怎么从做的标准品(10倍系列稀释)的色谱数据中找出目标峰。比如:标准品是原薯蓣皂苷,怎么找出原薯蓣皂苷的峰?
[color=#444444]HPLC测定人参皂苷Ra1对照品含量以色谱纯乙腈-水为流动相,为什么所得结果只有前五分钟的溶剂峰,却没有想要的保留时间为16分钟左右的峰呢?[/color]
不同产地百合药材中薯蓣皂苷元及总皂苷元的含量测定 帮忙下载下啦
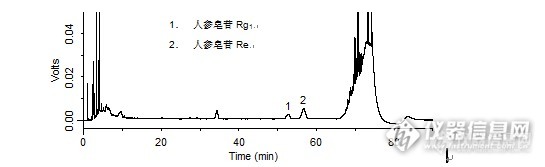
问题:启脾口服液中人参皂苷Rg1、人参皂苷Re的检测:对照品中人参皂苷Rg1、人参皂苷Re的分离度是多少?答案:2.467获奖名单:吕梁山(ID:shih20j07)dahua1981(ID:dahua1981)m3071659(ID:m3071659)http://ng1.17img.cn/bbsfiles/images/2016/02/201602231542_584931_708_3.jpghttp://ng1.17img.cn/bbsfiles/images/2016/02/201602231543_584932_708_3.jpghttp://ng1.17img.cn/bbsfiles/images/2016/02/201602231543_584933_708_3.jpg【活动奖励】幸运奖(2钻石币):抽奖软件,当天随机抽取3个回答正确的版友ID号(最后一个ID号,截止至下午3:00),每人奖励2个钻石币积分奖励:所有回答正确的版友奖励10个积分(幸运奖获得者除外)。【注意事项】同样的答案,每人只能发一次PS:该贴浏览权限为“回贴仅作者和自己可见”,回复的版友仅能看到版主的题目及自己的回答内容,无法看到其他版友的回复内容。下午3点之后解除,即可看到正确答案、获奖情况及所有版友的回复内容。启脾口服液中人参皂苷Rg1、人参皂苷Re的检测样品制备 制备方法1. 对照品:取人参皂苷Rg1对照品、人参皂苷Re对照品适量,精密称定,加甲醇制成每1 mL各含0.25 mg的混合溶液,摇匀,即得。2. 供试品:精密量取本品50 mL,加三氯甲烷振摇提取3次,每次30 mL,弃去三氯甲烷提取液,水液加水饱和正丁醇振摇提取5次(50 mL、30 mL、30 mL、20 mL、20 mL),合并正丁醇提取液,加氨试液洗涤4次,每次50 mL,弃去氨试液,再加正丁醇饱和的水轻轻振摇洗涤2次,每次50 mL,弃去水洗液,正丁醇液回收溶剂至干,残渣加甲醇溶解并转移至5 mL量瓶中,加甲醇稀释至刻度,摇匀,滤过,取续滤液,即得。分析条件 色谱柱Diamonsil C18(2) 250 × 4.6 mm,5 μm (Cat#:99603) 流动相A:水 B:乙腈 梯度流速1.0 mL/min 柱温35 ℃ 检测器UV 203 nm 进样量5 μL 色谱图对照品http://ng1.17img.cn/bbsfiles/images/2016/02/201602231201_584890_708_3.png 峰号 保留时间 min 峰面积 μV*s 峰高 μV 理论塔板数* N USP拖尾因子 分离度 1 52.603 387347 6585 18460.678 1.019 -- 2 56.668 262702 4169 16820.782 0.971 2.467 *药典要求理论板数按人参皂苷Re峰计算应不低于2500 供试品http://ng1.17img.cn/bbsfiles/images/2016/02/201602231202_584891_708_3.png 峰号 保留时间 min 峰面积 μV*s 峰高 μV 理论塔板数* N USP拖尾因子 分离度 1 52.664 117855 2108 18875.714 0.982 -- 2 56.654 291453 4502 18266.289 1.019 2.486 *药典要求理论板数按人参皂苷Re峰计算应不低于2500本品种同时使用了Leapsil C18色谱柱,在药典规定条件下进行人参皂苷Rg1[/sub
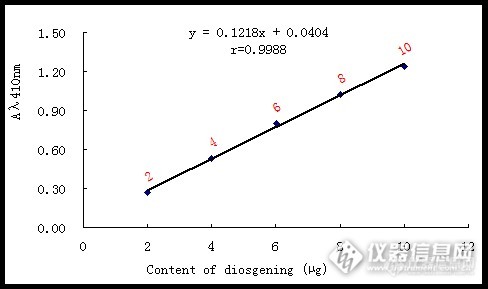
1. 实验材料薯蓣皂苷元标准品母液的配置:精确称取薯蓣皂苷元标准品(纯度98%)5mg,溶于10ml甲醇中,母液浓度为0.5mg/ml(0.5μg/μL),现用现配;2. 实验方法标准曲线的建立:分别吸取0、4、8、12、16、20μL 薯蓣皂苷元标准母液于新的96微孔板的每个孔中,每个浓度3个重复,室温条件下挥干溶剂,然后每个孔内添加200μL 70-72%的高氯酸,然后迅速将微孔板放入经预热的微孔板分光光度计内,35℃,摇振2min,静止10min后,在350nm~700nm波长范围内扫描其最大吸收波长,并在最大吸收波长下测定吸光值,根据最大吸收波长及系列标准溶液中的diosgenin的含量,绘制标准曲线。 图1为薯蓣皂苷元在在350nm~700nm波长范围内的光谱扫描图,根据图1所示,可确定其最佳吸收波长为410nm。 图2为根据高氯酸显色分光光度法最终得到的标准曲线,线性回归方程为y=0.1218x+0.0404,r=0.9988。y代表410nm处的吸光值,x代表反应体系中的diosgenin含量(μg)。http://ng1.17img.cn/bbsfiles/images/2015/09/201509171254_566402_3033448_3.png图1 薯蓣皂苷元的光谱扫描图http://ng1.17img.cn/bbsfiles/images/2015/09/201509171255_566403_3033448_3.png图2 薯蓣皂苷元的标准曲线3.结果与讨论样品中薯蓣皂苷元含量的确定:将提取制备的薯蓣皂苷元粗提物用甲醇完全溶解,然后吸取体积的样品溶液至新的96-微孔板的孔内,室温下挥干溶剂,然后按照上述实验条件进行检测,最后根据其在410nm处的吸光值和线性回归方程y=0.1218x+0.0404计算,可得出薯蓣皂苷元的含量。
大家都用那些容积配置过黄芩苷对照品啊?怎么我们用稀乙醇溶液溶解的时候不易溶解啊,超声半小时还会有很多不容物?
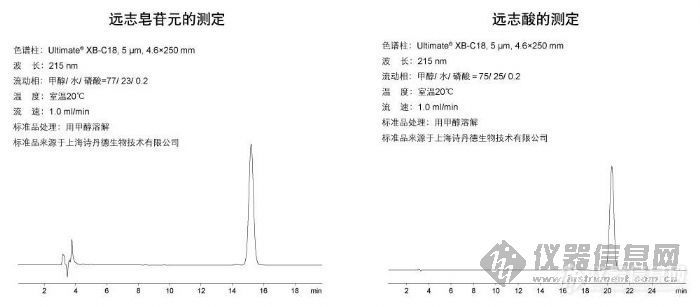
远志皂苷元的测定和远志酸的测定http://ng1.17img.cn/bbsfiles/images/2009/10/200910130307_175402_1896702_3.jpg
目的:建立麝香接骨胶囊的含量测定方法。方法:采用高效液相色谱法。色谱柱:Agilent Technologies ZORBAX Extend-C18 4.6×250mm, 5μm,流动相:乙腈-水(21:79 V/V),流速:1.0mlmin-1,检测波长:203nm,柱温:30℃。结果:三七皂苷R1在0.005~0.25mgml-1范围内线性关系良好(r=0.9998);平均加样回收率为97.6%, RSD=1.0%(n=6)。结论:该方法简便易行,结果准确可靠,可适用于麝香接骨胶囊的质量控制。 【关键词】高效液相色谱法 麝香接骨胶囊 三七皂苷R1 含量测定 Determination of Forsythin content in Shexiang Jiegu Jiaonang by HPLC 【Abstract】 OBJECTIVE:To set up a method for quality control of Shexiang Jiegu Jiaonang METHOD HPLC method was developed to quantitative determination. COLUMN Agilent Technologies ZORBAX Extend-C18 4.6×250mm, 5μm.The mobilic phase was acetonifrile-water (21:79V/V).The column temperature was 30℃,the wavelength for detection was 203nm,flow rate was 1.0mlmin-1. RESULTS The paeonol average of recovery rate was 97.6%, and RSD was 1.0%(n=6). CONCLUSION The method is simple, accurate and suitable for its assaying. 【Key word】 HPLC Shexiang Jiegu Jiaonang Notoginsenoside R1 determination 麝香接骨胶囊为《卫生部药品标准》第五册(中药成方制剂)收载的品种,由赤芍、三七、当归、续断、苏木、麝香等22味中药组成,具有散瘀止痛,续筋接骨之功效。临床用于跌打损伤,筋伤骨折,瘀血凝结,闪腰岔气[1]。处方中三七为主药之一,三七皂苷R1为其主要有效成分,原标准中无含量测定方法。为了更好地控制制剂的内在质量,本文采用高效液相色谱法测定麝香接骨胶囊中三七皂苷R1的含量,以期为该制剂的质量提供快速、准确的测定方法,现报告如下。 1 仪器与试药 LC-2010A 高效液相色谱仪,CLASS-VP 色谱工作站,紫外检测器;三七皂苷R1对照品(批号:110745-200312),由中国药品生物制品检定所提供,供含量测定用;麝香接骨胶囊为市售样品(辽源市亚东中药有限责任公司,规格0.3g粒-1,批号:060801,061102,060912)。乙腈为色谱纯;水为超纯水;其他试剂均为分析纯。 2 方法与结果 2.1 色谱条件与系统适用性试验 色谱柱:Agilent Technologies ZORBAX Extend-C18 4.6×250mm, 5μm;流动相:乙腈-水(21:79, V/V);检测波长:203nm;流速:1.0mlmin-1;柱温:30℃。理论板数按三七皂苷R1峰计应不低于4000。 2.2 溶液制备 2.2.1 对照品溶液的制备 精密称取经五氧化二磷减压干燥12小时以上的三七皂苷R1对照品适量,加甲醇制成0.25 mgml-1的对照品贮备液;再精密量取对照品贮备液适量,加甲醇制成0.05mgml-1的对照品溶液,即得。 2.2.2 供试品溶液的制备 取装量差异项下的麝香接骨胶囊内容物,研细,取约1g,精密称定,置具塞锥形瓶中,精密加入甲醇50ml,密塞,称定重量,超声处理(功率250W,频率50 kHz)1小时,取出,放冷,再称定重量,用甲醇补足减失的重量,摇匀,滤过,精密量取续滤液25ml,置蒸发皿中,蒸干,残渣加水饱和的正丁醇30ml使溶解,加氨试液振摇提取3次,每次15ml,合并氨试液提取液,再用水饱和的正丁醇振摇提取2次,每次15ml,与上述正丁醇液合并,蒸干,残渣加甲醇适量使溶解,转移置10ml量瓶中,加甲醇稀释置刻度,摇匀,用微孔滤膜(0.22μm)滤过,取续滤液,即得[2]。 2.2.3 阴性对照溶液的制备 取除三七皂苷R1以外的处方中其余药材的十分之一量,按法制成胶囊,再按供试品溶液制备方法,制成阴性对照溶液。 2.3 专属性试验 分别精密吸取供试品溶液、阴性对照溶液和对照品溶液各10μl注入液相色谱仪,记录色谱图(图1)。由图1可见,供试品色谱中,在与对照品色谱相应的位置上,有相同保留时间(三七皂苷R110.971min)的色谱峰,阴性试验无干扰,证明本法可行。2.4 线性关系考察 精密吸取三七皂苷R1对照品贮备液(浓度:0.25mg.ml-1)适量,加甲醇配成浓度分别为0.005、0.01、0.05、0.10、0.15、0.25mgml-1的溶液6份,摇匀,用0.22μm微孔滤膜过滤。精密吸取续滤液各10μl,注入液相色谱仪,测得峰面积。以峰面积(A)为纵坐标,其浓度(C)为横坐标,得三七皂苷R1的回归方程为:A=392250.2 C +8620.1,r =0.9998(n=6)。结果表明,三七皂苷R1在0.005~0.25mgml-1范围内峰面积与其浓度呈良好线性关系。 2.5 精密度试验 精密吸取三七皂苷R1对照品溶液(0.05mgml-1)10μl,在上述色谱条件下重复进样6次,求得三七皂苷R1溶液峰面积的相对标准偏差(RSD)为0.3%(n=6),表明精密度较好。 2.6 稳定性试验 精密吸取三七皂苷R1对照品溶液(0.05mgml-1)10μl,在0、4、8、12、24、48h分别进行测定,结果表明三七皂苷R1对照品溶液在48h内稳定, RSD为0.4%(n=6)。 2.7 重复性试验 取供试品(批号061101),共6份,分别按“2.2.2供试品溶液的制备”方法制备供试品溶液,进行测定,求得三七皂苷R1含量的RSD为1.6%(n=6),表明重现性较好。 2.8 加样回收率试验 精密称取已知含量的样品(批号061101,三七皂苷R1含量0.93mgg-1,平均装量0.2996g粒-1)适量,共6份,分别置具塞锥形瓶中,分别精密加入三七皂苷R1对照品贮备液(0.25mgml-1)各3ml,按“2.2.2供试品溶液的制备”方法操作,得回收率试验溶液,依法测定,结果见表1。表1 三七皂苷R1加样回收率试验(略) 2.9 样品含量测定 分别精密吸取供试品溶液与对照品溶液各10μl,注入液相色谱仪,测定三批样品中三七皂苷R1峰面积,按外标法计算其含量(n=5),结果见表2。表2 3批样品含量测定结果(略) 3 讨论 3.1 通过对3批样品中三七皂苷R1的测定,结果表明最高含量为0.31mg粒-1,最低为0.27mg粒-1,综合考虑确定限量每粒含三七皂苷R1不得低于0.20mg。 3.2 流动相的选择 试验中采用不同的流动相甲醇-水(25︰75),乙腈-水-冰醋酸(25︰75︰0.5),结果表明采用流动相乙腈-水(21︰79)的色谱图基线较稳。 3.3 本方法结果准确,方法简便,重现性及回收率均理想,可以作为该制剂的质量控制方法。
摘要:目的:利用薄层扫描法分别对薯蓣科植物粉背薯蓣及绵萆薜药材进行薯蓣皂苷元的含量测定。方法 分别取粉苹薜、绵萆薜各3批经提取后点样,以氯仿:丙酮(9.7:0.3)为展开剂,采用薄层扫描法,检测波长λs=480nm,λR=700nm。结果平均回收率为100.1%和99.64%,RSD为1.42%和2.10%,稳定实验RSD=2.26%。结论本方法方便、简单、准确,可用于该品种的质量控制。 粉萆薜和绵萆薜均为少常用中药《中国药典2000年版有收载。粉萆藓为薯蓣科植物粉背薯蓣Dioicorea hypoglauca Palibin的干燥根茎。具有利湿去浊,祛风除痹之功效。用于膏淋,白浊,白带过多,风湿痹痛,关节不利,腰膝疼痛。绵萆薜为薯蓣科植物绵萆藓Dioscorea sepemloba Thunb的干燥根茎。具有利湿去浊,祛风痛通痹之功效,用于淋病白浊,白带过多,湿热疮痛,腰膝痹痛。二者均为秋冬二季采挖,除去须根,洗净,切片,晒干。 1 仪器与材料 CS-9301PC型薄层色谱扫描仪(日本岛津公司);939型薄层制板器(重庆南岸贝尔德仪器技术厂);定量点样毛细管(USA Drummond Scientific Co.);硅胶G(青岛海洋化工有限公司);羧甲基纤维素钠(上海化学试剂采购供应站);薯蓣皂苷元(中国药品生物制品检定所200001含量测定用);粉萆薜和绵萆薜均由广西中医药研究所赖茂祥副研究员鉴定后提供。所用试剂均为分析纯。
配制两个黄芩苷对照品溶液第一个是用50%的甲醇溶解,我感觉这个好难溶,超声了也不见得全部溶解,如果全部溶解配制出来的应该是澄清的吧?该如何处理好呢??第二个要用减压干燥器60度干燥4小时再配制,我觉得涂了凡士林在60度会溶了吧,就不能减压了,我可以直接打开对照品瓶盖直接在烘箱里烘4小时不?这样做会有很大的影响吗?
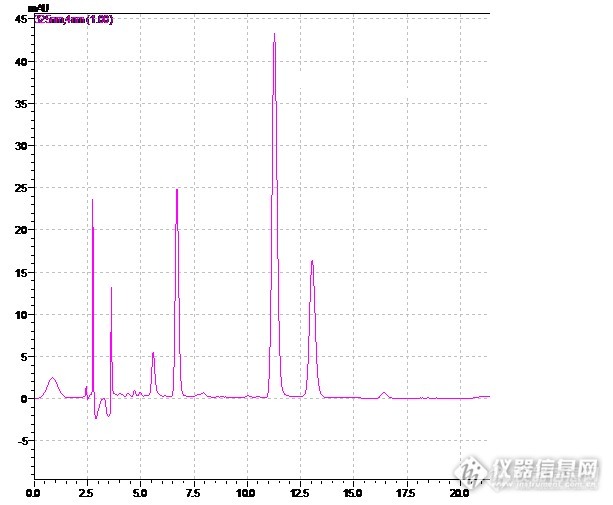
摘要: 目的 建立茵栀黄颗粒中栀子苷和绿原酸的含量测定方法。方法 采用高效液相色谱法。色谱柱:Kromasil C18柱(4.6mm×250mm,5μm);流动相:乙腈-0.1%甲酸(10:90);流速1.0ml/min;柱温:35℃;检测波长:238nm、325nm。结果:对茵栀黄颗粒中栀子苷和绿原酸进行含量测定。 栀子苷在10.43~52.15μg范围内呈线性关系(r=0.9999 ,n=6),线性方程为y=123721821.7x-6951.8(r=0.9999)。平均加样回收率为97.21% RSD为1.62% (n=6) 绿原酸在10.74~53.70μg范围内呈线性关系(r=0.9999 ,n=6),线性方程为y=29447356x-25436(r=0.9999)。平均加样回收率为96.61% RSD为1.35% (n=6)结论 该方法操作简便,结果可靠,可用于茵栀黄颗粒中栀子苷和绿原酸的含量测定。 清茵栀黄颗粒,清热解毒,利湿退黄。有退黄疸和降低谷丙转氨酶的作用。用于湿热毒邪内蕴所致急性、慢性肝炎和重症肝炎(I型)。也可用于其他型重症肝炎的综合治疗。由陈提取物、栀子提取物、黄芩苷、金银花提取物组成。为了进一步控制药品质量,我们对其中的进行了栀子苷和绿原酸含量测定,以期为后续进一步研究奠定基础!关键词:HPLC;DAD;茵栀黄颗粒;栀子苷 ;绿原酸1 仪器与试药1.1 仪器 岛津LC-20AT高效液相色谱仪,SPD-M20A检测器, LCsolution色谱工作站, Metteler AB265S(0.01mg);Sartorius BS124S(0.1mg)),必能信超声波清洗器(必能信超声有限公司)1.2 试药 绿原酸对照品(批号110753-201314)、栀子苷对照品(批号:110749-201115),均购自中国食品药品检定研究院;乙腈(色谱纯);其它试剂均为国产分析纯,蒸馏水自制。2 实验方法与结果2.1 色谱条件与图谱 色谱柱: Kromasil C18柱(4.6mmx250mm,5um); 检测波长:238nm;325nm 流速:1.0ml.min-1; 流动相:乙腈-0.1%甲酸(10:90); 柱温:35℃。2.2 溶液制备2.2.1对照品溶液 取栀子苷和绿原酸对照品适量,精密称定,加甲醇制成每1ml 分别含0.1043mg和0.1074mg 的混合溶液,即得。2.2.2供试品溶液 取本品,研细,取约0.5g,精密称定置50ml量瓶中,精密加入甲醇40ml,称定重量,超声处理30min ,再称定重量,用甲醇补足减失的重量,定容至50ml,摇匀,滤过,即得。2.3 线性关系考察 精密量取各对照品溶液,制得5个系列不同浓度的对照品溶液,各取10微升依次进样,按上述色谱条件测定峰面积,以对照品峰面积Y为纵坐标,浓度X(mg·mL )为横坐标,绘制标准曲线,结果栀子苷在10.43~52.15μg范围内呈线性关系(r=0.9999 ,n=6),线性方程为y=123721821.7x-6951.8(r=0.9999)。绿原酸在10.74~53.70μg范围内呈线性关系(r=0.9999 ,n=6),线性方程为y=58134683.4x-7988.3(r=0.9999) http://ng1.17img.cn/bbsfiles/images/2015/10/201510020006_569010_1839779_3.pnghttp://ng1.17img.cn/bbsfiles/images/2017/10/2015100200003568_01_1839779_3.png2.4精密度试验 精密吸取同一供试品溶液,重复进样6次,计算栀子苷和绿原酸对照品 峰面积的RSD分别为0.4% 和1.4% 。2.5 稳定性试验 精密吸取同一供试品溶液,分别于0,2,4,8,16,24 h测定,结果表明:栀子苷和绿原酸对照品在24 h内稳定,RSD分别为1.1% 和1.5% 。2.6 重复性实验 取同一批次的茵栀黄颗粒平行6份,制成供试品溶液,在上述色谱条件下分别进行HPLC分析,计算栀子苷和绿原酸对照品峰面积的RSD分别为1.2%和1.4% 。2.7 回收试验 取同一批茵栀黄颗粒,约取0.25g,共计6份,精密称定,按上述色谱条件进样测定,用回归方程计算回收率。2.8 样品含量测定 取3个批号的茵栀黄颗粒,配制成供试品溶液,在上述色谱条件下进行测定,栀子苷和绿原酸对照品的含量,结果见表。样品编号栀子苷(mg/g)20150729 2.54201410032.61201502162.65样品编号绿原酸(mg/g)20150729 1.04201410031.16201502161.12http://ng1.17img.cn/bbsfiles/images/2017/10/2015100111035245_01_1839779_3.bmp 图1 茵栀黄颗粒的HPLC图谱(238nm)http://ng1.17img.cn/bbsfiles/images/2017/10/2015100111125995_01_1839779_3.bmp 图2 栀子苷的HPLC图谱(238nm)http://ng1.17img.cn/bbsfiles/images/2017/10/2015100111105517_01_1839779_3.png 图3 茵栀黄颗粒的HPLC图谱(325nm)http://ng1.17img.cn/bbsfiles/images/2017/10/2015100111150072_01_1839779_3.bmp 图4 绿原酸的HPLC图谱(325

问题:颈痛颗粒中三七皂苷、人参皂苷的检测使用了哪几款液相色谱柱?答案:Platisil ODS、Leapsil C18、Spursil C18-EP【活动奖励】幸运奖(2钻石币):抽奖软件,当天随机抽取3个回答正确的版友ID号(最后一个ID号,截止至下午3:00),每人奖励2个钻石币mengzhaocheng(ID:mengzhaocheng)999youran(注册ID:999youran)WUYUWUQIU(注册ID:wulin321)http://ng1.17img.cn/bbsfiles/images/2016/01/201601291556_583923_1610895_3.pnghttp://ng1.17img.cn/bbsfiles/images/2016/01/201601291556_583924_1610895_3.png积分奖励:所有回答正确的版友奖励10个积分(幸运奖获得者除外)。【注意事项】同样的答案,每人只能发一次PS:该贴浏览权限为“回贴仅作者和自己可见”,回复的版友仅能看到版主的题目及自己的回答内容,无法看到其他版友的回复内容。下午3点之后解除,即可看到正确答案、获奖情况及所有版友的回复内容。=======================================================================颈痛颗粒中三七皂苷、人参皂苷的检测样品制备制备方法1. 对照品:取人参皂苷Rg1对照品、人参皂苷Rb1对照品和三七皂苷R1对照品适量,精密称定,加甲醇制成每 1 mL含人参皂苷Rg1 0.5 mg、人参皂苷Rb1 0.5 mg、三七皂苷R1 0.1 mg的混合溶液,即得。2. 供试品:取装量差异项下的本品适量,研细,取约1.3 g,精密称定,加乙醚40 mL,加热回流30分钟,滤过,弃去乙醚液,药渣及滤纸挥尽乙醚,再精密加入甲醇40 mL,称定重量,加热回流1小时,放冷,再称定重量,用甲醇补足减失的重量,摇匀,滤过,精密量取续滤液20 mL,回收溶剂至干,残渣加水10 mL使溶解,用水饱和的正丁醇振摇提取5次,每次10 mL,合并正丁醇提取液,用2%碳酸钠溶液洗涤2次,每次20 mL,再用正丁醇饱和的水洗涤2次,每次20 mL,取正丁醇液回收溶剂至干,残渣用甲醇溶解,转移至10 mL量瓶中,加甲醇稀释至刻度,摇匀,即得。分析条件色谱柱Platisil ODS 250 x 4.6 mm,5 μm (Cat#:99503)流动相A:水 B:乙腈 梯度流速1.0 mL/min柱温30 ℃检测器UV 203 nm进样量10 μL色谱图对照品 http://ng1.17img.cn/bbsfiles/images/2016/01/201601290942_583886_1610895_3.jpg 峰号 保留时间 min 峰面积 μV*s 峰高 μV 理论塔板数* N USP拖尾因子 分离度 1 36.408 264983 25847 265850.102 1.018 -- 2 38.915 1775914 178004 318998.439 0.998 8.983 3 54.506 1316933 127848 597818.242 0.870 55.926 *药典要求理论板数按三七皂苷 R1峰计算应不低于3000供试品http://ng1.17img.cn/bbsfiles/images/2016/01/201601290943_583887_1610895_3.jpg 峰号 保留时间 min 峰面积 μV*s 峰高 μV 理论塔板数* N USP拖尾因子 分离度 1 36.344 250230 25147 274216.739 0.996 --
葛根配方颗粒特征图谱黄豆苷元对照品配制时,在水浴上加热溶解了,但是放冷就析出了,有大神可以指导一下吗?
请问一下各位:车前醚苷对照品哪里有卖呢?
小弟在测甾体皂苷,用的是已腈-磷酸水(酸万分之一),梯度洗脱,DAD检测。待测样品用普通分析纯的甲醇溶解,微孔滤膜过滤,进样分析。做了好几次,发现皂苷的峰不明显,并且更要人命的是,谱图中有很多溶剂峰,强度很大很高,现在才意识到,以前都当成皂苷的吸收峰,因为不像溶剂峰,都是末端吸收,没办法区分。今天,在进对照品时,发现对照品进入液相后,有好几个末端吸收峰,发现问题比较严重,对照品纯度没问题的。我怀疑:1、普通分析甲醇溶解对照品和样品后,才导致出现谱图中不同位置出现大小不一样的溶剂峰。我马上做了空白对照,确实甲醇单独进入液相分析,有很多溶剂峰,末端吸收,真是无语。2、考虑到溶剂问题,我又做了已腈空白对照,发现还是有溶剂峰,只不过少了很多,只有两个看起来比较高的峰。一个出峰在10分钟,一个在很靠后。接下来,我想做下面尝试:1、用流动相来溶解样品,就是已腈-磷酸水,同时,先做溶剂空白。2、考虑用已腈溶解样品紫外检测,感觉皂苷的峰不灵敏,但是比较实用,所以还是选择用紫外检测。对以上问题,请各位朋友多多指导。十分感谢!!![em09508]
目的:建立HPLC-ELSD 检测酸枣仁制剂中酸枣仁皂苷a的含量方法。方法:采用Diamonsil C18色谱柱以乙腈-水(25∶75) 为流动相;ELSD:流速1.0mL/min,ELSD漂移管温度110 ℃,载气(N2)流速2.8 L/min结果:酸枣仁皂苷A进样量在0.104~1.04μg范围内线性良好( r= 0.9998); 平均回收率为95.6%。结论:本法准确、灵敏、重复性好,为酸枣仁制剂的质量控制提供了依据。1.仪器与试药1.1仪器 安捷伦1100高效液相色谱色谱仪,Alltech 2000ES型蒸发光散射检测器,KQ250超声波清洗器(江苏昆山超声仪器厂),高速离心机(上海飞鸽)十万分之一分析天平(梅特勒公司)。1.2试药桔梗药材:由市场购买提供。经鉴定符合2010版中国药典规定。对照品:酸枣仁皂苷对照品(购自中国食品药品鉴定研究院,批号:111851-201003)乙腈为色谱纯,其他试剂均为分析纯。2 方法与结果2.1 色谱条件 Diamonsil C18色谱柱 (5um, 4.6*250mm),流动相乙腈-水( 25:75) ,柱温30℃,流速1.0 mL·min - 1 ,进样量10μL[size=12pt
用药典方法测定柴胡药材中柴胡皂苷a、d的含量,刚开始预实验,对照品和样品的峰型都挺好,后来平衡好色谱柱,直接批量进针,问题就出现了,无论是对照还是样品,柴胡皂苷a的峰高变低,峰型变胖,保留时间也提前了4分钟,但是柴胡皂苷d没有什么变化,求助大家,问题在哪?而且每个样品进3针,第1针与第2、3针的柴胡皂苷a在保留时间上也有0.5分钟的差别...
[font=宋体]【[b]含量测定[/b]】 照高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]法(通则0512)测定。[/font][b][font=宋体]色谱条件与系统适用性试验[/font][/b][font=宋体] 以十八烷基硅烷键合硅胶为填充剂;以乙腈为流动相A,以0.1%甲酸为流动相B,按下表中的规定进行梯度洗脱;乌药碱检测波长为227nm,木兰花碱、酸枣仁皂苷A、酸枣仁皂苷D为蒸发光散射检测器检测。理论板数按斯皮诺素和酸枣仁皂苷A峰计算均应不低于2000。[/font][table][tr][td=1,1,201][font=宋体]时间(分钟)[/font][/td][td=1,1,185][font=宋体]流动相A(%)[/font][/td][td=1,1,182][font=宋体]流动相B(%)[/font][/td][/tr][tr][td=1,1,201][font=宋体]0~25[/font][/td][td=1,1,185][font=宋体]10→20[/font][/td][td=1,1,182][font=宋体]90→80 [/font][/td][/tr][tr][td=1,1,201][font=宋体]25~30[/font][/td][td=1,1,185][font=宋体]20→23[/font][/td][td=1,1,182][font=宋体]80→77[/font][/td][/tr][tr][td=1,1,201][font=宋体]30~42[/font][/td][td=1,1,185][font=宋体]23→26[/font][/td][td=1,1,182][font=宋体]77→74[/font][/td][/tr][tr][td=1,1,201][font=宋体]42~45[/font][/td][td=1,1,185][font=宋体]26→37[/font][/td][td=1,1,182][font=宋体]74→63[/font][/td][/tr][tr][td=1,1,201][font=宋体]45~47[/font][/td][td=1,1,185][font=宋体]37[/font][/td][td=1,1,182][font=宋体]63[/font][/td][/tr][tr][td=1,1,201][font=宋体]47~54[/font][/td][td=1,1,185][font=宋体]37→39[/font][/td][td=1,1,182][font=宋体]63→61[/font][/td][/tr][tr][td=1,1,201][font=宋体]54~65[/font][/td][td=1,1,185][font=宋体]39→100[/font][/td][td=1,1,182][font=宋体]61→0 [/font][/td][/tr][/table][b][font=宋体]对照品溶液的制备[/font][/b][font=宋体] 取乌药碱对照品、木兰花碱对照品、酸枣仁皂苷A对照品、酸枣仁皂苷D对照品适量,精密称定,加甲醇制成每1 mL分别含100 μg、2mg、100 μg和100 μg的混合溶液,即得。 [/font][b][font=宋体]供试品溶液的制备[/font][/b][font=宋体] 取本品粉末(过四号筛)约1g,精密称定,加石油醚(60~90℃)50ml,冷浸过夜,超声处理30分钟,弃去石油醚液,药渣挥去溶剂,转移至锥形瓶中,加入70%乙醇20ml,加热回流2小时,滤过,滤渣用70%乙醇5ml洗涤,合并洗液与滤液,回收溶剂至干,残渣加甲醇溶解,转移至5ml量瓶中,加甲醇至刻度,摇匀,滤过,取续滤液,即得。 [/font][b][font=宋体]测定法[/font][/b][font=宋体] 分别精密吸取对照品溶液5μl、20μl,供试品溶液10μl,注入[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱仪[/color][/url],测定,用外标两点法对数方程计算木兰花碱、酸枣仁皂苷A、酸枣仁皂苷,用外标法测定乌药碱,即得。[/font][font=宋体]按干燥品计算,含酸枣仁皂苷A(C58H94O26)不得少于0.030%,乌药碱(C17H19NO3)不得少于0.008%,木兰花碱(C20H24NO4)不得低于0.20%,酸枣仁皂苷D(C58H94O26))不得低于0.02%。[/font]
http://ng1.17img.cn/bbsfiles/images/2012/09/201209272130_393425_2255248_3.gifHPLC测定木犀草苷对照品为什么峰前面有个小峰????
完全按照药典标准做的三七总皂苷,可是对照品和样品的Re均不出峰,其他几个峰都出得很好

【作者】 李玉英;【机构】 广东省惠州市中药厂 广东惠州516001;【摘要】 目的:探讨护肝片中绿原酸的定性鉴别方法。方法:采用高效液相色谱法(HPLC法)鉴别绿原酸成分,以DiamonsilC18柱(250mm×4.6mm,5μm)为色谱柱,乙腈-0.4%磷酸溶液(13∶87)为流动相,检测波长为327nm。结果:样品峰的保留时间与绿原酸对照品峰的保留时间保持一致。结论:HPLC法专属性强,可用于护肝片的质量控制。 更多还原http://ng1.17img.cn/bbsfiles/images/2012/08/201208271538_386433_2379123_3.jpg
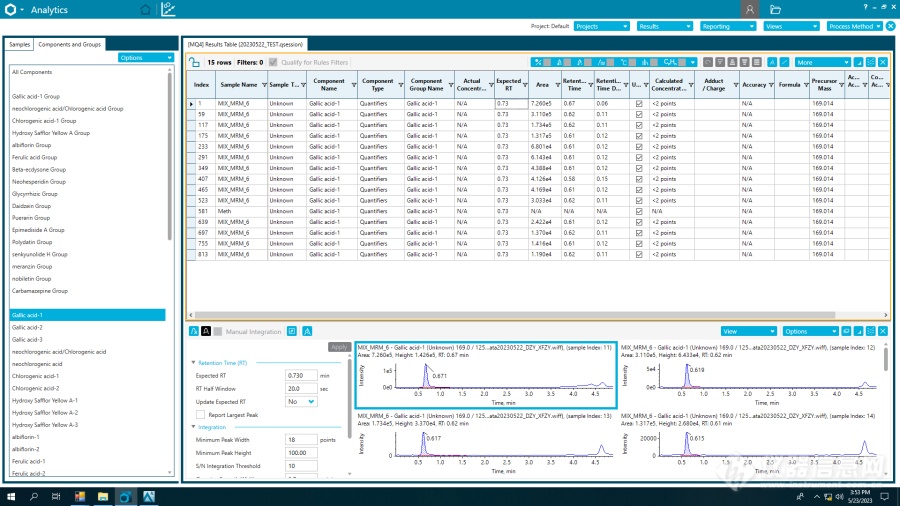
建了[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url]/MS分析方法,想同时检测没食子酸、新绿原酸和绿原酸和其他几种成分,如羟基红花黄色素A、阿魏酸等,但对照品建立方法时连续进样6针,没食子酸、新绿原酸和绿原酸的峰面积逐渐降低,怀疑是降解,但拿到HPLC上检测是没有问题的, 峰面积与新配制的相同。请问是什么原因导致的,而且对对照品为混标,其他成分都没有降解的情况,很稳定,可以排除进样的原因,如图[img=,690,387]https://ng1.17img.cn/bbsfiles/images/2023/05/202305281454099518_1703_3962371_3.png!w690x387.jpg[/img][img=,690,387]https://ng1.17img.cn/bbsfiles/images/2023/05/202305281454257768_9259_3962371_3.png!w690x387.jpg[/img][img=,690,387]https://ng1.17img.cn/bbsfiles/images/2023/05/202305281454341112_6631_3962371_3.png!w690x387.jpg[/img]
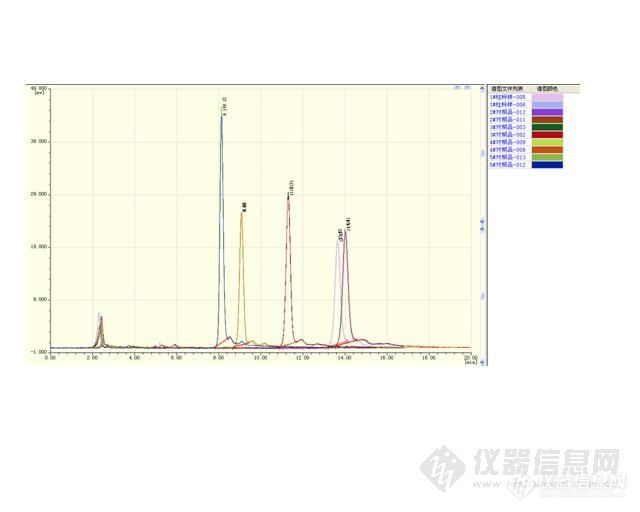
应雪妖版主的客户要求,对月旭不同型号的C8色谱柱进行知母皂苷BⅡ的测试,最终选择一款适合知母皂苷BⅡ测试的柱子。前言很快我就收到了雪妖寄来的快递,里面装了满满5根柱子,当然还有知母皂苷BⅡ对照品溶液和知母样品溶液。因为当时氮气用完了,中间等待了两天才开始实验,实验总共花了3天时间。1、实验部分1.1、仪器和和设备LC310 液相色谱仪-ELSD 蒸发光散射检测器(LC310 为江苏天瑞仪器股份有限公司生产,ELSD 为美国奥泰公司提供);超纯水机(南京易普易达科学发展有限公司);超声波清洗机(张家港市神科超声电子有限公司)1.2 试剂氮气(购于昆山当地,纯度大于99.999%);乙腈(色谱纯,美国TEDIA 公司生产);超纯水:自制;色谱柱(C8)共5根,均为月旭材料科技(上海)有限公司提供。5根色谱柱型号如下:1#:Xtimate C8,编号:22904 ,4.6mm*250mm,5um;2#:XB-C8,编号:Ult 101202c1,4.6mm*250mm,5um;3#:Ultimate LP-C8,编号:631001522,4.6mm*250mm,5um;4#:Xtimate C8,编号:4311015264.6mm*250mm,5um;5#:Topsil C8,4.6mm*250mm,5um。1.3 色谱条件知母药材中知母皂苷BⅡ的含量测定在中国药典2010 版一部正文198 页中规定:以辛烷基硅烷键合硅胶为填充剂,以乙腈:水=25:75 为流动相,蒸发光散射检测器检测。理论板数按照知母皂苷BⅡ峰计算应不低于10000。备注:因客户按药典方法制备提供的对照品和供试品溶液,因此制备溶液部分不做深究。 2 实验数据按液相色谱仪器操作规程打开仪器、氮气钢瓶、调整流动相比例,同时参照相关网络文献,首先将流速设置为1.0ml/min,色谱柱温:35℃,氮气压力设置为0.3MPa, 漂移管温度70℃。用1#色http://ng1.17img.cn/bbsfiles/images/2011/09/201109191501_317791_1628076_3.jpg在此色谱条件下对照品溶液进样10ul,色谱图如下:http://ng1.17img.cn/bbsfiles/images/2011/09/201109191503_317792_1628076_3.jpghttp://ng1.17img.cn/bbsfiles/images/2011/09/201109191503_317793_1628076_3.jpg按照同样的色谱条件,对供试品进样测试,谱图如下:http://ng1.17img.cn/bbsfiles/images/2011/09/201109191504_317795_1628076_3.jpghttp://ng1.17img.cn/bbsfiles/images/2011/09/201109191505_317796_1628076_3.jpg对照品与供试品谱图叠加如下:http://ng1.17img.cn/bbsfiles/images/2011/09/201109191505_317797_1628076_3.jpg备注:在此之前,对氮气压力做过调整,试过0.25MPa,漂移管温度试过65℃,与0.3MPa和70℃基本没有什么区别,故将色谱条件设置为0.3MPa 的氮气压力和70℃的漂移管温度。且在此色谱条件下,色谱图能满足测试要求,理论板数都大于10000。同样的色谱条件,对其他几根色谱柱进行试验。2#色谱柱试验谱图基线谱图:http://ng1.17img.cn/bbsfiles/images/2011/09/201109191507_317798_1628076_3.jpghttp://ng1.17img.cn/bbsfiles/images/2011/09/201109191508_317799_1628076_3.jpghttp://ng1.17img.cn/bbsfiles/images/2011/09/201109191509_317800_1628076_3.jpg供试品溶液色谱图如下:http://ng1.17img.cn/bbsfiles/images/2011/09/201109191509_317801_1628076_3.jpghttp://ng1.17img.cn/bbsfiles/images/2011/09/201109191510_317802_1628076_3.jpg对照品和供试品谱图叠加如下:http://ng1.17img.cn/bbsfiles/images/2011/09/201109191511_317803_1628076_3.jpg备注:用2#色谱柱进行测试,理论板数均大于10000,且峰形对称。3#色谱柱,色谱条件同前:空跑基线谱图:http://ng1.17img.cn/bbsfiles/images/2011/09/201109211054_318270_1639959_3.jpg两针对照品溶液色谱图:http://ng1.17img.cn/bbsfiles/images/2011/09/201109211055_318274_1639959_3.jpghttp://ng1.17img.cn/bbsfiles/images/2011/09/201109211055_318275_1639959_3.jpg供试品溶液色谱图如下:http://ng1.17img.cn/bbsfiles/images/2011/09/201109211057_318276_1639959_3.jpg对照品与供试品色谱图叠加如下:http://ng1.17img.cn/bbsfiles/images/2011/09/201109211057_318278_1639959_3.jpg备注:3#色谱柱测试谱图,理论板数均大于10000。4#色谱柱,色谱条件同前:空跑基线谱图:http://ng1.17img.cn/bbsfiles/images/2011/09/201109211058_318279_1639959_3.jpg对照品色谱图:http://ng1.17img.cn/bbsfiles/images/2011/09/201109211059_318280_1639959_3.jpghttp://ng1.17img.cn/bbsfiles/images/2011/09/201109211100_318281_1639959_3.jpg供试品色谱图:http://ng1.17img.cn/bbsfiles/images/2011/09/201109211100_318282_1639959_3.jpg对照品与供试品色谱图叠加如下:http://ng1.17img.cn/bbsfiles/images/2011/09/201109211101_318283_1639959_3.jpg5#[size
各位老师、朋友 我使用waters的仪器检测三七,在换了新批号(110704-200324)的人参皂苷Rg1对照品之后,发现峰面积降了20~30%,进样量和柱子都没换,原来使用的对照品批号是(110704-200323),而检测其他物质都没有发现此种问题。是仪器的问题还是对照品的问题呢? 色谱条件:流动相是乙腈-0.05%磷酸(20:80),波长203nm。 请各位老师、朋友指教,不胜感谢!

全自动固相萃取-比色法测定保健食品中总皂苷 作者:赵晓冬http://ng1.17img.cn/bbsfiles/images/2015/11/201511261708_575182_2904170_3.jpg目的:建立保健食品中总皂苷的含量测定方法。方法:采用Sepline全自动固相萃取装置对保健食品中的总皂苷富集,通过70%乙醇回收,以香草醛-高氯酸为显色剂,采用比色法测定。结果:比色法测定的线性范围为0.038~0.268mg,相关系数 r=0.9997; 在不同保健食品中的平均加样回收率均在98.3%~102.4%,RSD〈2.2% (n=6)。结论:本法简便、准确、灵敏度高、重复性好,可用于不同类型保健食品中总皂苷的含量测定。 目前保健食品中的总皂甙测定方法尚无国家标准,大多都是按照《保健食品检验与评价技术规范 (2003年版) 》“保健食品中总皂甙的测定”方法检测,该方法采用人参皂苷 Re为对照,用香草醛比色法测定总皂苷。其显色体系为:香草醛-冰醋酸-高氯酸,但在实际操作中和据相关文献报道中,但采用该方法检测结果误差较大,尤其是不同实验室间测定结果差异更大,不利于产品的质量控制。有文献报道,卫生部食品卫生监督所2000年组织全国22个省、市、自治区省级疾病预防控制中心对这一方法进行比对试验,结果显示:离散度较大,有的浓度水平还出现离群值,各实验室间的RSD达80%,因此需要对测定方法加以改进。本文采用莱伯泰科公司Sepline全自动固相萃取提取装置 具有洗脱液流速恒定可控、大批量产品自动化处理,保证试验系统的密闭性等优点可保证结果的平行性和准确性。在对大孔树脂进行品牌筛选的基础上,定制统一规格固相萃取柱、对总皂苷测定前处理过程进行了优化,整套方法简单,数据结果离散性小,平行性得到提高。1材料与方法1.1 仪器 UV-2450 紫外可见分光光度计(日本,岛津公司),CP2250分析天平,AS10200ADT超声波清洗仪,TLL-DCⅡ型氮吹仪,Sepline全自动固相萃取系统(北京莱伯泰科有限公司),固相萃取柱(底层有砂芯隔板,内装XAD-2大孔树脂2.7g,Alumina-N中性氧化铝,6ml,北京莱伯泰科有限公司)。1.2 试药 人参皂苷Re对照品(中国食品药品检定研究院,含量92.7%),冰醋酸、香草醛、高氯酸、乙醇均为分析纯,水为纯化水。1.3 供试品 上普牌西洋参含片 (厦门上普药业有限公司,批号14040201);天信减肥茶( 福建安溪馨隆保健品有限公司,批号: 20140504 ) ;辅助降血脂片(哈尔滨久盛医药科技开发有限公司,批号:20140714)珍迪牌黄芪阿胶口服液(南昌健民营养补品厂,批号: 20130701)2方法与结果2.1 标准溶液制备精密称取20mg人参皂苷Re对照品置于100mL量瓶中,用甲醇溶解并稀释至刻度,摇匀。吸取10mL人参皂苷Re标准溶液(0.2mg/ml)放蒸发皿中,放在水浴挥干(低于60℃),准确加入10ml水溶解,作为标准溶液2.2 样品溶液制备 精密称取适量样品,置于50mL容量瓶中,加水适量溶解,超声30min,放置至室温,用水稀释至刻度,摇匀, 3000r·min-1离心5min,0.45um微孔滤膜过滤。若为非乙醇类液体试样,先经0.45um微孔滤膜过滤,取精密过滤后样品1mL (根据试样含总皂苷量定) 进行固相萃取。2.3 样品溶液吸附及洗脱 取样品溶液上清液置于收集管中(大于2ml), 放置于Sepline全自动固相萃取系统中,按以下步骤进行吸附及洗脱(详见图1)。2.3.1 活化:在固相萃取柱中,以2.5 ml·min-1 流速冲洗70%乙醇25ml、然后以2.5 ml·min-1 流速冲洗水25ml。2.3.2 上样:以1ml·min-1 流速加1ml样品溶液于固相萃取柱中。2.3.3 洗脱:以0.8ml·min-1 流速, 依次用25ml水,25ml70%乙醇进行洗脱,收集70%乙醇洗脱液,在收集结束时氮气吹扫管理残留洗脱液50s。2.3.4 挥干:将收集的洗脱液转入50ml的离心管中,在60℃下氮气吹干。https://mmbiz.qlogo.cn/mmbiz/1M6WGftQWKcMCtAjjEeTO31icqxEfIgZXHUdyj5UNWFsWqjnVFOfw77OyyjdE8AtsH0V1ibNylQgZibv2FialaXicibA/0?wx_fmt=jpeg2.4 显色与测定 在上述已挥干的洗脱液中准确加入0.2ml5%香草醛冰乙酸溶液,使残渣都溶解,再加0.8ml 高氯酸,混匀后60℃水浴上加热 10min,冰浴冷却后,准确加入冰乙酸 5.0ml,摇匀后,以 1cm 比色池于 560nm 波长处与标准管一起进行比色测定。2.5 线性关系考察 精密吸取“2.1”项标准溶液0、0.2、0.5、0.8、1.0、1.2、、1.4人参皂苷Re标准溶液(0.2mg/ml),按 “2. 3”“2.4”项下方法进行前处理并测定。以吸光度值为横坐标, 以人参皂苷Re含量 (mg) 为纵坐标进行回归计算,回归方程为y =0.3521x+0.0018,r=0.9997,结果表明,总皂苷( 以人参皂苷Re计) 在0.038~0.268mg 范围内线性关系良好。2.6 加样回收率试验 采用加样回收率法,精密称取4类保健品适量,分别按样品量的100%的比例各6份加入对照品 ,按“2.3”项下和“2.4”项下进行测定,并分别计算回收率(见表1-表5),结果各品种平均加样回收率均在98.3%~102. 4% ,RSD<2.2% ,并与传统方法(手填层析柱、人工进行柱洗脱)进行比较,表明该方法准确度良好,且离散度低于传统方法。https://mmbiz.qlogo.cn/mmbiz/1M6WGftQWKcMCtAjjEeTO31icqxEfIgZXpKdklF1UoeMzvU6eQJEseF6P0shyEyUViacyO2M0iaBmcQmLVtq4YUIQ/0?wx_fmt=jpeghttps://mmbiz.qlogo.cn/mmbiz/1M6WGftQWKcMCtAjjEeTO31icqxEfIgZXcxdobibU0cPzwap5qsdShaQtxlrmMbDxOG6AKyWSTxYHbwu085loQog/0?wx_fmt=jpeghttps://mmbiz.qlogo.cn/mmbiz/1M6WGftQWKcMCtAjjEeTO31icqxEfIgZXudGTrHbh0Eu8R6zY3PNo4iaHwJMh58wAxhK00QPk76bje6KXTQ6LWAA/0?wx_fmt=jpeghttps://mmbiz.qlogo.cn/mmbiz/1M6WGftQWKcMCtAjjEeTO31icqxEfIgZXO3ae9XyfEiaaUU6Qkibib1WgWDiaAVh9AQEFhV6tJ8oJxfsibuHW7tMWHUw/0?wx_fmt=jpeghttps://mmbiz.qlogo.cn/mmbiz/1M6WGftQWKcMCtAjjEeTO31icqxEfIgZXfVUKQfmpzF7ootVP2rp4VREoKIGLKDM54tMwEK2GyZ8DazgJL6tfug/0?wx_fmt=jpeg2.7 稳定性试验 将显色后样品分别放置 0、0.5、1、1.5、2、2.5、3h, 测定吸光值。结果表明该显色体系在1 h内吸光度基本不变,1h后吸光度有所降低,故在显色后1 h内测定。2.8 样品含量测定 取4类保健食品 ,按“2.3”项下和“2.4”项下进行测定,于 560nm 波长处测定吸光度值, 根据所得到的标准曲线计算样品中总皂苷的含量(见表6) 。[align
在我国制造业向高质量发展的过程中,高技术制造业发挥着重要作用。高技术制造业属于技术密集型产业,通常依托高新技术进行研发、生产,近年来催生出新能源汽车等新兴行业。在智联研究院“寻找下一个风口”潜力行业调研项目中,这些新兴行业受到众多职场人的关注。本次,智联招聘结合平台数据以及调研项目的统计结果,将15个行业作为研究范围(具体包括:人工智能、计算机硬件、通信/网络设备、船舶/航空/航天/火车制造、电气机械/器材制造、智能设备制造、工业自动化、新能源汽车、电子设备制造、仪器仪表制造、通用设备制造、专用设备制造、云计算/大数据、区块链、物联网)[b]行业平均薪酬差异较大,人工智能平均月薪高达13076元[/b]智联招聘数据显示,高技术制造业的平均薪酬为9935元/月,略高于全行业的9889元/月。 但是具体到各行业来看,平均薪酬仍存在较大差距。其中人工智能行业平均招聘薪酬最高,达到了13076元/月,其次是新能源汽车(12848元)、云计算/大数据(12176元)、物联网(11383元)、区块链(11187元)。虽然工业自动化行业人才需求量增长最快,但是普工/技工的招聘岗位需求占比更大,这类岗位目前的薪资相对较低,因此该行业整体平均薪酬低于行业平均薪酬,为9910元。排在末位的是通用设备制造,平均月薪为8251元。[img=智联招聘:高技术制造业人才需求与发展环境报告]https://accesspath.com/wp-content/uploads/2022/06/2284602352781864614.jpeg?imageMogr2/format/webp[/img][b]半导体/芯片、人工智能平均薪酬最高,超2万元[/b]具体职业方面,半导体/芯片的平均招聘薪酬最高为21960元/月,其次是人工智能,达到21146元/月,平均薪酬均超过2万元。近年来,国家对半导体/芯片、人工智能等行业资金扶持力度加大,同时吸引了大量资本进行投资。政府的扶持加上资本的聚拢加速行业发展的同时,也自然导致从业人员的薪酬待遇水涨船高。[img=智联招聘:高技术制造业人才需求与发展环境报告]https://accesspath.com/wp-content/uploads/2022/06/3525948857031611492.jpeg?imageMogr2/format/webp[/img][b]薪酬排行前3名的技能是ASIC、ISP、CAMERA[/b]掌握什么样的技能,可以在高技术制造业获得较高的薪酬呢?从数据分析来看,ASIC、ISP、CAMERA排在前三位。其中掌握ASIC技能的人才可以获得平均38119元/月的高薪。除了以上三种技能,排进薪酬TOP50的系统搭建、技术研发管理、IC设计等,也都是“学习门槛”颇高的技能。
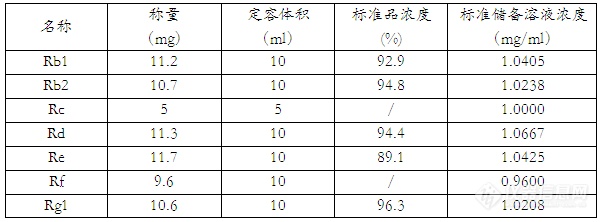
一、实验目的及原理:参照药典中人参皂苷的检测,使用超高效液相色谱,对于人参中7种皂苷成分进行检测,大大提高了检测速度,并拓展了检测的皂苷种类。人参中的皂苷成分通过超声波萃取,高校液相色谱柱进行分离,采用紫外检测器进行检测,外标法定量。二、实验仪器及试剂:仪器:UPLC(waters H-Class),检测器:TUV紫外检测器、超声波仪、超纯水机、离心机、涡旋振荡仪、50ml离心管、20ml胖肚移液管试剂:人参皂苷标准品Rb1、Rb2、Rd、Re、Rf、Rg1购自食品药品检定院, 人参皂苷标准品Rc购自百灵威、乙腈(HPLC)、甲醇(HPLC)三、色谱条件:色谱柱:BEHC18 2.1mm×50 mm×1.7um 柱温:35℃ 样品温度:25℃进样体积:5µL 流速:0.4ml/min 波长:203nm 采样速率:20点/秒流动相配比:时间水乙腈曲线类型08218/1082186185050618.1821862082186四、实验步骤4.1、标准储备溶液配制:称取人参皂苷标准品,用甲醇溶解定容,具体见下表:http://ng1.17img.cn/bbsfiles/images/2012/10/201210241734_399095_1854832_3.jpg4.2、样品处理称取样品约0.4g(精确到0.0001g)于50ml离心管中,加入20.0ml超纯水,涡旋15s,超声波萃取30min,5000转/min离心5min,过0.22um水膜,待测。五、计算5.1样品中人参皂苷浓度的计算: X=C*100/m式中:X——样品中人参皂苷的浓度(mg/g) m——样品称量(g) C——待测溶液中人参皂苷的浓度(ug/mL)六、标准曲线与线性相关系数取标准储备溶液混合,用水稀释至约5、10、20、50、100ug/ml,进行2次平行测定,得到相应的峰面积平均值。以浓度为横坐标,以峰面积为纵坐标,绘制标准曲线,结果如下:http://ng1.17img.cn/bbsfiles/images/2012/10/201210241728_399091_1854832_3.jpg七、样品测定及回收率实验(以6年红参体为样本)http://ng1.17img.cn/bbsfiles/images/2012/10/201210241730_399092_1854832_3.jpg八、对照与样品测定的色谱图http://ng1.17img.cn/bbsfiles/images/2012/10/201210241731_399093_1854832_3.jpg九、结果与讨论本实验尝试多个色谱条件, Re、Rg1组分性质非常接近较难分离,选择梯度洗脱的方式加快其他组分的分离,相对于传统液相方法需要至少一个小时的进样时间,UPLC的分离时间在20min,具有较大的优势。
对照品甘草苷 后面出杂峰 是怎么回事[img]https://ng1.17img.cn/bbsfiles/images/2018/08/201808221432219383_6429_3461163_3.jpeg[/img]
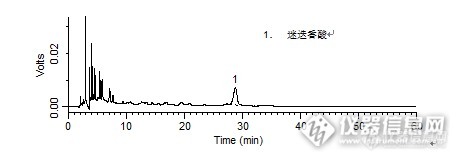
问题:对照品中迷迭香酸的理论塔板数是多少呢?答案:9201.310【活动奖励】幸运奖(2钻石币):抽奖软件,当天随机抽取3个回答正确的版友ID号(最后一个ID号,截止至下午3:00),每人奖励2个钻石币积分奖励:所有回答正确的版友奖励10个积分(幸运奖获得者除外)。【注意事项】同样的答案,每人只能发一次PS:该贴浏览权限为“回贴仅作者和自己可见”,回复的版友仅能看到版主的题目及自己的回答内容,无法看到其他版友的回复内容。下午3点之后解除,即可看到正确答案、获奖情况及所有版友的回复内容。获奖名单有:dahua1981(ID:dahua1981)999youran(ID:999youran)牛一牛(ID:v2700892)http://ng1.17img.cn/bbsfiles/images/2015/12/201512091543_577030_1987954_3.jpghttp://ng1.17img.cn/bbsfiles/images/2015/12/201512091544_577031_1987954_3.jpghttp://ng1.17img.cn/bbsfiles/images/2015/12/201512091544_577032_1987954_3.jpghttp://ng1.17img.cn/bbsfiles/images/2015/12/201512091544_577033_1987954_3.jpg夏桑菊颗粒中绿原酸、迷迭香酸、蒙花苷的检测样品制备 制备方法指纹图谱:取绿原酸对照品、迷迭香酸对照品和蒙花苷对照品适量,精密称定,分别加甲醇制成每1 mL含绿原酸25 μg、迷迭香酸15 μg和蒙花苷25 μg的溶液,即得。1. 对照品:取迷迭香酸对照品适量,精密称定,加甲醇制成每1 mL含25 μg的溶液,即得(10℃以下棕色瓶保存)。2. 供试品:取装量差异项下的本品内容物,混匀,研细,取约1.25 g,精密称定,置具塞锥形瓶中,精密加入75%甲醇25 mL,密塞,称定重量,超声处理(功率250 W,频率33 kHz)30分钟,取出,放冷至室温,再称定重量,用75%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。指纹图谱分析条件 色谱柱Platisil ODS 250 x 4.6 mm,5 μm (Cat#:99503)流动相A:乙腈 B:1%冰醋酸溶液 梯度流速1 mL/min柱温30 ℃检测器UV 320 nm 进样量10 μL 含量测定分析条件 色谱柱Platisil ODS 250 x 4.6 mm,5 μm (Cat#:99503)流动相乙腈:1%醋酸溶液=19:81流速1 mL/min柱温30 ℃检测器UV 329 nm 进样量:10 μL 色谱图指纹图谱http://ng1.17img.cn/bbsfiles/images/2015/12/201512091015_576966_1987954_3.png 峰号 保留时间 min 峰面积 μV*s 峰高 μV 理论塔板数* N USP拖尾因子 分离度 1 16.809 952883 50605 18864.347 0.919 -- 2 40.298 651301 39185 137816.388 0.978 50.856 3 47.797 311451 23679 315547.837 0.923 19.364 *药典要求理论板数按迷迭香酸峰计算应不低于20000对照品http://ng1.17img.cn/bbsfiles/images/2015/12/201512091027_576967_1987954_3.png 峰号 保留时间 min 峰面积 μV*s 峰高 μV 理论塔板数* N USP拖尾因子 分离度 1 28.8
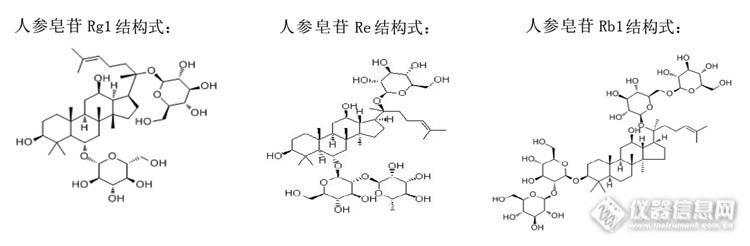
人参皂苷检测 人参大家都知道,是好东西,是一种大补的药材。它的功效非常神奇,具有抗癌奇效,增强机体免疫力,快速增强或恢复体质,兴奋中枢神经,缓解或抗疲劳,改善或提高记忆力,延迟衰老,镇定、安神、解热、放松、催眠等多种功效,是一种非常昂贵和神奇的药材。 人参的主要营养成分是人参皂苷,然而人参皂苷也分很多种,有人参皂苷Rh2、Rg、Rg1、Rg2、Rg3、Rb1、Rb2、Rc、Rb3、Rh、Rh1、Ro、Rbt等。 大家可能也知道人参皂苷不好检测,一是检测时间长,一般都需一个多小时,二是准确度、精密度很难保证,三是有几种不好分离,四是检测波长低,一般都采用203nm,五是对流动相、色谱柱要求高等难题。 下面我们就看看该方法,该色谱柱检测人参皂苷Rg1、Re、Rb1的效果吧。http://ng1.17img.cn/bbsfiles/images/2014/03/201403302025_494728_2369266_3.png色谱条件:色谱柱:Welchrom-C18 ( 250 mm ×4.6 mm 5μ )Serial Number:W13212255流动相:以乙腈为流动相A,水为流动相B,按下表中的规定进行梯度洗脱: 时间(min)流动相A(%)流动相B(%)0~35198135~5519→2981→7155~70297170~10029→4071→60流速:1 mL/min进样量:10 μL检测波长:203 nm柱温:30℃色谱图:对照品色谱图:http://ng1.17img.cn/bbsfiles/images/2014/03/201403301542_494709_2369266_3.png供试品色谱图:http://ng1







 我要推广仪器
我要推广仪器
 下载APP
下载APP