六环峰高应为基线噪音的5倍以上,分别取供试品溶液及对照品溶液顶空进样,重复进样至少3次。环氧乙烷峰面积的相对标准偏差应不得过15%,二氧六环峰面积的相对标准偏差应不得过10%,按标准加入法计算,含环氧乙烷不得过0.0001%, 含二氧六环不得过0.001%。
各位有没有做药品乙二醇残留的,求乙二醇的[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]分析方法!
求助: 精制的乙二醇,需要测试里面的磷P 钴Co 锑Sb 等杂质含量。 请问哪里可以分析,宁波附近的优先。 联系方式:15058480703
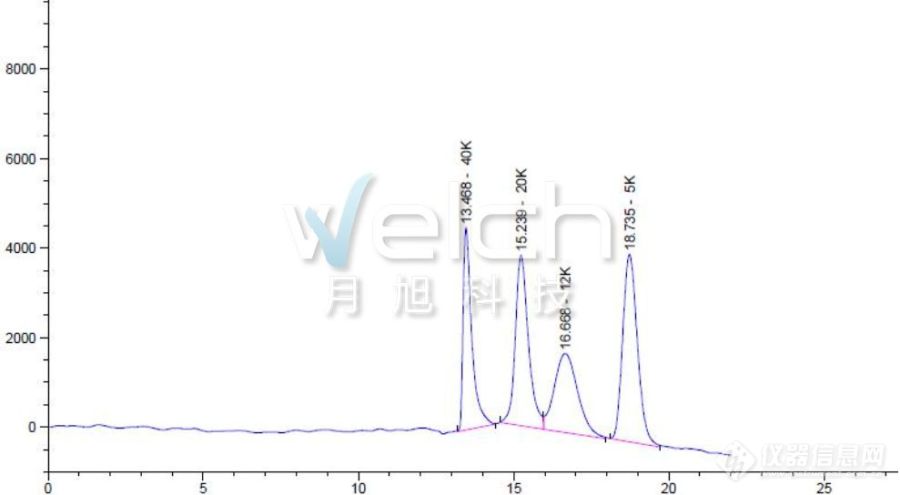
[b][/b][align=center][b]新增项目[/b][/align]最近在浏览国家药典委员会官网的时候,惊奇的发现聚乙二醇300,400,1000……全部的聚乙二醇品种药典方法都要修订了!在浏览了公示方法,发现原来是新增了一个项目,从2020版药典开始要标明重均分子量及分子量分布系数的标示值(按所附测定方法测定),那各位小伙伴了解分子量及分子量分布测定方法吗?这里就给大家详细讲讲。[b]2020版中国药典征求意见稿之分子量及分子量分布测定方法[/b]分别称取聚乙二醇600、聚乙二醇1000、聚乙二醇4000、聚乙二醇7000、聚乙二醇10000分子量对照品适量,加流动相溶解并稀释制成每1ml中约含2mg的溶液作为对照品溶液。称取样品适量,加流动相溶解并稀释制成每1ml中约含2mg的溶液作为供试品溶液。照分子排阻色谱法(通则0514)测定,采用适宜分离范围的凝胶色谱柱,以0.1mol/L硝酸钠溶液(含0.02%抑菌剂)为流动相,示差折光检测器;检测器温度35℃,柱温35℃,取对照品溶液各100μl注入液相色谱仪,记录色谱图,由GPC软件计算回归方程,线性相关系数R应不得小于0.99。取供试品溶液100μl,同法测定,根据回归方程计算供试品的重均分子量及分子量分布。供试品的重均分子量应为标示值的90%~110%,分布系数应为产品标示值的90%~110%。[b]什么是分子排阻色谱法[/b]分子排阻色谱法是根据待测组分的分子大小进行分离的一种液相色谱技术。分子排阻色谱法的分离原理为凝胶色谱柱的分子筛机制。色谱柱多以亲水硅胶、凝胶或经过修饰的凝胶如葡聚糖凝胶(Sephadex)和琼脂糖凝胶(Sepharose)等为填充剂,这些填充剂表面分布着不同孔径尺寸的孔,药物分子进入色谱柱后,它们中的不同组分按其分子大小进入相应的孔内,大于所有孔径的分子不能进入填充剂颗粒内部,在色谱过程中不被保留,最早被流动相洗脱至柱外,表现为保留时间较短;小于所有孔径的分子能自由进入填充剂表面的所有孔径,在色谱柱中滞留时间较长,表现为保留时间较长;其余分子则按分子大小依次被洗脱。[b]分子排阻色谱法应用案例[/b]月旭科技Xtimate SEC色谱柱是硅胶基质的分子排阻色谱柱,其色谱填料为高纯度、具有良好稳定性的硅胶微球表面键合亲水性聚合物。月旭科技采用特殊的表面修饰技术,确保了该填料具有良好的稳定性和批次重现性。Xtimate SEC色谱填料采用独特的化学键合技术,在硅胶表面键合了亲水性聚合物以及亲水性二醇基官能团,双重键合机制使水溶性高分子聚合物蛋白、生物酶、多肽等生物样品的非特异性吸附极小,因而可广泛应用于水溶性聚合物及生物大分子的分离和测定。月旭科技采用Xtimate SEC-300 (7.8*300mm,5μm)两根色谱柱串联的方式成功分离检测聚乙二醇40k,20k,12k,5k含量。[b]色谱柱:[/b]月旭Xtimate SEC-300 (7.8*300mm,5μm)两根色谱柱串联[b]流动相:[/b]高纯水[b]检测波长:[/b]示差检测器[b]柱温:[/b]柱温40℃,检测器40℃[b]流速:[/b]1.0ml/min[b]进样量:[/b]20μl[align=center][img=,690,379]https://ng1.17img.cn/bbsfiles/images/2019/10/201910160944591635_8915_932_3.jpg!w690x379.jpg[/img][/align][align=center][img=,414,40]https://ng1.17img.cn/bbsfiles/images/2019/10/201910160946105076_5570_932_3.png!w414x40.jpg[/img][/align]各位小伙伴想了解更多有关分子排阻色谱法的相关信息,请咨询月旭当地销售人员或拨打400-810-6969垂询。
[b]求教在做呕吐毒素时加聚乙二醇8000的作用 是不是 不加也没什么问题[/b]
易制毒化学品的分类和品种目录: 第一类 1.1-苯基-2-丙酮 2.3,4-亚甲基二氧苯基-2-丙酮 3.胡椒醛 4.黄樟素 5.黄樟油 6.异黄樟素 7. N-乙酰邻氨基苯酸 8.邻氨基苯甲酸 9.麦角酸* 10.麦角胺* 11.麦角新碱* 12.麻黄素、伪麻黄素、消旋麻黄素、去甲麻黄素、甲基麻黄素、麻黄浸膏、麻黄浸膏粉等麻黄素类物质* 第二类 1.苯乙酸 2.醋酸酐 3.三氯甲烷 4.乙醚 5.哌啶 第三类 1.甲苯 2.丙酮 3.甲基乙基酮 4.高锰酸钾 5.硫酸 6.盐酸 说明: 一、第一类、第二类所列物质可能存在的盐类,也纳入管制。 二、带有*标记的品种为第一类中的药品类易制毒化学品,第一类中的药品类易制毒化学品包括原料药及其单方制剂。[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=13318]易制毒化学品管理条例[/url]
生姜是餐桌上不可或缺的养生食材,除了能调味、温中,还有着极好的杀菌、消炎、解暑、解毒效果。所谓月满则亏,凡事过度都会适得其反,食用生姜过度,或者吃法不对皆会影响身体健康,甚至会毒如砒霜、致癌。下面就给大家介绍几种错误的生姜吃法,希望大家在平时的饮食中加以注意。 生姜是药食同源的典型代表 生姜是日常生活中非常普遍的一种食材,亦可称之为中药材,其是药食同源的典型代表,无论是食用价值还是药用价值都非常高。生姜是我国烹饪文化中非常重要的一种调味料,无论是烧荤菜还是素菜,生姜的辛辣味和特殊芳香味都可为菜肴增加另一番风味,有些菜的烹饪甚至离不开生姜。不仅如此,生姜还可制成酱菜,单独成菜品为我们食用。 另一方面,生姜亦是一款中药材。很多女性朋友都有这样的经历,就是经期时会喝生姜红糖水来缓解痛经,而很多风寒感冒的朋友亦会用生姜红糖水来驱寒,而这就是生姜的药用价值之一。生姜性温味辛,归脾、肺经,具有发汗解表、温中健脾、止咳止吐、温肺强心等功效。对于风寒感冒、女性经痛、畏寒呕吐、过食寒凉之物引起的肠胃不适等有极好的缓解作用。 生姜中含黄樟素,多食可致癌 生姜中除了含有糖类、蛋白质、氨基酸、维生素、矿物质等常见营养素,其还含有很多特殊的营养成分,如具有杀菌作用的植物抗菌素、抑制胆固醇吸收的挥发油、具有抗氧化作用的姜辣素等。但是值得大家注意的是,生姜除了拥有以上几种有助于人体健康的营养元素,其还含有一种成分,名叫黄樟素。 黄樟素是一种食品添加剂,为无色或浅黄色液体,有樟木气味,天然存在与生姜、茴香、八角、桂皮、花椒等香辛作料中,低毒,一定量级可致癌。所以过食生姜有可能增加癌症的患发率,尤其是肝癌。美国食品药物管理局(FDA)的研究显示:黄樟素可引起肝癌,在小鼠的饲料中添加0.04%~1%的黄樟素,150天到2年,可诱导小鼠产生肝癌,若黄樟素在体内遇到氧化剂,则会产生更强致癌活性的环氧黄樟素,亦大幅增加人体罹患肝癌的可能性。所以在平时的烹饪过程中,一定要适度使用生姜,切忌无限制的使用。 怎样缓解黄樟素对人体的危害 过食生姜以后怎样中和黄樟素对人体造成的危害呢?最好的方法就是吃水果和蔬菜,新鲜的果蔬能有效地抑制黄樟素与氧化剂结合,从而降低黄樟素转化为环氧黄樟素的几率,从而降低黄樟素的致癌效应,所以大家吃完生姜以后一定要多食果蔬。 烂姜可产生的毒素,亦会致癌 在生活中,很多人都认为“烂姜不烂味”,即使生姜有些烂了,也会把其削掉,继续使用,其实这是一种非常危险的做法,长期如此或可致癌。腐烂的生姜亦会产生毒素,即使切除,仍然会有致癌的可能,严重时甚至会导致机体罹患食道癌和肝癌。所以,生姜一旦出现腐烂,不要舍不得扔掉,一定要杜绝吃腐烂的生姜。 要想身体好,生姜要这么吃 第一,留皮 有过烧菜经验的朋友一定会有这样的经历,就是使用生姜调味的时候,会去皮,其实这样做会大大削减生姜的营养价值。其实生姜皮与生姜肉的性能是完全不同的,一个性凉,一个性温。《医林纂要》中曾记载“姜皮辛寒”,而中医理论也有留姜皮则凉,去姜皮则热”之说。所以在食用生姜的时候尽量留皮,从而发挥出生姜更为全面的营养价值。 第二,上火时别吃 上火别吃生姜大家都知道,但是很多阴虚火旺的朋友并不知道自己不宜吃生姜。生姜性温,所以天气炎热时、机体上火时亦不宜吃生姜。 结语:生姜的食用价值、营养价值大家并不陌生,只是过犹不及,生姜虽好,吃不对就会如砒霜,吃多了或者吃烂掉的生姜还会致癌。所以在平时的生活中,不要误解“夏吃姜”这一养生论据,盲目多食生姜,一定要适度适量。
领导让做聚丙烯酰胺的离子度,标准17514-2017里面提到甲基乙二醇甲壳素(MGC)标准溶液0.005mol/L,请问我要怎么采购,供应商那里说没有这东西,只有甲基乙二醇壳聚糖
找标样的问题(说是广告,不让发帖)http://ng1.17img.cn/bbsfiles/images/2011/09/201109162331_317252_1615838_3.jpg







 我要推广仪器
我要推广仪器
 下载APP
下载APP