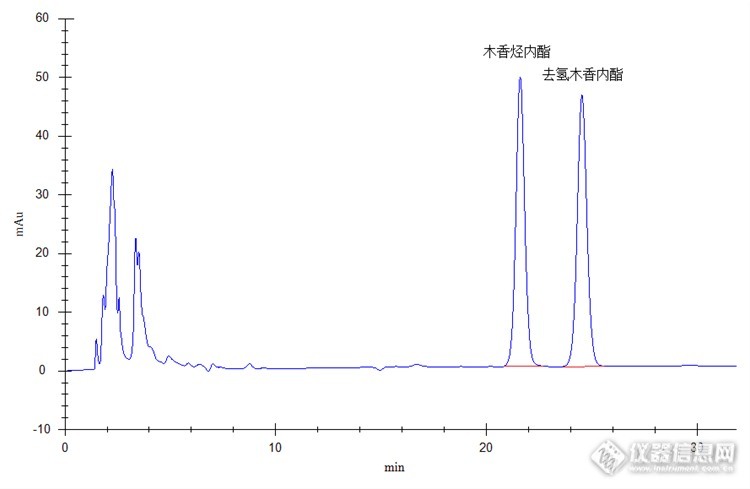
高效方法检测名贵药材 名贵药材很多种,有一种叫木香。 它花色艳丽,形状各异,香味迷人,漂亮极致。根、茎、叶也有着非常不菲的药用价值和营养成分。 药用部分主要来自根部,它气香特异,味微苦,含有丰富的去氢木香内酯,木香烯内脂,木香萜醛,木香内酯,二氢木香内酯,α-环木香烯内酯,β-环木香烯内酯,土木香内酯等药物成分。另外根部还含天冬氨酸,谷氨酸,甘氨酸,甘氨酸,瓜氨酸等20多种氨基酸,营养成分也非常丰富。 叶含蒲公英甾醇,α-香树精硬脂酸酯,β-香树精棕榈酸酯,羽扇醇棕榈酸酯等多种香精、香料成分。 木香具有行气、止痛、止血、健脾、消炎、抗菌、降压等多种药物功效,可用于多种疑难病症治疗。http://ng1.17img.cn/bbsfiles/images/2017/10/2015081418455848_01_0_3.jpghttp://ng1.17img.cn/bbsfiles/images/2017/10/2015081418460455_01_0_3.jpghttp://ng1.17img.cn/bbsfiles/images/2017/10/2015081418462259_01_2536753_3.jpeghttp://ng1.17img.cn/bbsfiles/images/2015/08/201508141901_560867_2536753_3.png说明:以上几张图片是网上截图,不是本人所拍,在这只是为了说明下木香药材的外观及美感,希望大家能够理解。 既然木香价值这么高,检测它就显得尤为重要,方法选择尤为关键。下面我们就介绍几种高效液相色谱法检测该药材中木香烃内酯、去氢木香内酯方法。1.实验部分1.1原理 精密称取该药品适量,超声波提取后经进样器注入高效液相色谱系统,通过C18色谱柱分离,紫外检测器检测,外标法(保留时间定性,峰面积定量)计算,得出所测成分含量。1.2 仪器及试剂 仪器:高效液相色谱仪(紫外检测器+高压恒流泵+柱温箱),四号筛(药典筛),超声波清洗器,溶剂过滤器,电子天平(0.0001级)等。 试剂:甲醇(色谱纯),超纯水1.3样品制备 对照品溶液制备:准确称取木香烃内酯对照品、去氢木香内酯对照品各5mg于50ml容量瓶中,加甲醇至刻度,配制成含木香烃内酯对照品、去氢木香内酯各0.1mg/ml的混合对照品溶液,备用。 供试品溶液制备:取本品适量充分粉碎后过四号筛,准确称取过筛粉末0.3g,置于具塞锥形瓶中,精密加入甲醇50ml,密塞,称定重量,放置过夜后超声处理30分钟,放冷,再次称定重量,用甲醇补足减少的重量,摇匀,0.45μm微膜滤过,待测。1.4 色谱条件检测器:紫外检测器检测波长:225nm色谱柱:Promosil C18,4.6 X 250mm,5μm流动相:甲醇-水(65:35)流速:1.0ml/min柱温:室温进样量:10μl2、实验过程及色谱图对照品色谱图http://ng1.17img.cn/bbsfiles/images/2017/10/2015081418542201_01_0_3.pnghttp://ng1.17img.cn/bbsfiles/images/2017/10/2015081418545304_01_2536753_3.png供试品色谱图:http://ng1.17img.cn/bbsfiles/images/2017/10/2015081418542563_01_0_3.pnghttp://ng1.17img.cn/bbsfiles/images/2017/10/2015081418545925_01_2536753_3.png 计算公式:样品中各物质的含量计算公式:http://ng1.17img.cn/bbsfiles/images/2017/10/2015081418541866_01_2536753_3.pngD----供试品中被测物含量,以(%)表示V----供试品稀释的总体积,单位(mL)P3----供试品溶液被测物峰面积值P4-----对照品溶液被测物峰面积值ρ2----对照品被测物浓度。单位为mg/mlm2----样品质量,单位(mg) 通过计算木香烃内酯的含量大概是0.8%,去氢木香内酯的含量大概是1.6%,总含量2.4%,远大于药典中要求的1.8%,该木香药材满足药典要求,属合格品。 这个检测时间有点长,我们不防改变下色谱条件,缩短些时间。那就把柱温调整到40℃,看下结果。对照品色谱图:http://ng1.17img.cn/bbsfiles/images/2017/10/2015081418542990_01_2536753_3.png供试品色谱图:http://ng1.17img.cn/bbsfiles/images/2017/10/2015081418543464_01_2536753_3.png 看来有效果,检测时间是短了,但分离度也相对差了点,不够理想。那我们再不防改变下色谱条件,争取把分离度效果弄好些。那就调整下流动相,甲醇-水(60:40),看下结果。对照品色谱图: http://ng1.17img.cn/bbsfiles/images/2017/10/2015081418544178_01_2536753_3.png供试品色谱图:http://ng1.17img.cn/bbsfiles/images/2017/10/201508141854474
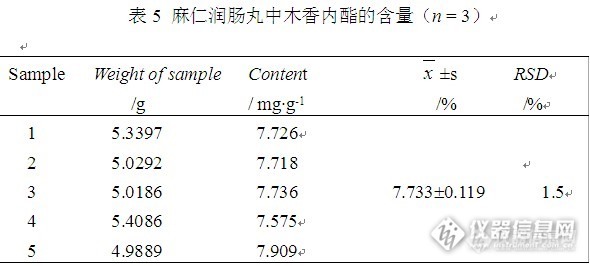
麻仁润肠丸中木香内酯的提取和含量测定方法的建立 麻仁润肠丸收载于2010年版中国药典,为黄褐色的大蜜丸;气微香,味苦、微甘。成份有火麻仁、炒苦杏仁、大黄、木香、陈皮、白芍。辅料为赋形剂蜂蜜。功能主治为润肠通便,用于肠胃积热,胸腹胀满,大便秘结。药典中仅对麻仁润肠丸中大黄中的成分大黄素和大黄酚的含量做了标准要求,为了进一步保证药品质量,完善检测指标,我们探索并建立了麻仁润肠丸中木香内酯成分的提取和含量测定。目的:建立HPLC法测定麻仁润肠丸中木香内酯含量的方法。方法:采用岛津Wondasil C18色谱柱(4.6mm×200mm,5μm)。流动相:甲醇:乙腈:水(50:10:40),柱温38℃,流速1.0 mL/min,检测波长224nm。结果:木香内酯在31.56μg·mL-1~1010μg·mL-1呈良好的线性关系(r = 0.9996),平均回收率为100.9%,RSD =1.6%,麻仁润肠丸药材中木香内酯的含量为7.733mg g-1。结论:本法灵敏,准确,重现性好,可作为麻仁润肠丸中木香内酯的含量测定方法。材料与方法1 材料1.1 仪器Agilent 1100 高效液相色谱仪,紫外检测器,安捷伦化学工作站。梅特勒-托利多电子分析天平(1/10万,德国)。超声波清洗器(昆山)。紫外可见分光光度计(850,美国PE)。1.2 试剂木香内酯标准品(中国药品生物制品鉴定所提供),木香对照药材(中国药品生物制品鉴定所提供),麻仁润肠丸(实验室自制及其他四个厂家)。甲醇、乙腈为色谱纯,所用水为去离子水,其他试剂均为分析纯。2 方法2.1 实验部分2.1.1色谱条件Agilent 1100 高效液相色谱仪,岛津Wondasil C18(4.6mm×200 mm, 5μm)色谱柱,紫外检测器。流动相:甲醇:乙腈:水(50:10:40)。流速:1.0mL/min。柱温:38℃。进样量:20μL。检测
【作者】 宋粉云; 梁从庆; 毋福海; 钟兆健;【机构】 广东药学院; 广东药学院 广东广州510224; 广东广州510224;【摘要】 目的:建立拈痛丸中木香烃内酯与去氢木香内酯含量测定的方法。方法:采用RP-HPLC法,Diamonsil-C18柱,甲醇-水(67∶33)为流动相;流速1.0 ml/min;检测波长225 nm;柱温30℃。结果:木香烃内酯的平均回收率为98.8%,方法精密度(RSD)为1.62%(n=6);去氢木香内酯的平均回收率为100.6%,方法精密度(RSD)为1.46%(n=6)。结论:所建方法可用于拈痛丸中木香烃内酯与去氢木香内酯的含量测定。 更多还原【关键词】 拈痛丸; 木香烃内酯; 去氢木香内酯; HPLC; 含量测定; 文献没有图谱
木香和土木香怎么薄层鉴别? 什么成分? 什么展开剂?
对照品4个,第2(延胡索乙素)、3(盐酸小檗碱)个色谱峰前出现了小峰,不知道什么原因引起的,之前用相同色谱条件跑过,也没有小峰出现,现在连续进了五针都是主峰前有小峰。请各位帮我分析分析!流动相是0.1%的磷酸水:乙腈,对照品用甲醇溶解。色谱条件如图[img=,690,284]https://ng1.17img.cn/bbsfiles/images/2022/11/202211180044560750_5451_5351399_3.png[/img][img=,690,517]https://ng1.17img.cn/bbsfiles/images/2022/11/202211180044563666_9373_5351399_3.png[/img][img=,690,517]https://ng1.17img.cn/bbsfiles/images/2022/11/202211180044551717_7304_5351399_3.png[/img][img=,690,618]https://ng1.17img.cn/bbsfiles/images/2022/11/202211180044568830_7416_5351399_3.png[/img]
做一个甾体皂苷的含量测定,已经做第4次了,问题始终没解决,遂请教。具体要求如下:色谱条件要求:C8的柱子;流动相是乙腈-水(30:70);检测波长210nm。样品处理:取一定量,加75%乙醇超声10分钟,放冷,补足减失重量,滤过,即可。 对照品也是用75%乙醇溶解。测试经过如下:有同一厂家同一品种不同批号两批样品,用C8的柱子做,计算了下,含量合格,但峰实在太难看。怕峰形太差影响结果的准确性,但只有一根这C8的柱子,只好换根C18的柱子,调整流动相试试;峰形很好,进了一针其中一批的样品,计算了也合格,就编个序列继续做下去了。第四天才发现另一批不合格,只好第五天返工。第二次,问题出现了。返工时,同样的仪器,色谱柱,色谱条件,用原来配的对照品溶液,重新处理样品,不合格的那一批还是不合格,结果比上次测的还低一点,合格的那批也不合格了。又另配了对照品溶液,与之前配的浓度相差不大,但峰面积小多了。但是之前配的对照品峰面积反而变大了,估计是因为流动相微调,把之前没分开的小峰分开了,不至于斜着积分的原因吧。怀疑过对照品峰面积小,是不是冷冻(对照品要求冷冻保存哈)后没放至室温,直接称,导致称不准。样品怀疑过的原因 A:是不是不合格那一批取样量大了,浓度高了,导致过饱和,未全溶 B:样品是不是未混匀,取样代表性不够 C:超声时间,功率是不是有问题,是不是超声发热导致样品降解(对照品要冷冻保存嘛)E:是不是第二次配的溶样溶剂有问题第三次做,更蹊跷了。之前怀疑的样品的原因都排除了。减少,增加取样量;混匀样品;超声不同时间,用不同超声设备(不同功率);超声中换水,防止温度升高,水浴上回流处理样品;重配75%乙醇几次,用无水乙醇配也试了,还换用甲醇,样品溶液居然都没有目标峰。但之前两次配的对照品溶液峰高变化不大,高的还是高,低的还是低。第四次,就是今天。又重配了对照品溶液,换甲醇(色谱纯)溶解,居然对照品也没峰了,用75%乙醇溶解也是。之前配的对照品峰高还是老样子。将第一次配的对照品溶液跟这次提取的测不到目标峰的样品溶液混合,也能出目标峰。这根柱子换走另一个对照品测试,峰好得很。再换另一根柱,走样品和对照品,还是老样子,之前能出的还出,出不了的还是还是出不了。总结:一共配了三次对照品,浓度差异不大,但峰面积在变小,第三次就没了。样品也是,第一次合格的,第二次提取也不合格了,以后干脆目标峰也不出了。 分析:应该不是目标物不稳定的原因,因为第一次配的对照品,隔了一周,峰高依然变化不大,更何况新配的对照品溶液。 几次都是我操作,对照品溶液都超声助溶过,应该也是溶了的。而且用紫外扫过,稀释后吸收值会降低,虽然最大吸收波长在约205nm。其实用甲醇比用75%乙醇溶解要好些,几乎振摇就能溶,用75%乙醇必须稍超声一下。[/fo
请问哪里销售的可以用作液相色谱试验的对照品的?我需要购置一批对照品
[b]Q:[b][b][b][/b][/b]木香烃内酯、去氢木香内酯的测定,木香烃内酯的理论塔板数是?[/b]A:19900.521===============================================================【活动内容】1、每个工作日上午10:00左右发布一个关于应用数据库的应用问答题,版友根据题目给出自己理解的答案。2、每个工作日下午15:10公布参考答案。【活动奖励】幸运奖:抽奖软件,当天随机抽取3个或5个回答正确的版友ID号(最后一个ID号,截止至下午15:00),每人奖励[color=#ff0000]2钻石币[/color](抽奖人数≤10,抽取3个版友;抽奖人数>10,抽取5个版友);中奖名单:yifan1117(注册ID:yifan1117)dadgoh(注册ID:dadgoh)sixingxing(注册ID:v2889187)莫名其妙(注册ID:moyueqiu)WUYUWUQIU(注册ID:wulin321)[img=,690,388]https://ng1.17img.cn/bbsfiles/images/2018/11/201811211510283867_9448_1610895_3.png!w690x388.jpg[/img][img=,690,388]https://ng1.17img.cn/bbsfiles/images/2018/11/201811211510301867_942_1610895_3.png!w690x388.jpg[/img]积分奖励:所有回答正确的版友奖励[color=#ff0000]10个积分[/color](幸运奖获得者除外)。【注意事项】同样的答案,每人只能发一次[/b][align=left][color=#ff0000][b]PS:该贴浏览权限为“回贴仅作者和自己可见”,回复的版友仅能看到版主的题目及自己的回答内容,无法看到其他版友的回复内容。[/b][/color][/align][align=left][color=#ff0000][b] 下午3点之后解除,即可看到正确答案、获奖情况及所有版友的回复内容。[/b][/color][/align][align=center]=======================================================================[/align]方法:HPLC基质:标准溶液应用编号:103178化合物:木香烃内酯、去氢木香内酯色谱柱:[url=http://www.dikma.com.cn/product/details-219.html]Diamonsil C18(2) 5μm 250 x 4.6mm[/url]样品前处理:取适量标品,用甲醇将其溶解(0.1 mg/mL)。色谱条件:色谱柱:Diamonsil C18 (2) 5μm 250*4.6 mm (Cat#:99603)流动相:A:水 B:甲醇 A:B=35:65流速: 1.0 mL/min柱温:30 ℃检测器:UV 225 nm进样量:10 μL文章出处:天津应用实验室关键字:木香烃内酯、去氢木香内酯、HPLC、Diamonsil C18(2)摘要:参照2015年药典图谱:[img=,603,445]http://www.dikma.com.cn/Public/Uploads/images/011.PNG[/img]

【作者中文名】陈正收; 周应军; 胡达; 徐瑾; 曾光尧;【作者英文名】CHEN Zheng-shou1; 2; ZHOU Ying-jun2; HU Da1; XU Jin2; ZENG Guang-yao2(1.Hunan Jinsha Pharmaceutical CO.; LTD.; Changsha 410007; 2.College of Pharmacy; Central South University; Changsha 410013);【作者单位】湖南金沙药业股份有限公司; 中南大学药学院; 中南大学药学院 长沙;【摘要】目的建立高效液相色谱法测定调经活血片中木香烃内酯和去氢木香内酯的含量方法。方法Diamonsil C18色谱柱(250 mm×4.6 mm,5μm);流动相:甲醇-0.2%磷酸溶液(65∶35);检测波长:225 nm。结果木香烃内酯和去氢木香内酯的平均回收率分别为101.6%和98.4%,RSD(n=6)分别为2.34%和2.27%。结论方法简便、准确,可作为调经活血片的定量控制方法。http://ng1.17img.cn/bbsfiles/images/2012/08/201208061327_381850_2379123_3.jpg
三黄片的实验条件:乙腈:水(1:1)(每1000ml中加磷酸二氢钾3.4g,十二烷基硫酸钠1.7g)流动相,波长265nm,对照品的峰面积不稳定,每五针的RSD值大于2%。且样品的峰面积稳定是什么原因?对盐酸小檗碱发生变化。对照品的溶剂是甲醇。

[size=4][b] 小卢推荐:一种标定二级对照品的方法[/b][/size]对照品作为实验室(制药行业)一种常用的、重要的试剂,根据其类型,可分为:一级对照品,即为从中国药品生物制品检定所(简称:中检所)购买后直接使用的对照品;二级对照品,由一级对照品标定原料药得到的对照品。由于一级对照品的规格小、价格高、购买周期长的缺点,对于实验室对照品用量大的企业来说,使用二级对照品成了实验室的首选。现在,我就介绍一种标定二级对照品的方法,供大家参考一下。[b]第一,选定样品[/b]一般来说,选择自己生产的原料药价格便宜,不需要外购,且取用方便,是我们的首选。如果我们的生产工艺不好、稳定相差,最好选择外购知名企业的原料药。但要注意,要选择作为对照品的原料药一定是相对其他批次各检验项目都比较好的同一批原料药。[b]第二,标定方法[/b]现以高效液相测定法检测含量为例,来表述其测定方法。由于要严格保证所标定原料药的含量,因此采用3人、3份样品的方法进行测定,即:每个人称取2份对照品、3份样品进行测定;共有3人进行测定。如果有条件,3个人可以选择3台不同的液相色谱仪进行实验。在这里要求2份对照品共进样5针,计算校正因子,并求RSD应小于0.5%,3份样品各进1针,求平均值。方法和要求如下表:[img]http://ng1.17img.cn/bbsfiles/images/2009/11/200911090939_182733_1622024_3.jpg[/img]按照表格内容,由3人得到的3个不同的含量,最后求得均值,即得样品(二级对照品)的含量,并要求3者的RSD≤1%。[b]第三,分装[/b]使用抗生素瓶分装,装量按照每次使用量(如60mg,则装入80-100mg即可)为标准,即使每个抗生素瓶中的对照品只使用一次。这样既能避免对照品被污染,又能使其少吸潮。如果为了节省抗生素瓶,采用大装量,即一瓶中的对照品可以使用多次,那么,建议在使用3-6次后就报废本瓶对照品。因为每次打开瓶口称取对照品都是对该瓶对照品的一次污染,尤其是空气中水分对它的影响,这样会是对照品的含量发生变化,原来的标定也就失去了意义。分装环境:建议在层流罩下进行,严格控制温湿度(建议温湿度:18-24℃,45-65%)。封口步骤:分装后,用橡胶盖盖紧,再用封口膜封好后,用铝盖压实即可。[b]第四,制定有效期[/b]一般比较稳定的样品制定2年,不是很稳定的样品制定1年。但是这个有效期不能超过该样品本身法定的有效期。[b]第五,贴签[/b]制定好了有效期就可以把样品(二级对照品)的标签贴上去了,标签格式如下:[img]http://ng1.17img.cn/bbsfiles/images/2009/11/200911090939_182734_1622024_3.jpg[/img][b]第六,储存[/b]不管原来样品法定的存储温度是多少,都建议保存的温度最好在2-10°,即冰箱中的冷藏温度。根据资料研究,2°是药品的最佳保存温度,因为这个温度下药品的降解速度最慢。[b]第七,复核[/b]我们制定了有效期后,并不是就完成了所有的工作。我们要在有效期的一半时,对二级对照品进行复核,检验方法同本法中第二步骤,所取样品则是从原标定的二级对照品中抽取。如果复核结果没有变化,则继续使用;如果复核结果发生了变化,那就按照复核的含量,从新贴签标示。通过以上7步就完成了对照品的标定工作,大家有什么看法可以回帖说明,我们共同讨论![em09505][em09505](全文完!)
大家平时做实验的时候都带标准对照品吗?我们半年期间核查的时候用对照品,平时就做加标回收的,不知大家怎么样,欢迎讨论!
植物成分标准品、对照品、单体、http://hi.baidu.com植物提取物http://hi.baidu.com植物提取物标准品1加兰他敏、石蒜碱,丹皮酚 Paeonol、光甘草定、丹参酮系列(丹参酮ⅡA、隐丹参酮、丹参酮Ⅰ),齐墩果酸,白黎芦醇(RESV),叶黄素、红景天苷、原花青素B2 Procyanidin B2金丝桃苷,金丝桃素、辣椒素、Asiaticoside(积雪草苷)Astragaloside IV(黄芪甲苷)系列等等。 联系方式:jiehua0501@yahoo.com.cn 刘 推荐,请告电话联系方式。1到2天回复。 qq37144588(请注明事由)。MSN:jiehuahua0501@hotmail.com 13482587565 植物提取物:单体白黎芦醇、绿原酸、加兰他敏、石蒜碱、盐酸青藤碱,二十八烷醇,丹皮酚,、丹参酮ⅡA,葛根素,番茄红素、莽草酸、5-HTP(五羟色氨)、青蒿素、二氢杨梅素、獐牙菜苦苷,鬼臼毒素,冬凌草甲素,熊果酸等等。以上产品提供20%-99%的产品,大量供应,包装大小根据您的需要。提取物:葡萄籽提取物(原花青素opc95%)、茶多酚(tp),红景天(甙)提取物,枇杷叶提取物,锯叶棕提取物,葛根提取物等,以及各种比例提取物。 标准品2木犀草素,甘草酸单铵,异欧前胡素,Vindoline(文多灵),Rosmarinic acid(迷迭香酸),Sailkosaponins D(柴胡皂苷D),Imperatorin(欧前胡素),Isoimperatorin(异欧前胡素),Vinblastine sulfate(硫酸长春碱),肉苁蓉苷A,芦荟大黄素,β-谷甾醇,秦皮甲素,Cichoric acid(菊苣酸),Mangiferin(芒果苷),α-Cyperone(α-香附酮),1-Deoxynojirimycin (1-脱氧野尻霉素),Sarsasapogenin(知母皂苷元),Nitidine Chloride(氯化两面针碱),10-Deacetylbaccatine III,Buddleoside(蒙花苷),Silybin(水飞蓟宾),6-Gingerol(6-姜酚),Catharanthine(长春质碱),Syringin(紫丁香苷),人参皂苷Rb3,三七皂苷R1,柴胡皂苷A,五味子丙素,佛手柑内酯,蛇床子素,白花前胡甲素,羽扇豆醇,Praeruptorin A,柴胡皂苷C,白头翁皂苷B4,积雪草苷,豆腐果苷,五味子酯甲,五味子甲素,大黄酸,五味子乙素,五味子醇甲,苍术素, Pseudohypericin(伪金丝桃素), 苍术素醇,安五脂素,细辛脂素,苦杏仁苷,Polydatin(虎杖苷),3,29-二苯甲酰栝蒌仁三醇,大黄素甲醚,薄荷醇,细辛脂素,鬼臼毒素,丁香苷,冬绿苷,豆腐果新苷A,B,C。menisdaurin,3,29-二苯甲酰栝蒌仁三醇,表木栓醇,柴胡皂苷D,毛花洋地黄苷C,黄芪皂苷II,对羟基苯甲酸乙酯,白花前胡丙素,桃叶珊瑚甙,胡黄连苦苷I,和厚朴酚,白花前胡丁素,秦皮乙素,没食子酸,芍药甙,补骨脂素,岑酮,白花前胡素E,胡黄连苦苷II,阿魏酸,龙胆苦苷,丹参素钠,水杨苷,木香烃内酯 Luteolin、穿心莲内酯,右旋比扣扣灵碱,槐果碱,乌头碱,槐胺碱青藤碱、姜黄素系列,靛玉红,豨莶精醇,异古伦宾 银杏系列(白果内酯、银杏内酯A、银杏内酯b)虎杖甙、、芹菜素、茄尼醇、芥子碱硫氰酸盐,常春藤皂苷元,木犀草素, 虫草素、EGCG(姜黄素 Curcumin,去甲氧基姜黄素 Curcumin2,去二甲氧基姜黄素 Curcumin 3、阿魏酸 Ferulic acid、积雪草苷,豨莶精醇 Asiaticoside、柴胡系列柴胡皂甙 A Saikosaponins A、柴胡皂甙 D Saikosaponins D、去氢木香内酯,异土木香内酯,土木香内酯,番泻苷A Sennoside A栀子苷 Caryptoside、山奈酚 Kaempferol、Hyperoside、根皮苷Phloridzin、氢溴酸槟榔碱Arecoline Hydrobromide、2-hydroxyeupatolide、阿卡宁 Alkannin、Salidroside、肉桂醇苷 Rosavin、酪醇 Tyrosol、奇任醇,辣椒素系列、苍术内酯Ⅲ,二氢辣椒素 Dihydrocapsaicin、番泻苷A,二氢辣椒素 Dihydrocapsaicin、乌药醚内酯,吉马酮,雄烯二酮 Androstenedione、10-脱乙酰巴卡丁 III10-DAB 10 III麻醉椒苦素 Methysticin 枸橼酸血根碱,醉椒素Kavain 二氢醉椒素Dihydrokavain吴茱萸碱Evodiamine1-乙酸基-5-去乙酰基-巴卡亭 I,吴茱萸次碱Rutaecarpine 水飞蓟宾 Silybin石衫碱甲 Huperzine-A哈巴饿甙,二氢丹参酮,Harpagoside水杨甙 20-羟基蜕皮甾酮β- 蜕皮甾酮吲哚- Ecdysone甘草酸 Glycyrrhizic acid、异鼠李素Nordihydrocapsaicin、N –Vanillylnonanamide、鸢尾苷,野黄芩苷,乙氧基血根碱,吲哚醇,乙氧基白屈菜红碱N –Vanillyldecanamide、秋水仙碱,白藜芦醇甙Polydatin、、白鲜碱dictamnine、山萘素、Kaempferol、异鼠李素Isorhamnetin、花椒毒酚、淫羊藿苷Icariin、染料木素,芝麻素, 川芎嗪,羟基吴茱萸碱,紫草氰苷,新橙皮甙, 橙皮甙,柚皮甙,橙皮甙二氢查尔酮、柚皮甙二氢查尔酮、东方唐松草苷、苦参碱、茵芋苷,胡黄连苷Ⅰ、氧化苦参碱、贝母甲素,贝母乙素,青蒿素、辛弗林、apiosylskimmin,格列风内酯、高良姜素,芝麻素, 黄芪甲苷、蜕皮激素, 阿马碱, 莽草酸,熊果酸、EGCE。穗花双黄酮,番茄红素(90~95%)5-HTP,大黄素、蜕皮激素(20-β-蜕皮甾酮)阿魏酸,黄芪甙,豆蔻明,甘草酸二铵盐,异甘草素,原花青素B2 Procyanidin B2、丹参酮ⅡA,番茄红素,绿原酸,叶黄素,钩腾碱,水杨甙,灵芝酸, 山奈酚-3-O-芸香糖苷、阔叶冬青苷G、迷迭香酸Rosmarinic, 齐墩果酸Oleanolic acid, 刺芒柄花素Formononetin( 98%美国进口/5mg) , 鞣花酸 Ellagic acid, 熊果酸 Ursolic acid, 连翘苷 phillyrin, 氢溴酸槟榔碱(97%)Arecoline Hydrobromide, 牛蒡子苷Arctiin, 栀子苷 Caryptoside,大黄素甲醚 Physcion, 大黄酚 Chrysophanol, 芦荟大黄素 Aloe_emodin, 金丝桃苷, 薯蓣皂苷元, 甘草酸单胺盐, 熊果苷, 梣酮、人参皂甙系列,ROSAVIN,五味子醇甲,五味子乙素,扁蓄苷、木香烃内酯、五味子甲素,柴胡皂甙A,柴胡皂甙B,银杏内酯A,Ginkgolide A,银杏内酯B,GinkgolideB,去二氢甲氧基姜黄素,去甲氧基姜黄素 ,Curcumin2,, 18-β甘草次酸 18β-Glycyrrhetinic acid ,甘草次酸,五味子酯A GomisinA,羟基吴茱萸碱、 五味子酯N Gomisin N ,白果内酯 Bilobalide,乙酰紫草素Acetylshikonin,苦杏仁苷Amygdalin、牛蒡子苷、苍术内酯Ⅲ、吴茱萸碱、异丁酰紫草素素,甘草酸单胺盐DENG,枸橼酸血根碱、儿茶酸(+)-Catechin,紫草素 Shikonin,咖啡酸、乌药醚内酯、柯里拉京,苦杏仁苷,辣椒素,龙胆苦甙,氯化两面针碱,夏无碱、落叶松树脂醇-吡喃糖苷,马兜铃酸,马钱素,马钱子碱,吲哚醇、拟人参皂苷F11,尿囊素,牛蒡子甙,欧前胡素,七叶甙,秋水仙碱,肉桂酸,异土木香内酯、三尖杉宁碱,山姜素,山奈酚,山奈素,麝香草酚,石杉碱甲,酸枣仁皂苷A,酸枣仁皂苷B,乙氧基白屈菜红碱、阿马碱、天麻素,甜菜碱,土大黄甙,乌索酸,五味子醇甲,西贝碱,延胡索乙素,左旋紫草素,对-香豆酸,枸橼酸血根碱、原人参二醇,南蛇藤素,雷公藤内酯A、雷公藤红素、番泻苷A、槐角苷,岩白菜素,千金子二萜醇,豨莶精醇、靛蓝,槐角苷、斑蝥素,羽扇豆醇,右旋比扣扣灵碱、异土木香内酯、羟基积雪草苷,鸢尾苷,8-Gingerol(8-姜酚),10-Gingerol(10-姜酚)等等。产品在不段更新中。jiehua0501@yahoo.com.cn 提供大部分产品(液相色谱hplc)检测条件(参考)。供应标准品,g级供应可获更大优惠。部分产品可以kg级供货(等)-价格有竞争力,多种规格。以上可以部分产品可以大量生产供货,还有多种的含量与规格。有需要,欢迎你联系。由于种类太多,请您先有邮件和我联系,标明你要的数量等具体要求,我会在最短的时间给你回复,推荐联系方式:jiehua0501@yahoo.com.cn qq: 37144588或MSN:jiehuahua0501@hotmail.com 13482587565
[size=20px][color=#93c6bc][b]鉴别[/b][/color][/size][size=16px][color=#e2a4a4]|[/color][/size] [font=宋体] (1)本品横切面:假种皮有时残存,为多角形薄壁细胞。种皮表皮细胞类圆形,壁较厚;下皮为1~3列薄壁细胞,略切向延长;色素层为数列棕色细胞,其间散有类圆形油细胞1~2列,直径约50[/font]μm[font=宋体];内种皮为1列栅状厚壁细胞,棕红色,内壁与侧壁极厚,胞腔小,内含硅质块。外胚乳细胞含淀粉粒和草酸钙方晶及少数细小簇晶。内胚乳细胞含糊粉粒。[/font] [font=宋体]粉末黄棕色。种皮表皮细胞表面观呈长条形,直径约至30[/font]μm[font=宋体],壁稍厚,常与下皮细胞上下层垂直排列;下皮细胞表面观长多角形或类长方形。色素层细胞皱缩,界限不清楚,含红棕色物,易碎裂成不规则色素块。油细胞散生于色素层细胞间,呈类圆形或长圆形,含黄绿色油状物。内种皮厚壁细胞黄棕色或红棕色,表面观多角形,壁厚,非木化,胞腔内含硅质块;断面观细胞1列,栅状,内壁及侧壁极厚,胞腔偏外侧,内含硅质块。外胚乳细胞充满淀粉粒集结成的淀粉团,有的包埋有细小草酸钙方晶。内胚乳细胞含糊粉粒和脂肪油滴。[/font] [font=宋体](2)取本品粉末1g,加甲醇5ml,置水浴中加热振摇5分钟,滤过,取滤液作为供试品溶液。另取山姜素对照品、小豆蔻明对照品,加甲醇制成每1ml各含2mg的混合溶液,作为对照品溶液。照薄层色谱法(通则0502)试验,吸取上述两种溶液各5[/font]μl[font=宋体],分别点于同一硅胶G薄层板上,以甲苯-乙酸乙酯-甲醇(15:4:1)为展开剂,展开,取出,晾干,在100℃加热至斑点显色清晰,置紫外光灯(365nm)下检视。供试品色谱中,在与山姜素对照品色谱相应的位置上,显相同的浅蓝色荧光斑点;喷以5%三氯化铁乙醇溶液,供试品色谱中,在与小豆蔻明对照品色谱相应的位置上,显相同的褐色斑点。[/font] [font=宋体][/font] [font=宋体][/font] [font=宋体][/font] [font=宋体][/font] [font=宋体] [/font] [size=20px][color=#93c6bc][b]含量测定[/b][/color][/size][size=16px][color=#e2a4a4]|[/color][/size] [font=宋体][/font] [b][font=宋体][/font] [font=宋体] 挥发油 [/font][/b][font=宋体] [/font][font=宋体]照挥发油测定法(通则2204)测定。[/font] [font=宋体]本品含挥发油不得少于1.0%(ml/g)。[/font] [b][font=宋体]山姜素、乔松素、小豆蔻明与桤木酮 [/font][/b][font=宋体] [/font][font=宋体]照高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]法(通则0512)测定。[/font] [b][font=宋体]色谱条件与系统适用性试验[/font][/b][font=宋体] [/font][font=宋体]以十八烷基硅烷键合硅胶为填充剂;以甲醇为流动相A,以水为流动相B,按下表中的规定进行梯度洗脱,检测波长为300nm。理论板数按小豆蔻明峰计算应不低于5000。[/font] [align=center] [/align] [b][font=宋体]对照品溶液的制备[/font][/b][font=宋体] [/font][font=宋体]取山姜素对照品、乔松素对照品、小豆蔻明对照品、桤木酮对照品适量,精密称定,加甲醇分别制成每1ml含山姜素、乔松素、小豆蔻明各40[/font]μg[font=宋体],桤木酮80[/font]μg[font=宋体]的溶液,即得。[/font] [b][font=宋体]供试品溶液的制备[/font][/b][font=宋体] [/font][font=宋体]取本品粉末(过三号筛)约0.5g,精密称定,置具塞锥形瓶中,精密加入甲醇50ml,称定重量,超声处理(功率250W,频率40kHz)30分钟,放冷,再称定重量,用甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。[/font] [b][font=宋体]测定法[/font][/b][font=宋体] [/font][font=宋体]分别精密吸取对照品溶液与供试品溶液各5[/font]μl[font=宋体],注入[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱仪[/color][/url],测定,即得。[/font] [font=宋体]本品按干燥品计算,含山姜素[/font][font=宋体]([/font]C[sub]16[/sub]H[sub]14[/sub]O[sub]4[/sub][font=宋体])[/font][font=宋体]、乔松素[/font][font=宋体]([/font]C[sub]15[/sub]H[sub]12[/sub]O[sub]4[/sub][font=宋体])[/font][font=宋体]和小豆蔻明[/font][font=宋体]([/font]C[sub]16[/sub]H[sub]14[/sub]O[sub]4[/sub][font=宋体])[/font][font=宋体]的总量不得少于1.35%,桤木酮[/font][font=宋体]([/font]C[sub]19[/sub]H[sub]18[/sub]O[font=宋体])[/font][font=宋体]不得少于0.50%。[/font] [font=宋体][/font][font=宋体][/font] [font=宋体][/font]

土木香原料检测国家药典上写的方法是气相色谱检测,用PEG-20M色谱柱,柱温箱才升到240℃,可是土木香内酯沸点有275℃,极性色谱柱承受温度比较低,好久都没有出峰,故提高柱前压,和升温速率,柱前压为0.15KPa,升温速率为20℃/min进行测试,缩短出峰时间,标样谱图和样品谱图如下http://ng1.17img.cn/bbsfiles/images/2014/12/201412282250_529740_2620176_3.pnghttp://ng1.17img.cn/bbsfiles/images/2014/12/201412282251_529741_2620176_3.png
最近在做高分辨率质谱(ESI源),但是遇到了对照品不出峰的情况,试了正负离子模式,流动相甲醇,乙腈,纯水,甲酸水也都试了。一共6个对照品,就是这一个找不到峰。对照品拿去测了HPLC和GC-MS都没有问题,分子式和质量数也确认了没有错误,加H和加Na的质量数也都找了,但是还是扫不到这个对照品的母离子。请问大家可能是什么原因造成的?有没有什么解决办法呢?谢谢了
请教:注射用水溶性维生素中关于烟酰胺、等5项的液相检测方法其标准为烟酰胺、盐酸吡哆辛、硝酸硫胺、泛酸钠、维生素C钠和核黄素磷酸钠 照高效液相色谱法(中国药典1995年版二部附录Ⅴ D)测定。 色谱条件与系统适用性试验 用氨基键合多孔硅胶为填料,以(0.02mol/L)磷酸二氢钾溶液-乙腈(27:73),用10%盐酸溶液调节pH为5.3的溶液为流动相,流速为1.5ml/min,检测波长:烟酰胺、盐酸吡哆辛、硝酸硫胺、泛酸钠、维生素C钠为214nm;核黄素磷酸钠用萤光检测λEX=445nm、λEM=520nm。各组分的分离度应符合要求。 对照品溶液的制备 (1)取烟酰胺对照品约150mg、硝酸硫胺对照品约12mg、盐酸吡哆辛对照品约18mg、泛酸钠对照品约62mg,分别精密称量置50ml量瓶中,加水溶解并稀释至刻度摇匀,精密量取2ml置50ml量瓶中,用流动相稀释至刻度,摇匀,即为对照品溶液(Ⅰ),此溶液置暗处充氮气于零下20℃可保存1个月。(2)取维生素C钠对照品约425mg、核黄素磷酸钠对照品约19mg,精密称定,置50ml量瓶中加水溶解并稀释至刻度摇匀,精密量取2ml置50ml量瓶中,用流动相稀释至刻度,摇匀即为对照品溶液(Ⅱ),此溶液必须临用新鲜配制,并于零下20℃保存,用前放置至室温。 等容混合对照品溶液(Ⅰ)和对照品溶液(Ⅱ)即为对照品溶液。 供试品溶液的制备 取装量差异项下的内容物约2瓶重量,精密称定,置100ml量瓶中,加水溶解并稀释至刻度,摇匀,精密量取15ml置200ml量瓶中,用流动相稀释至刻度。 测定法 取对照品溶液和供试品溶液各10μl,交替注入液相色谱仪,测定,用外标法计算各组分含量,即得。目前存在问题用紫外检测的分不开5种组分,大家有什么好办法,谢谢
请教各位大神,新配的对照品溶液,峰面积减小很快是什么原因呢?谢谢。
我的样品做高效液相-蒸发光检测器,用的是甲醇-水梯度洗脱,之前做的图谱都很好,最近几天做,对照品的图谱没问题,可是样品的峰都拖尾,有些很严重。开始我以为是柱子问题,后来换了一根新柱子,结果还是对照品的图谱还好,样品就出现拖尾,不知什么原因,所以请教大家有没遇过这种情况。(柱子在我做之后又做过另一个产品,流动相是乙腈-0.4%磷酸溶液,后来做我的样品就出现这个问题,再拿那根柱子做另一个产品也拖尾,对照品顶头裂成2个峰)。色谱条件都没变过,液相和检测器前后都是同一台机器。样品1 -前后次图谱http://ng1.17img.cn/bbsfiles/images/2012/08/201208282153_386877_2593542_3.jpghttp://ng1.17img.cn/bbsfiles/images/2012/08/201208282153_386879_2593542_3.jpg样品2 -前后图谱http://ng1.17img.cn/bbsfiles/images/2012/08/201208282155_386880_2593542_3.jpghttp://ng1.17img.cn/bbsfiles/images/2012/08/201208282155_386881_2593542_3.jpg对照品:http://ng1.17img.cn/bbsfiles/images/2012/08/201208282158_386883_2593542_3.jpg
我第一次测土木香含量,哪位做过这个,土木香内酯和异土木香内酯的出峰时间是怎样的?色谱图是什么样的?请各位老师帮帮忙,谢谢
中药对照品称量的时候静电太多,,粉末状的对照品粉末,比如陈皮苷,用了除静电仪,也没有用,大佬们有什么办法解决吗(???.???)????
岛津液相色谱,对同一个对照品溶液进行检测,手动进样,但是每次得到主峰的响应值都不一样,而且相差很大,到底是怎么回事啊?因为怕是溶液混合不均匀,特意超声了10分钟,并且,每次进样都摇匀,请高手赐教!!同一个溶液,响应值不会差那么多啊!谢谢各位~~!
本人刚上手气相,想请教一下:气相的对照品是不是不能用普通的化学试剂的,而是要买对照品试剂的,就像苯的话就要苯对照品而不是化学纯的或是优级纯的化学试剂
公司自己标定的工作对照品放在冰箱储存,如何保持干燥,放干燥剂的话,有什么合适的方法放置吗?对照品的种类比较多,目前是放在盒子里,每个小瓶都封蜡,但湿度还是很大,怕影响含量,求提议~
问题: 各位老师,有人做过北豆根的含量测定吗?对照品蝙蝠葛苏林碱用甲醇很难溶,试了用流动相也几乎不溶,请问有什么好建议吗
木香板中游离甲醛的测定,9-11L干燥法和40L干燥法测定结果差距大,标准上也说的含糊,这是什么原因,鉴于这种情况,如何取舍或修正?
我是做中药检验的,以前用的对照品溶液都经0.45滤膜过滤后使用,还做过某两种对照品溶液的测试,峰面积差异不大。但最近我使用黄芩苷对照品时发现检品含量总是很高,反复测试才找到原因,过滤后对照品的峰面积为:3249725.000,没过滤的对照品峰面积为:4933873.000,差异很大,不知道各位朋友使用对照品溶液时是否也经过0.45滤膜过滤?有没有文件规定对照品溶液能否过滤?
各位前辈,小妹有个问题请教,我用的是安捷伦的高效液相色谱仪,最近做黄柏中盐酸小檗碱的含量测定,347nm,对照品不出峰,样品也就前四分钟内有点杂质峰。很奇怪,以前做的都很正常,这次流动相,对照品以及样品的处理方法都没有变。怀疑是流动相得原因,所以让同事帮忙复检,结果也一样。怀疑是柱子的原因,换了三根柱子,一根做的都有峰,但是个很奇怪的肩峰,另外两根都无峰。又怀疑是检测器的原因,但用它做其他的品种都很好。很是纳闷。想问一下各位前辈在工作中有没有遇到过这种类似的问题,最后是怎么解决的。感激不尽![em09508]
中药标准品等中英文名称对照货号 英文名称 中文名称019-14621 Aconitine std. 乌头碱012-14091 Albiflorin std 芍药内酯苷018-13231 Alisol B acetate,98.0% 乙酸泽泻酯B010-13431 Alisol B std 泽泻醇B015-11921 Arbutin std 熊果苷012-14611 Atractylenolide III std. 苍术内酯016-11691 Atropine sulfate std. 硫酸阿托品016-10351 Aucubin std. 桃叶珊瑚苷027-07751 Baicalein std. 黄岑素020-07741 Baicalin std. 黄岑苷022-08161 Barbaloin std. 芦荟苷022-07681 Berberine chloride std. 氯化黄连素028-12051 Bergenin std. 岩白菜内酯025-10121 Bufalin std. 蟾毒灵024-10691 Bufotalin std. 蟾毒它灵030-10611 Capillarisin std. 茵陈色原酮036-10613 Capillarisin std. 茵陈色原酮031-15141 Capsaicin std. 辣椒素032-10551 Catalpol std. 梓醇038-13711 Cinobufagin std. 华蟾毒精035-13721 Cinobufotalin std. 华蟾毒它灵036-11311 Coptisine chloride 氯化黄连碱036-14971 Corydaline std. 紫堇碱032-13731 Costunolide std. 木香烃内酯043-27611 Dehydrocorydaline nitrate std. 硝酸脱氢紫堇碱040-21881 Dehydrocostuslactone std. 脱氢广木香内酯044-25321 Dihydrocapsaicin std. 二氢辣椒素042-18911 Dimethylesculetin std. 二甲基七叶树内酯052-04921 Ergosterol std. 麦角甾醇056-05161 β-Eudesmol std. β-桉油醇050-04601 Evodiamine std. 吴茱萸碱070-02241 Geniposide std. 京尼平苷078-02301 Geniposidic acid std. 京尼平苷酸077-02871 6-Gingerol std. 6-姜辣醇079-02191 Ginsenoside Rb1 std. 人参皂苷Rb1078-03261 Ginsenoside Rc 人参皂苷Rc072-03301 Ginsenoside Rd std. 人参皂苷Rd075-03271 Ginsenoside Re std. 人参皂苷Re072-02201 Ginsenoside Rg1 std. 人参皂苷Rg1071-02271 Glycyrrhizin std. 甘草甜素089-04951 Honokiol std. 和厚朴酚081-06851 (E)-10-Hydroxy-2-decenoic acid std. (E)-10-羟基-2-癸烯酸084-07061 Hypaconitine 次乌头碱099-03651 Isofraxidine std. 125-03361 Liquiritin std. 甘草甙125-03621 Loganin std. 番木鳖苷137-09081 Magnolol std. 厚朴酚130-12261 Mesaconitine 中乌头碱140-06211 Naringin std. 柚皮甙151-01801 Osthole std. 甲氧基欧芹酚150-01511 Oxymatrine std. 氧化苦参碱167-11711 Paeoniflorin std. 芍药苷163-11713 Paeoniflorin std. 芍药苷169-12871 Paeonol std. 牡丹酚166-17641 Palmatine chloride std. 氯化巴马亭164-14091 (+)-phyllodulcin std. (+)-叶甜素162-14071 Puerarin std. 葛根素184-00951 Resibufogenin std. 残余蟾蜍配基183-00921 Rutaecarpine std. 吴茱萸次碱190-08411 Saikosaponin a std. 柴胡皂甙a194-10021 Saikosaponin b2 std. 柴胡皂甙b2197-08421 Saikosaponin c std. 柴胡皂甙c194-08431 Saikosaponin d std. 柴胡皂甙d192-10441 Schizandrin std. 五味子素197-09261 Scopolamine hydrobromide, 99.0% 氢溴酸莨菪胺190-08531 Sennoside A std. 番泻苷A194-09271 Sennoside B, 96% 番泻苷B199-11811 Sennoside B std. 番泻苷B191-08681 Shikonin std. 紫草素193-09361 Sinomenine std. 青藤碱197-08541 Swertiamarin std. 獐牙菜苦苷231-00811 Wogonin std. 汉黄岑素
做含量测试中,由于购买标准品的成本比较高,很多公司采用自己生产的产品作为工作对照品使用。我想请问一下,这个工作对照品的使用之前,是否需要做一些稳定性考察,以确定其保存环境和有效期?具体还需要做怎么样的考察?一般都如何考察?有没有相关的明确的规定?谢谢。







 我要推广仪器
我要推广仪器
 下载APP
下载APP