[color=#444444]请问[/color][color=#444444]GB/T5009.84-2003 [/color][color=#444444]食品中硫胺素(维生素[/color][color=#444444]B1[/color][color=#444444])的测定中所用的硫胺素标准品指的是硝酸硫胺素还是盐酸硫胺素?这两种标准品都有的卖。[/color][color=#444444]但就是找不到[/color][color=#444444]“[/color][color=#444444]硫胺素[/color][color=#444444]”[/color]
维生素B1(硫胺硝酸盐) 标准品CDCT-C17455500 CAS 532-43-4 纯品型,有证书,0.25g价格 360RMB因有效期将近,03/2014,现5折清仓,库存有限,可直接回复联系我,也可联系安谱总部 021-54890099
检测VB1,根据5413.11-2010,标准品称相当于硫胺素50mg,我们用的标准品是硝酸硫铵(分子量327.37)或盐酸硫铵。硫胺素的分子量是300.81,换算系数应该是327.37/300.81=1.088,在网上查的换算系数是:1mg硫胺素=1.3379mg维生素B1盐酸盐,1mg硫胺素=1.2336mg维生素B1硝酸盐。我该用哪个呢?谢谢了!
大家好 我 分析硫氰酸铵,有点不 明白, 该方法的国家 标准是将0.3g的硫氰酸铵样溶到过量的硝酸银标准溶液里,用硫氰酸铵标准溶液标定过量的硝酸银,而该硫氰酸铵的标准溶液则是由分析纯的硫氰酸铵配制的,将他滴定标准硝酸银溶液来的,为什么不直接自己配样品的硫氰酸铵溶液来直接滴定硝酸银标准溶液呀。
[em04] 我要测定降水中硫酸根和硝酸根离子的含量,谁能帮我这方面的分析标准,谢谢
大家好 请教个问题 我做液相色谱分析标准曲线,用邻氯苯胺盐酸盐做标准曲线,但是样品是邻氯苯胺硫酸盐,怎么计算邻氯苯胺的含量?谢谢
及需硝酸铝、硝酸铁和碳酸铵的定量分析标准,哪位大侠有啊,急需
食品安全现场快速检测分析方法 编号索引 标准溯源GB/T 中华人民共和国国家标准分析方法CDC/SB 中国疾病预防控制中心营养与食品安全所 现场快速检测分析方法CDC/SB 101 农药有机磷类(国内允许使用的30 多种)和氨基甲酸酯类(国内允许使用的10 多种)的快速检测:采用国家标准快速检测方法GB/T5009.199-2003。适用于蔬菜、水果以及其它食品中农药残毒的快速检测,也适用于食物中毒物质中农药的快速筛选定性。现场使用,20 分钟出结果。CDC/SB 102 鼠药毒鼠强的快速检测:中国疾病预防控制中心营养与食品安全所现场快速检测方法。主要用于预防性监测和中毒物的筛选、定性鉴别。检出限5ug/ml。现场使用,20 分钟出结果。CDC/SB 103 鼠药敌鼠钠盐的快速检测:中国疾病预防控制中心营养与食品安全所现场快速检测方法。主要用于预防性监测和中毒物的筛选、定性鉴别。检出限45ug/ml。现场使用,10 分钟出结果。CDC/SB 104 鼠药安妥的快速检测:中国疾病预防控制中心营养与食品安全所现场快速检测方法。主要用于预防性监测和中毒物的筛选、定性鉴别。检出限20ug/ml。现场使用,10 分钟出结果CDC/SB 105 鼠药氟乙酰胺的快速检测:中国疾病预防控制中心营养与食品安全所现场快速检测方法。主要用于预防性监测和中毒物的筛选、定性鉴别。检出限50ug/ml。现场使用,15 分钟出结果。CDC/SB 106 亚硝酸盐的快速检测:中国疾病预防控制中心营养与食品安全所现场快速检测方法。本方法是在国家标准GB/T5009.33-2003 盐酸萘乙二胺检测方法基础上改进的现场快速检测方法--速测管法。定性兼半定量检测。可用作卫生指标检测、投毒监测和食物中毒物质的快速筛选、定性定量。最低检出量为0.025mg/L。现场使用,15 分钟出结果。
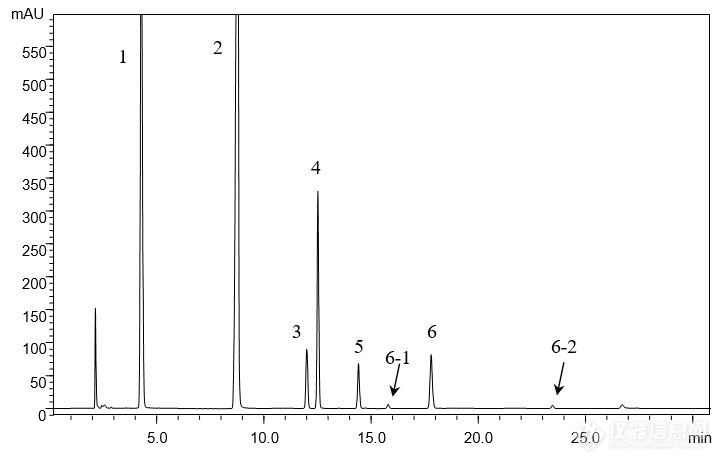
[align=center][b]注射用水溶性维生素的分析(1)[/b][/align][align=center][b]——维生素C钠、烟酰胺、泛酸钠、盐酸吡哆辛、硝酸硫胺、核黄素磷酸钠[/b][/align][align=center][b][/b][/align][align=left]客户提供了注射用水溶性维生素粉针剂及对照品(维生素C钠、烟酰胺、泛酸钠、盐酸吡哆辛、硝酸硫胺、核黄素磷酸钠),要求本实验室依据客户所提供方法筛选合适色谱柱,实现粉针剂中各组分物质的良好分离,同时满足方法中对分离度及理论塔板数的要求。[/align][align=left][/align][align=left]首先,依据客户提供的色谱条件,对对照品溶液进行分析。尝试使用不同填料类型的色谱柱,包括CAPCELL PAK C[sub]18[/sub] MGII,AQ S5,AQ S3,SUPERIOREX ODS,CAPCELL PAK ADME及PFP色谱柱。[/align][align=left][/align][align=left]由于不同色谱柱的分离选择性不同,最终发现CAPCELL PAK C[sub]18 [/sub]MGII色谱柱在完全按照客户提供方法的前提下能够得到最佳分离结果,对照品溶液中各组分均能够得到良好分离(如图1),分离度均在3.0以上,满足客户1.5要求;理论塔板数按核黄素计为103983,满足客户大于15000的要求(见表1)。[/align][align=left][/align][align=center][img=,690,443]http://ng1.17img.cn/bbsfiles/images/2018/01/201801041022_5661_2222981_3.jpg!w690x443.jpg[/img][/align][align=center]图1 CAPCELL PAK C[sub]18[/sub] MGII分析对照品溶液结果[/align][align=center] 1. 维生素C钠 2. 烟酰胺 3. 泛酸钠 4. 盐酸吡哆辛 5. 硝酸硫胺 6, 6-1, 6-2 核黄素磷酸钠[/align][align=left][img=,690,270]http://ng1.17img.cn/bbsfiles/images/2018/01/201801041022_1444_2222981_3.jpg!w690x270.jpg[/img][/align][align=left][/align][align=left][/align][align=center]表1 CAPCELL PAK C[sub]18 [/sub]MGII分析对照品溶液结果详表[/align][align=center][img=,458,316]http://ng1.17img.cn/bbsfiles/images/2018/01/201801041022_2894_2222981_3.jpg!w458x316.jpg[/img][/align][align=center][/align][align=center]在上述色谱条件下,继续使用CAPCELL PAK C[sub]18[/sub] MGII色谱柱对粉针剂供试品溶液进行分析,各待测组分及相邻杂质均能够得到良好分离,结果见图2、图3及表2。[/align][align=center][/align][align=center][img=,682,427]http://ng1.17img.cn/bbsfiles/images/2018/01/201801041023_7282_2222981_3.jpg!w682x427.jpg[/img][/align][align=center]图2 CAPCELL PAK C[sub]18[/sub] MGII分析供试品及空白溶液结果[/align][align=center]1. 维生素C钠 2. 烟酰胺 3. 泛酸钠 4. 盐酸吡哆辛 5. 硝酸硫胺 6, 6-1, 6-2 核黄素磷酸钠[/align][align=center][/align][align=left][img=,690,200]http://ng1.17img.cn/bbsfiles/images/2018/01/201801041023_5300_2222981_3.jpg!w690x200.jpg[/img][/align][align=left][/align][align=center][img=,690,455]http://ng1.17img.cn/bbsfiles/images/2018/01/201801041024_9636_2222981_3.jpg!w690x455.jpg[/img][/align][align=center]图3 CAPCELL PAK C[sub]18[/sub] MGII分析供试品溶液结果放大图[/align][align=center] [/align][align=center]表2 MGII分析供试品溶液结果详表[/align][align=center][img=,487,538]http://ng1.17img.cn/bbsfiles/images/2018/01/201801041024_7687_2222981_3.jpg!w487x538.jpg[/img][/align][align=center][/align]
2010年11月10日,日本厚生劳动省发布食安发1110第1号通知,设定部分农药和添加剂的标准:(1)根据食品卫生法第11条第1项的规定设定农药亚胺唑、氟哇唑以及异丙甲草胺在食品中的残留标准。(2)根据食品卫生法第11条第1项的规定设定苯乙胺、丁胺的使用标准以及成分规格。详细内容见附件。
中华人民共和国国家标准 GB\T 5009.48--1996蒸馏酒及配制酒卫生标准的分析方法 代替 GB 5009.48--85Method for analysis of hygienic standardof distilled wines and mixed wines____________________________________________________________________________1 主题内容与适用范围 本标准规定了以含糖或淀粉的物质为原料,经糖化发酵蒸馏而制得的白酒及以发酵酒或蒸馏酒作酒基,经添加可食用的辅料制成的配制酒中各项卫生指标的分析方法。 本标准适用于蒸馏酒和配制酒中各项卫生指标的分析。2 引用标准 GB 2757 蒸馏洒和配制洒卫生标准 GB 5009.2 食品中相对密度的测定方法。 GB 5009.11 食品中总砷的测定方法。 GB 5009.12 食品中铅的测定方法。 GB 5009.36 粮食卫生标准的分析方法。 GB 5009.35 食品中着色剂的测定方法。 GB 12396 食品中铁、锰的[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]测定方法3 感官检查3.1 量取 30mL 样品,倒入 50mL 清洁干燥无色玻璃烧杯中,观察其颜色,应透明,无沉淀或杂质。3.2 尝其味应有该种酒特有的芳香味和滋味,不应有霉味、酸味、异味。应符合GB 2757的规定。4 理化检验4.1 乙醇浓度(比重计法)4.1.1 原理 同GB 5009.2的原理。4.1.2 仪器 酒精比重计。4.1.3 分析步骤 吸取 100mL样品于 250mL或 500mL全玻璃蒸馏器中,加 50mL 水,再加入玻璃珠数粒,蒸馏,用100mL 容量瓶收集馏出液 100mL。 将蒸馏后的样品倒入量筒中,将洗净擦干的酒精计缓缓沉入量筒中,静止后再轻轻按下少许,待其上升静止后,从水平位置观察其与液面相交处的刻度,为乙醇浓度, 同时测定温度,按测定的温度与浓度,查《酒精计温度浓度换算表》,换算成温度为20℃时的乙醇浓度(%)。4.2 甲醇4.2.1 原理 甲醇经氧化成甲醛后,与品红亚硫酸作用生成蓝紫色化合物,与标准系列比较定量。最低检出量为 0.02g/100mL。4.2.2 试剂4.2.2.1 高锰酸钾-磷酸溶液:称取 3g 高锰酸钾,加入15mL磷酸 (85%)与 70mL水的混合液中,溶解后加水至 100mL。贮于棕色瓶内,防止氧化力下降,保存时间不宜过长。4.2.2.2 草酸-硫酸溶液:称取 5g 无水草酸(H2C2O4) 或7g含 2分子结晶水草酸(H2C2O4.2H2O ), 溶于硫酸 (1+1)中至 100mL. 4.2.2.3 品红- 亚硫酸溶液 :称取 0.1g碱性品红研细后,分次加入共60mL 80℃的水,边加入水边研磨使其溶解,用滴管吸取上层溶液滤于100mL 溶量瓶中,冷却后加10mL亚硫酸钠溶液(100g/L), 1mL盐酸 ,再加水至刻度, 充分混匀,放置过夜,如溶液有颜色,可加少量活性炭搅拌后过滤,贮于棕色瓶中,置暗处保存,溶液呈红色时应弃去重新配制。4.2.2.4 甲醇标准溶液:称取 1.000g 甲醇,置于 100mL 容量瓶中, 加水稀释至刻度。此溶液每毫升相当于 10mg 甲醇。置低温保存。4.2.2.5 甲醇标准使用液:吸取 10.0mL 甲醇标准溶液,置于 100mL容量瓶中,加水稀释至刻度。再取 10.0mL 稀释液置于 50mL 容量瓶中,加水至刻度,该溶液每毫升相当0.50mg甲醇。4.2.2.6 无甲醇的乙醇溶液:取 0.3mL按操作方法检查,不应显色。如显色需进行处理。取 300mL乙醇 (95%),加高锰酸钾少许,蒸馏,收集馏出液。在馏出液中加入硝酸银溶液(取1g硝酸银溶于少量水中)和氢氧化钠溶液(取 1.5g 氢氧化钠溶于少量水中),摇匀,取上清液蒸馏,弃去最初50mL馏出液,收集中间馏出液约200mL,用酒精比重计测其浓度,然后加水配成无甲醇的乙醇(60%)。4.2.2.7 亚硫酸钠溶液(100g/L)。4.2.3 仪器 分光光度计。
在今年年初的肉制品中亚硝酸盐检测的能力验证中,我们的结果是离群值,而且离得很离谱,在分析原因的过程中我们考虑到了标准物质的使用是否正确,当时我们是购买了液体的20ml一瓶的标准溶液,名称是亚硝酸盐标准物质以亚硝酸根计,标准上说用固体的亚硝酸钠配制,还有我在购买这个标准物质的时候还有一种标准溶液是亚硝酸盐以氮计,这让我很迷惑,到底哪种标准物质适合这个检测方法呢?希望前辈指点。以亚硝酸根计、以氮计、亚硝酸钠计之间是该怎么换算呢?
据新西兰初级产业部(MPI)消息,针对新西兰牛奶中检出低含量双氰胺的报道,新西兰两家最大的肥料公司已暂停双氰胺的销售,新西兰初级产业部对两家公司采取的行动表示支持。 双氰胺是肥料辅助品,用于牧场可增加牧草的产量,还可防止肥料的副产品硝酸盐流入河流和湖泊,并减少温室气体的排放。 新西兰初级产业部表示,目前双氰胺在食品中的限量尚无国际标准,使用双氰胺不存在食品安全风险,而且双氰胺毒性较低,未发现高剂量使用会出现不良反应。 然而新西兰MPI担心,由于目前尚无国际标准,消费者不能接受牛奶中检出双氰胺的事实,这可能会对新西兰的乳品贸易产生影响。 原文链接:据了解,现在有很多第三方机构及政府机构提供双氰胺检测的方法有HPLC \LC-MS\LC-MS-MS ,后者居多,本机构也是提供LC-MS-MS 检测方法检测
《食品安全国家标准 食品中农药最大残留限量》2016版正式颁布实施,这一农药残留的新国标,在标准数量和覆盖率上都有了较大突破,规定了433种农药在13大类农产品中4140个残留限量,较2014版增加490项,基本涵盖了我国已批准使用的常用农药和居民日常消费的主要农产品。 食品伙伴网标法中心结合2016版标准前言部分内容与2014版进行相应的对比分析,供参考: 1、对原标准中氟唑磺隆、甲咪唑烟酸、氟吡菌胺、三唑酮和三唑醇等5种农药残留物定义,敌草快等5种农药每日允许摄入量等信息进行了核实,修订了敌草快、三环锡等5种农药的ADI值。 2、增加了2,4-滴异辛酯等46种农药;增加了490项农药最大残留限量标准 2014版规定了食品中2,4-滴等387种农药3650项最大残留限量,2016版规定了433种2,4-滴等农药4140项最大残留限量。增加了46种农药:2,4-滴异辛酯、2甲4氯异辛酯、苯嘧磺草胺、苯嗪草酮、吡唑草胺、丙硫多菌灵、除虫菊素、毒草胺、多抗霉素、呋虫胺、氟吡菌酰胺、复硝酚钠、甲磺草胺、井冈霉素、抗倒酯、苦参碱、醚苯磺隆、嘧啶肟草醚、扑草净、嗪草酸甲酯、氰氟虫腙、氰烯菌酯、炔苯酰草胺、噻虫胺、三苯基乙酸锡、三氯吡氧乙酸、杀螺胺乙醇胺盐、莎稗磷、虱螨脲、特丁津、调环酸钙、五氟磺草胺、烯丙苯噻唑、烯肟菌酯、烯效唑、辛菌胺、辛酰溴苯腈、溴氰虫酰胺、唑胺菌酯、唑啉草酯、啶菌噁唑、丁吡吗啉、噁唑酰草胺、甲哌鎓、丁酰肼、唑嘧菌胺。 3、增加 12 项检测方法标准,删除1项检测方法标准 增加了SN/T 0162、SN 0198、SN/T 0931、SN/T 1624、SN/T 1989、SN/T 2229、SN/T 2231、SN/T 2237、SN/T 2323、SN/T 2387、SN/T 2795、SN/T 2807,删除了SN/T 0711,其中SN 0198标准已于2015年12月31日被认监委废止,废止依据为《国家认监委办公室关于公布2015年检验检疫行业标准复审结论的通知》。 4、修改了丙环唑等8种农药的英文通用名 修改了丙环唑、六六六、烯肟菌胺、氯啶菌酯、杀虫双、四氯苯酞、氯氟吡氧乙酸和氯氟吡氧乙酸异辛酯 5、将苯噻酰草胺和灭锈胺的限量值由临时限量修改为正式限量;对资料性附录 A 进行了修订,增加了干制蔬菜等3种食品名称,修改1项作物名 食品伙伴网对附录A部分内容的对比发现如下变化: 1)水果(核果类)的类别说明增加了青梅,枣修改为枣(鲜)。 2)水果(浆果和其他小型水果)的类别说明中露莓增加了备注:包括波森莓和罗甘莓。 3)水果(热带和亚热带水果)的类别说明中将大型果的木瓜修改为番木瓜。 4)干制水果的类别说明中增加了枣(干)等。 5)食品类别名称修改:将饮料修改为饮料类。 6、食品伙伴网在对比过程中发现,2016版标准除了以上所列变化外,还修正了其他一些内容: 1)引用的标准名称的修正,如GB/T 19648、GB/T 19469等部分标准的名称中 “兽”字已删除。 2)引用的作废标准的修正,如2014版标准中引用的是2006版GB/T 20770的标准名称,2016版标准已经修正为2008版GB/T 20770的标准名称。 3)农药中文名称修改:2014版标准中的2甲4氯(钠)修改为2甲4氯钠。 4)附录A中动物源食品部分类别的测定部位描述进行了修正。
做SPS的分析,但是没有标准品,只好用国外产品做标样。但是老板不认可国外产品含量标注。无法,只好上来求助。SPS:http://www.chemyq.com/xz/xz1/2062nilyd.htm,这个是化工引擎上的介绍。聚二硫二丙烷磺酸钠,结构式 NaSO3(CH2)3-S-S-(CH2)3SO3Na。我目前使用C18柱,uv检测器,50%甲醇加离子对试剂。
[color=#444444]用高效液相色谱分析生物胺标准品,梯度,温度,衍生方法等条件都用的一样。流动相是乙腈和超纯水。怎么跑出来的图差别这么大。求助大家,谢谢。两张图不一个时间[/color][color=#444444][img=,690,517]https://ng1.17img.cn/bbsfiles/images/2019/05/201905201035452605_6441_1676638_3.png!w690x517.jpg[/img][img=,690,517]https://ng1.17img.cn/bbsfiles/images/2019/05/201905201035471785_2554_1676638_3.png!w690x517.jpg[/img][/color]
[b]我国香蕉农药残留标准与国外标准的比较分析[/b][i]李玉萍,方佳,梁伟红,董定超[/i] 1 我国香蕉农药残留国家标准现状 在我国,水果农药残留国家标准开始于20世纪70年代末,起步较晚。经过20余年的努力,取得了可喜的成绩,截至到1999年9月底,我国已发布18个与水果有关的农药最大残留限量强制性国家标准,涉及50种农药。2005年1月我国又颁布新的国家标准《食品中农药最大残留限量》(GB2763-2005),将原先的18个标准代替。新标准规定了水果中70种农药的最大残留限量,其中仅有腈苯唑、咪鲜胺、丙环唑、戊唑醇、噻菌灵5种农药对香蕉的残留限量值作了专门规定;溴氰菊酯、乙烯利、代森锰锌3种农药对皮不可食的热带与亚热带水果作了专门规定;乙酰甲胺磷等13种农药对所有水果规定了统一的最大残留限量;多菌灵除对梨果类水果、葡萄作出专门规定外,对其他水果也都规定了相同的残留限量值。此外,在我国现行的《农产品安全质量无公害水果安全要求》(GB18406.2-2001)强制性国家标准中,也规定了无公害水果中22 种农药的最大残留限量值。综合上述国家标准,直接或间接涉及香蕉农药残留最大限量指标共33项,涉及农药33种。其中杀虫剂23种,杀菌剂8种,除草剂1种,植物生长调节剂1种。 2 国际及国外先进国家香蕉农药残留最大限量标准现状 食品法典委员会(Codex Alimentarius Commission,简称CAC)是世界上唯一的政府间协调国际食品标准法规的国际组织,成立于1962年,至今已拥有163个成员国。到目前为止,CAC 已规定了香蕉、葡萄、苹果等63种(类)水果及其产出品的最大农药残留限量,涉及农药100余种,其中有24种农药对香蕉作了专门规定(其中除草剂2种,杀虫剂9种,杀菌剂12种,杀螨剂1种)。 欧盟制定的香蕉农药最大残留限量标准涵盖180种农药。其中除草剂39种,杀虫剂73种,杀菌剂49种,杀螨剂9种,熏蒸剂2种,植物生长调节剂8种。 美国在美国联邦法规(CFR)第40篇第180部分对香蕉规定了详细的农药最大残留限量,涵盖农药47种,有些农药还分别对香蕉全果、果肉进行了规定。在47种农药中,包括除草剂10种,杀虫剂11种,杀菌剂23种,杀螨剂1种,熏蒸剂1种,植物生长调节剂1 种。 日本有91种农药对香蕉的农药最大残留限量进行了规定。其中除草剂16种,杀虫剂40种,杀菌剂27种,植物生长调节剂2种,薰蒸剂2种,杀螨剂4种。 3 我国与国外香蕉农药残留指标比对分 析3.1 我国香蕉农药残留指标与 CAC 比对分析 目前我国香蕉农药残留指标33项,涉及33种农药,CAC 香蕉农药残留指标24项,涉及24种农药。我国香蕉农药残留指标与CAC 相比,有6种农药与 CAC都有限量要求,其中指标相同的有腈苯唑、丙环唑、戊唑醇、噻菌灵4种,占我国香蕉指标的12.1%,占 CAC指标的16.7%;比 CAC 严的有克百威1种;比 CAC 宽的有百菌清1种。有27种农药我国有限量要求而CAC 却没有,有18种农药 CAC 有限量要求而我国却没有。
本人需要TS-Amine-011A标准的具体内容如果能够提供乙二胺纯度的其它气相色谱分析方法也可以,谢谢最后顺便求一下乙二胺水分的分析方法,什么方法都可以,不一定非得是气相的,拜谢!~~~http://simg.instrument.com.cn/bbs/images/brow/em09506.gif
各位大侠:那位知道最新纯品三甲胺的国家标准?纯品三甲胺(99%以上)的分析测试方法?请给本人一份!不胜感谢!在线等!
食品安全国家标准GB2762-2012《食品中污染物限量》,将于2013年6月1日实施,本标准与GB 2762—2005相比,主要变化如下: ——修改了标准名称; ——增加了可食用部分的定义; ——增加了应用原则; ——取消了硒、铝、氟的限量规定; ——增加了锡、镍、3-氯-1,2-丙二醇及硝酸盐的限量规定; ——将N-亚硝胺限量指标由N-二甲基亚硝胺和N-二甲基乙硝胺调整为N-二甲基亚硝胺,并将N-亚硝胺限量指标名称修改为N-二甲基亚硝胺; ——增加了附录 A; ——稀土限量指标按原GB 2762—2005执行。与我们相关的就是,其中明确规定,铅的限量:酒类(蒸馏酒、黄酒除外) 0.2mg/kg ;蒸馏酒、黄酒 0.5mg/kg取消了酒类中无机砷的限量大家来讨论下在自己的领域中的变化!
[摘要] 叙述了以盐酸-氨水缓冲溶液为介质,以磺胺和N-1-萘基-乙二胺二盐酸盐为显色剂直接测定乳与乳制品中亚硝酸盐的双光束分光光度法。由光谱分析得最大吸收波长为538nm,以滤液为参比,有效的消除了干扰,方法简便、快速。对6个样品进行了测定,测定结果与国标方法基本一致。相对标准偏差为4.76%~2.50%之间,回收率在95.36%~104.63%之间。 关键词:双光束分光光度 亚硝酸盐 乳与乳制品亚硝酸盐广泛存在于自然界,在乳与乳制品中都有一定的含量。现已证明,亚硝酸盐与食品中固有的胺类化合物是产生致癌物质-亚硝胺的前体物质,是潜在的致癌物质,亚硝酸盐含量过高会引起高铁蛋白症。因此,对乳与乳制品亚硝酸盐含量的检测和控制是非常有必要的。在乳与乳制品测定亚硝酸盐的过程中,将样品沉淀脂肪和蛋白质后,进行过滤。在滤液中加入磺胺和N-1-萘基-乙二胺二盐酸盐,使其显粉红色,然后用分光光度计在其最大吸收波长538nm下测定其吸光度。将测得的吸光度与亚硝酸钠标准系列溶液的吸光度进行比较定量。1 主要试剂和仪器1.1仪器:UV-2550型紫外可见分光光度计(岛津有限公司)1.2试剂:标准亚硝酸盐溶液:GBW(E)0802230504水中亚硝酸盐-氮,浓度:100mg/mL,并逐级稀释为0.9857μg/mL的标准贮备液;硫酸锌溶液:535g/L 亚铁氰化钾溶液:172g/L 盐酸-氨水缓冲溶液:pH9.6~9.7;显色液1:盐酸:水= 450:550(V:V)盐酸溶液;显色液2:5g/L磺胺溶液;显色液3:1g/L萘胺盐酸盐溶液。试验中所用试剂均为分析纯,所用水为去离子水。2 试验方法2.1入射光波长的选择国标《GB/T5413.32-1997 乳粉 硝酸盐、亚硝酸盐的测定》中规定最大吸收波长为538nm,但在实验过程中,做光谱扫描时,最大吸收波长往往因为样品的不同而发生变化,发上红移或蓝移的现象。考虑到测定的灵敏度,应采用最大吸收波长作为入射光波长。所以,在进行定量分析之前,应先才用光谱扫描进行峰形和峰位的确认。以表1的仪器条件,对亚硝酸盐标准系列进行扫描,光谱图如图1。图1中曲线均比较平滑,为了消除干扰组分的吸收,采用“吸收最大,干扰最小”的原则,即所选择的测定波长处待测组分的吸收最大,但干扰组分的吸收最小;从消除非单色光引起的对郎伯比尔定律的偏离角度考虑,应选用吸收曲线比较平坦部分对应波长的光作为入射光波长。综上所述,应选择图1中538nm作为入射光波长,与国家标准一致。表1 仪器条件波长扫描范围/nm420~650光谱带宽/nm0.5采样间隔0.5扫描速度中速 图1 标准系列吸收光谱图 2.2标准曲线 标准曲线的绘制:取 6个50mL容量瓶,分别加入浓度为0.9857μg/mL的亚硝酸钠标准贮备液0.0,0.5,1.0,2.0,5.0,10.0mL,加水至20mL,然后依次加入3mL显色液1,2.5mL显色液2,混合均匀后,静置5min,加1mL显色液3, 静置5min,用去离子水定容至刻度,此溶液浓度分别为0.00、9.86、19.72、59.14、98.56、197.14μg/L。静置5min后于538nm处,用1cm比色皿进行测定,并以吸光度为纵坐标,浓度为横坐标绘制标准曲线(见图2)。 图2 亚硝酸盐标准曲线 标准曲线方程为A=0.0011C-0.0005,相关系数r2=0.9999。2.3样品测定2.3.1 样品前处理称90.0g左右牛乳样品,按顺序加入25mL硫酸锌溶液,25mL亚铁氰化钾溶液,40mL盐酸-氨水缓冲溶液,每加一种试剂充分混合,用去离子水定容于250mL容量瓶中,静置40min,用定性中速滤纸过滤后收集滤液。2.4.2样品测定移取20mL滤液于50mL容量瓶中,加3mL显色液1,2.5mL 显色液2,摇匀,静置5min;加1mL显色液3,静置5min,定容后静置5min,上机测定。3结果与讨论干扰消除一般有两种方法,一类是不分离的情况下消除干扰,另一类是分离杂质消除干扰。因为在分离过程中,会造成硝酸盐和亚硝酸的损失,所以一般在不分离的情况下消除干扰。本试验采用双光束分光光度法,在不分离的情况下消除干扰。3.1在样品前处理后,在滤液中的某些成分影响被测组分吸光度时,将构成干扰,影响测量结果的准确性。干扰离子与试剂生成有色配合物,使测定结果偏高;干扰离子与试剂反应,生成的配合物虽然无色,但消耗大量的显色剂,使被测离子的显色反应不完全,使测定结果偏低;与被测离子结合成离解度小的另一种化合物,使被测离子与显色剂不反应,使测定结果偏低。加入盐酸-氨水缓冲溶液,控制溶液的酸度,从而有效的消除了干扰。因为控制溶液酸度在一定范围,可以使亚硝酸盐与显色剂反应,干扰离子不能生成有色化合物。3.2选择适当的参比溶液,消除显色剂本身颜色和某些共存的有色离子的干扰。干扰离子本身有颜色,使测定结果偏高; 本试验选择溶剂参比和试液参比分别进行考察:溶剂参比:当显色剂及其他溶剂均无色,被测溶液中又无其他有色离子时,可用溶剂-去离子水做参比溶液。试液参比:显色剂无色,被测溶液中有其他有色离子,可采用不加显色剂的被测溶液做参比溶液。 以去离子水作溶剂参比,只能消除吸收池壁对光的反射和散射所带来的干扰;以被测溶液作试液参比,不仅能消除吸收池壁对光的反射和散射带来的干扰,还能消除滤液中共存离子对光的选择性吸收所带来的干扰。所以采用不加显色剂的滤液作试液参比。 3.3选择适当的波长消除干扰。入射光波长的选择必须从灵敏度与选择性两方面来考虑,从吸收曲线的峰形来看,无杂峰干扰,应选择最大吸收波长进行测定,这样灵敏度高,偏离郎伯-比尔定律的程度较小。本实验通过光谱扫描,从吸收光谱图上测定最大吸收波长后进行定量分析,避免了波长偏差带来的误差和干扰。3.4精密度试验分别称取纯牛乳﹑原味酸牛乳﹑全脂乳粉和乳清蛋白粉样品,用上述方法进行测定,计算结果见表2:(单位:μg/L)表2精密度试验 n=6样品名称测定值范围/mg• kg-1平均值/mg• kg-1标准偏差S相对标准偏差RSD/%纯牛奶0.21~0.220.210.0104.76原味酸牛奶0.31~0.350.330.0144.06全脂奶粉1.08~1.171.120.0312.79乳清蛋白粉2.41~2.592.500.0622.503.5加标回收率试验在纯牛乳﹑原味酸牛乳﹑全脂乳粉和乳清蛋白粉样品中,分别吸取0.50,1.00,1.50,2.00mL 浓度10mg/mL的标准亚硝酸盐溶液进行加标回收试验,结果见表3。表3加标回收率试验样品名称本底值/ mg• kg-1样品质量/g加标量/ mg• kg-1测定值/ mg• kg-1回收率/ %纯牛奶0.21 90.200 0.055 0.268 104.63原味酸牛奶0.33 71.300 0.140 0.471 97.68全脂奶粉1.12 10.234 1.466 2.624 102.34乳清蛋白粉2.50 5.145 3.887 6.203 95.364.结论双光束分光光度法测定纯牛奶中的亚硝酸盐,灵敏度高,检出限低,选择性强,准确可靠,有效的消除了共存离子干扰,测定方法简便,快速。参考文献:[1] 国标GB/T5413.32-1997 乳粉 硝酸盐、亚硝酸盐的测定[2] 国标GB/T5009.33-2003 食品中亚硝酸盐与硝酸盐的测定[3] 杜岱春,分析化学,复旦大学出版社,1993[4] 黄伟坤等,食品检验与分析,中国轻工业出版社,1989.1[5] 张振辉,张改兰,痕量分析,1989.1
刚开始做化学分析,以前是玩生物的。例如:滴定氯化钠用的0.1mol硝酸银标准液一定要用2级标准品配制吗?配制完了还得用基准氯化钠标定。那么硝酸银标准液能用一般的AR硝酸银配制吗?然后用基准氯化钠标定
这几天用岛津的碳18分析氨基酸,用的异硫氰酸苯酯为衍生剂。 用水代替氨基酸标准品,会出现很多杂峰,所以想应该是衍生过程引进的。衍生的方法是 标准品+三乙胺乙腈溶液+异硫氰酸苯酯乙腈溶液 反应一个小时候用正己烷萃取各位大虾有更好的衍生方法么?本实验室没有氮吹仪,没法吹干哦
己二胺纯度分析 有关的所有标准
哪位有红外碳硫仪分析标准?
各位同行: 在农药残留分析中,一个讨论较多的话题就是关于基质效应的问题,一般采用添加基质的标准品来补偿基质效应,但是在具体的配制过程中,现在尚为有一个统一规范的标准,请大家都来讨论一下,各自在平时的实验中,是怎么样来操作的! 此贴只为抛砖引玉,希望大家踊跃讨论,有好的操作,我们一起学习!
1、硫酸浓度测定1.1 方法提要 以甲基红—次甲基兰为指示剂,用氢氧化钠标准滴定溶液中和滴定以测得硫酸含量。1.2 试剂和材料1.2.1 氢氧化钠标准滴定溶液:C(NaOH)=0.5 mol/L1.2.2 甲基红—次甲基兰混合指示剂1.3 分析步骤1.3.1 用一称量的带有磨口的小称量瓶,称取约0.7g浓硫酸试样或1.5g左右50%硫酸试样(精确至0.0001g),小心移入盛有50mL水的250ml锥形瓶中,冷却至室温,备用。1.3.2 试液中,加2—3滴混合指示剂(1.2.2),用氢氧化钠标准滴定溶液(1.2.1)滴定至溶液呈灰绿色为终点。1.4 分析结果的表述 硫酸的质量分数W1=C*V*(M/1000)*100/m=4.904*C*V/m式中:V:滴定耗用的氢氧化钠标准滴定溶液的体积的数值,mL;C:氢氧化钠溶液的实际浓度的数值,mol/L;m:试料的质量的数值,g;M:硫酸的摩尔质量的数值,g/mol(M=49.04)2、灰分2.1 方法提要 试料蒸发至干,灼烧,冷却后称量。2.2 仪器、设备2.2.1 铂皿(或石英皿,瓷皿);容量60mL—100mL。2.2.2 高温电炉:可控制温度(800±50)℃。2.3 分析步骤 将铂皿(2.2.1)于高温电炉(2.2.2)内,在800℃±50℃温度下灼烧15min,置于干燥器中,冷却至室温称量,精确至0.0001g。 称取约25g—20g试样于铂皿中(精确至0.01g),在可调温电炉或沙浴上,小心加热蒸发至干,移入高温电炉(2.2.2)内,在800℃±50℃温度下灼烧15min,取出铂皿,置于干燥器中,冷却至室温后称量,精确至0.0001g。2.4 分析结果的表述 硫酸灰分的质量分数W2(%)按下式计算: W2=m*100/m0 式中:m:灼烧后灰分的质量的数值,g;m0:试料的质量的数值,g。3、氯化物3.1 方法提要 试样用水稀释,加硝酸及硝酸银与氯离子反应生成氯化银浑浊液,与标准色阶比较,求算出氯含量。3.2 试剂和材料3.2.1 无氯硫酸:将硫酸(GB/T 625)加热至冒白烟赶Cl﹣后使用。3.2.2 硝酸(GB/T 626)溶液:1+2。3.2.3 硝酸银(GB/T 670)溶液:C(AgNO3)=0.1mol/L;3.2.4 氯标准溶液:1mL含氯0.100mg;3.2.5 氯标准溶液:1mL含氯0.010mg; 用氯标准溶液(3.2.4)稀释而得,该溶液使用时配制。3.2.6 具塞玻璃比色管,容积50mL。3.3 分析步骤 称取约5g试样(精确至0.1g),在冷却条件下加入盛有25mL水的烧杯中,冷却后移入比色管中,加1mL硝酸溶液(3.2.2),2mL硝酸银(3.2.3),加水稀释至刻度,摇匀。于暗处静置20min。 按上述操作,用无氯硫酸(3.2.1)代替试样制备标准色阶,依次含氯0.020、0.040、0.060、0.080 mg。 用目视比浊法确定氯含量。3.4 分析结果的表述硫酸中氯的质量分数W3(%),按下式计算: W3=m*10﹣3*100/m0式中:m:与试料浊度相当的标准色阶中氯(Cl)的质量的数值,mg;m0:试料试样的质量的数值,g。4、铁含量的测定(邻菲啰啉分光光度法)4.1 方法提要 试料蒸干后,残渣溶解于盐酸中,用盐酸羟胺还原溶液中的铁,在PH为2—9条件下,二价铁离子与邻菲啰啉反应生成橙色络合物,对此络合物作吸光度测试。4.2 试剂和材料4.2.1 盐酸溶液:1+10。4.2.2 盐酸羟胺溶液:10g/L。4.2.3 乙酸—乙酸钠缓冲溶液:PH≈4.5。4.2.4 邻菲啰啉盐酸溶液:1g/L。4.2.5 铁标准溶液:0.100 mg/mL。4.2.6 铁标准溶液:0.010 mg/mL。4.2.7 分光光度计。4.3 分析步骤4.3.1 试料溶液的制备 称取10g—20g试样(精确至0.01g),置于50ml烧杯中,在可调温电炉上蒸发至干,冷却,加2ml盐酸溶液(4.2.1),和25ml水,加热使其溶解,移入100mL容量瓶中,用水稀释至刻度,摇匀,备用。4.3.2 工作曲线的制作4.3.2.1 标准显色溶液的制备 取6个50ml容量瓶,按表1所示,分别加入铁标准溶液(4.2.6)表1铁标准溶液体积 mL对应铁质量 ug0a02.0204.0406.0608.08010.0100a 空白溶液对每一容量瓶中的溶液作下述处理:加水至25mL,加2.5mL盐酸羟胺溶液(4.2.2)和5mL缓冲溶液(4.2.3),5min后加5mL邻菲啰啉溶液(4.2.4),用水稀释至刻度,摇匀,放置15min显色。4.3.2.2 吸光度的测量 在510nm波长处,用1cm吸收池,以水为参比,将分光光度计的吸光度调整到零后,测出标准显色溶液(4.3.2.1)的吸光度。4.3.2.3 工作曲线的绘制 从每一标准显色溶液的吸收度值,减去空白溶液的吸光度值(4.3.2.2),以所得吸光度值差为纵坐标,对应的铁质量为横坐标绘制工作曲线。4.3.3 试料的测定4.3.3.1 显色 取一定量的试液(4.3.1),置于50mL容量瓶中,加水至约25mL,然后按4.3.2.1中说述“加2.5mL盐酸羟胺溶液(4.2.2)……”的步骤进行。4.3.3.2 吸光度的测量 按4.3.2.2步骤,以水为参比,测量试液(4.3.3.1)的吸光度。4.3.4 空白实验 在测定试液(4.3.3.1)的同时,用同样的步骤,同样数量的试剂,不加试液作白色实验。4.4 分析结果的表述 从试液的吸光度值(4.3.3.2)减去空白实验的吸光度值(4.3.4),据所得吸光度值差从工作曲线(4.3.2.3)查出对应的铁质量,并按试液吸取比例计算出试样中的铁质量。 硫酸中铁的质量分数W4(%)按下式计算: W4=m*100/m0式中:m:试料中铁含量的质量的数值,g;m0:试料的质量的数值,g。

[size=16px] 食品安全综合分析仪主要标准是什么 食品安全综合分析仪的主要标准包括以下几个方面: 检测项目:食品安全综合分析仪的检测项目涵盖了多个方面,主要包括营养成分、添加剂、农药残留、重金属和微生物等。这些项目的检测是确保食品安全的重要手段,可以全面了解食品的质量和安全状况。 检测精度和效率:食品安全综合分析仪需要具备高检测精度和效率,以确保测试结果的准确性和可靠性。为实现这一目标,需要选择可实现自校的仪器,并设置合理的自校时间间隔,按期校对仪器。此外,在仪器操作前也需进行校对,以及定期对仪器进行检修与保养,降低检测出现误差的可能性。 农残检测标准:食品安全综合分析仪的农残检测标准通常是由国家食品药品监管总局制定的。对于农药残留限量,一般采用“最大残留限量(MRL)”作为标准。这个限度是由卫生、环境和经济等多方面考虑制定的。在农残检测过程中,除了检测仪器的准确性外,样品的采样和制备也是非常重要的。 国家和行业标准:食品安全综合分析仪的检测方法应符合国家和行业标准。例如,对于营养成分、添加剂、重金属和微生物等的检测,应采用国家规定的检测方法,确保数据的准确性和可靠性。 仪器性能:食品安全综合分析仪应具备良好的稳定性、重复性和灵敏度等性能指标,以确保在不同环境和条件下都能获得准确的检测结果。 综上所述,食品安全综合分析仪的主要标准包括检测项目、检测精度和效率、农残检测标准、国家和行业标准以及仪器性能等方面。这些标准共同构成了保障食品安全的重要技术支持。[img=,690,690]https://ng1.17img.cn/bbsfiles/images/2024/02/202402200937557726_9841_6098850_3.jpg!w690x690.jpg[/img][/size]

[align=center][b]GB/T 20442-2010 饲料添加剂亚硫酸氢烟酰胺甲萘醌的分析[/b][/align][align=right][b][/b][/align][align=center][b][img=,440,647]http://ng1.17img.cn/bbsfiles/images/2017/11/201711090926_01_2222981_3.png!w440x647.jpg[/img][/b][/align][align=right][b] [/b][/align]亚硫酸氢烟酰胺甲萘醌(简称MNB)在饲料工业中作维生素类饲料添加剂,其在水溶液中分解为亚硫酸甲萘醌和烟酰胺。[align=center][img=,520,238]http://ng1.17img.cn/bbsfiles/images/2017/11/201711090926_02_2222981_3.png!w520x238.jpg[/img][/align][align=center][/align][align=left]实验室严格依据国标《GB/T 20442-2010 饲料添加剂亚硫酸氢烟酰胺甲萘醌》方法,对客户提供亚硫酸氢烟酰胺甲萘醌原料样品进行分析,筛选合适的C[sub]18[/sub]色谱柱以实现亚硫酸氢烟酰胺甲萘醌的良好分析。[/align][align=left][/align][align=left]首先,使用资生堂中等极性色谱柱CAPCELL PAK C[sub]18[/sub] MGII,严格依照国标方法对MNB原料供试品溶液进行分析。结果如图1,所得谱图与国标方法中给出的标准色谱图(见附图)出峰行为基本一致,烟酰胺保留时间约为3 min,亚硫酸甲萘醌保留时间约为6 min,峰形良好,分离度为14.37,两组分能得到良好分离。[/align][align=left][/align][align=center] [img=,476,354]http://ng1.17img.cn/bbsfiles/images/2017/11/201711090928_01_2222981_3.png!w476x354.jpg[/img][/align][align=center]图1 MGII色谱柱分析MNB原料供试品溶液色谱图[/align][align=left]*注:峰上标数字为分离度(下同)。[/align][align=left][img=,509,147]http://ng1.17img.cn/bbsfiles/images/2017/11/201711090928_02_2222981_3.png!w509x147.jpg[/img][/align][align=center][/align][align=center][img=,690,577]http://ng1.17img.cn/bbsfiles/images/2017/11/201711090930_01_2222981_3.png!w690x577.jpg[/img][/align][align=center]附图 国标中标准色谱图[/align][align=center][/align][align=left]为使客户有更多色谱柱选择,我们也尝试了能在100%水系流动相下稳定使用的资生堂高极性C[sub]18[/sub] AQ S5色谱柱,以及具有高碳载量的SUPERIOREX ODS色谱柱对MNB原料供试品溶液进行分析,亦能得到与标准谱图基本一致结果,其中高极性的C[sub]18 [/sub]AQ色谱柱的保留更强(见图2、图3)。[/align][align=center][/align][align=center][img=,486,358]http://ng1.17img.cn/bbsfiles/images/2017/11/201711090929_01_2222981_3.png!w486x358.jpg[/img][/align][align=center]图2 C[sub]18 [/sub]AQ色谱柱分析MNB原料供试品溶液色谱图[/align][align=center][img=,486,365]http://ng1.17img.cn/bbsfiles/images/2017/11/201711090929_02_2222981_3.png!w486x365.jpg[/img][/align][align=center]图3 SUPERIOREX ODS色谱柱分析MNB原料供试品溶液色谱图[/align][align=left][img=,500,162]http://ng1.17img.cn/bbsfiles/images/2017/11/201711090937_01_2222981_3.png!w500x162.jpg[/img][/align]
如题,请论坛各位高人有谱库的帮我一下,求一个六水合硝酸镍的拉曼标准谱。有原始数据点最好,至少也得是个清晰的带刻度的谱图,不胜感激,谢谢了!!







 我要推广仪器
我要推广仪器
 下载APP
下载APP