求助,配制盐酸表小檗碱和盐酸黄连碱对照液时,发现其很难溶解,文献中溶剂用的甲醇或甲醇-盐酸,我试了,也很难溶,超声加热溶解性也不好,有什么办法可以帮助溶解啊?
求助,配制盐酸表小檗碱和盐酸黄连碱对照液时,发现其很难溶解,文献中溶剂用的甲醇或甲醇-盐酸,我试了,也很难溶,超声加热溶解性也不好,有什么办法可以帮助溶解啊?
三黄片的实验条件:乙腈:水(1:1)(每1000ml中加磷酸二氢钾3.4g,十二烷基硫酸钠1.7g)流动相,波长265nm,对照品的峰面积不稳定,每五针的RSD值大于2%。且样品的峰面积稳定是什么原因?对盐酸小檗碱发生变化。对照品的溶剂是甲醇。

[size=4][b] 小卢推荐:一种标定二级对照品的方法[/b][/size]对照品作为实验室(制药行业)一种常用的、重要的试剂,根据其类型,可分为:一级对照品,即为从中国药品生物制品检定所(简称:中检所)购买后直接使用的对照品;二级对照品,由一级对照品标定原料药得到的对照品。由于一级对照品的规格小、价格高、购买周期长的缺点,对于实验室对照品用量大的企业来说,使用二级对照品成了实验室的首选。现在,我就介绍一种标定二级对照品的方法,供大家参考一下。[b]第一,选定样品[/b]一般来说,选择自己生产的原料药价格便宜,不需要外购,且取用方便,是我们的首选。如果我们的生产工艺不好、稳定相差,最好选择外购知名企业的原料药。但要注意,要选择作为对照品的原料药一定是相对其他批次各检验项目都比较好的同一批原料药。[b]第二,标定方法[/b]现以高效液相测定法检测含量为例,来表述其测定方法。由于要严格保证所标定原料药的含量,因此采用3人、3份样品的方法进行测定,即:每个人称取2份对照品、3份样品进行测定;共有3人进行测定。如果有条件,3个人可以选择3台不同的液相色谱仪进行实验。在这里要求2份对照品共进样5针,计算校正因子,并求RSD应小于0.5%,3份样品各进1针,求平均值。方法和要求如下表:[img]http://ng1.17img.cn/bbsfiles/images/2009/11/200911090939_182733_1622024_3.jpg[/img]按照表格内容,由3人得到的3个不同的含量,最后求得均值,即得样品(二级对照品)的含量,并要求3者的RSD≤1%。[b]第三,分装[/b]使用抗生素瓶分装,装量按照每次使用量(如60mg,则装入80-100mg即可)为标准,即使每个抗生素瓶中的对照品只使用一次。这样既能避免对照品被污染,又能使其少吸潮。如果为了节省抗生素瓶,采用大装量,即一瓶中的对照品可以使用多次,那么,建议在使用3-6次后就报废本瓶对照品。因为每次打开瓶口称取对照品都是对该瓶对照品的一次污染,尤其是空气中水分对它的影响,这样会是对照品的含量发生变化,原来的标定也就失去了意义。分装环境:建议在层流罩下进行,严格控制温湿度(建议温湿度:18-24℃,45-65%)。封口步骤:分装后,用橡胶盖盖紧,再用封口膜封好后,用铝盖压实即可。[b]第四,制定有效期[/b]一般比较稳定的样品制定2年,不是很稳定的样品制定1年。但是这个有效期不能超过该样品本身法定的有效期。[b]第五,贴签[/b]制定好了有效期就可以把样品(二级对照品)的标签贴上去了,标签格式如下:[img]http://ng1.17img.cn/bbsfiles/images/2009/11/200911090939_182734_1622024_3.jpg[/img][b]第六,储存[/b]不管原来样品法定的存储温度是多少,都建议保存的温度最好在2-10°,即冰箱中的冷藏温度。根据资料研究,2°是药品的最佳保存温度,因为这个温度下药品的降解速度最慢。[b]第七,复核[/b]我们制定了有效期后,并不是就完成了所有的工作。我们要在有效期的一半时,对二级对照品进行复核,检验方法同本法中第二步骤,所取样品则是从原标定的二级对照品中抽取。如果复核结果没有变化,则继续使用;如果复核结果发生了变化,那就按照复核的含量,从新贴签标示。通过以上7步就完成了对照品的标定工作,大家有什么看法可以回帖说明,我们共同讨论![em09505][em09505](全文完!)
对照品4个,第2(延胡索乙素)、3(盐酸小檗碱)个色谱峰前出现了小峰,不知道什么原因引起的,之前用相同色谱条件跑过,也没有小峰出现,现在连续进了五针都是主峰前有小峰。请各位帮我分析分析!流动相是0.1%的磷酸水:乙腈,对照品用甲醇溶解。色谱条件如图[img=,690,284]https://ng1.17img.cn/bbsfiles/images/2022/11/202211180044560750_5451_5351399_3.png[/img][img=,690,517]https://ng1.17img.cn/bbsfiles/images/2022/11/202211180044563666_9373_5351399_3.png[/img][img=,690,517]https://ng1.17img.cn/bbsfiles/images/2022/11/202211180044551717_7304_5351399_3.png[/img][img=,690,618]https://ng1.17img.cn/bbsfiles/images/2022/11/202211180044568830_7416_5351399_3.png[/img]
黄连味苦性寒,具有清热燥湿、泻火解毒的功效。《中国药典》2020年版规定黄连为毛茛科黄连属植物黄连Coptis chinensis Franch.、三角叶黄连C. deltoidea C. Y. Cheng et Hsiao或云连C. teeta Wall.的干燥根茎。以上3种分别习称“味连”“雅连”“云连”,经课题组前期调研以味连产量最多,主产于我国重庆、湖北、四川等地[1]。现代研究表明,黄连含有多种活性成分,可发挥多种药理作用[2],包括抗炎、抗病毒、抗菌、抗癌、镇痛、抗抑郁、降血糖等作用,临床应用极广[3]。苦参性寒、味苦,为豆科苦参属植物苦参Sophora flavescens Ait.的干燥根,主产于我国内蒙古、河南、山东、安徽等地[1],具有抗菌、抗肿瘤、镇痛、抗炎、防治心力衰竭、心律失常及心肌缺血等多种功效[4-5]。 现代研究表明,生物碱类化合物是黄连及苦参的主要活性成分。苦参碱、氧化苦参碱可发挥抗炎、镇痛效果[6-7],其机制可能与降低促炎因子,升高抗炎因子有关;氧化苦参碱、苦参碱也可发挥抗肿瘤作用,其机制可能与抑制癌症基因表达,促进肿瘤细胞凋亡,抑制肿瘤细胞生长有关[8];而苦参碱、氧化苦参碱、槐定碱也可对多种菌株具有一定的抑菌作用[9]。木兰花碱可通过活性氧(reactive oxygen species,ROS)/鼠类肉瘤病毒癌基因(Kirsten rat sarcoma viral oncogene,KRAS)/单磷酸腺苷活化蛋白激酶(adenosine monophosphate activated protein kinase,AMPK)通路抑制结直肠癌SW480细胞的增殖和有氧糖酵解,从而发挥对结直肠癌的治疗效果[10];药根碱、巴马汀、表小檗碱、黄连碱、小檗碱可联合发挥降糖作用[11],其效果可能与调控丝氨酸-苏氨酸激酶1(serine/threonine kinase 1,LKB1)/ AMPK/CREB分子调节转录共激活剂2(CREB-regulated transcription coactivator 2,TORC2)信号通路抑制肝脏糖异生等有关[12];小檗碱具有抗炎作用,可保护螺旋神经节细胞免受巨细胞病毒诱导的凋亡作用,其机制与通过途径抑制线粒体活性氧的产生有关[13]。 除此之外,小檗碱、表小檗碱、巴马汀等生物碱类成分也可联合发挥抗心律失常作用[14]。基于此,选择苦参中苦参碱、槐定碱、氧化苦参碱及黄连中木兰花碱、非洲防己碱、药根碱、表小檗碱、黄连碱、巴马汀、小檗碱来作为黄连-苦参药对的代表性药效成分,用于研究该类成分溶出量与药对配比的关系。药对作为中药配伍的最小单元,是复方研究的重要组成部分之一[15]。用于不同疾病的治疗时,不同量的配比会有不同效果的相关呈现,因此,首先需要对黄连-苦参药对配比的不同物质基础,即量-质[16]相关性进行剖析比较,为进行量-效[17]相关性提供依据,为临床合理配比提供参考[18]。 1 仪器与试药 1.1 主要仪器 Waters e2695型高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]系统,Waters 2998型二极管阵列检测器(PDA),美国Waters公司;BBA224S-CW型电子天平,赛多利斯科学仪器(北京)有限公司;TGL-16C型离心机,上海安亭科学仪器厂;EPED-E2-20TS型超纯水一体机系统,南京易普易达科技发展有限公司;GM-0.5B型真空泵,天津市津腾实验设备有限公司;KH-500V型超声器,昆山禾创超声仪器有限公司。 1.2 药品及试剂 1.2.1 药材与饮片 本研究所选择黄连(产地重庆石柱黄水,批号20230411)及苦参(产地内蒙古赤峰市,批号2020121604)药材,均经南京中医药大学药学院刘圣金教授鉴定,分别为毛茛科黄连属植物黄连C. chinensis Franch.的干燥根茎和豆科苦参属植物苦参S. flavescens Ait.的干燥根。 1.2.2 对照品 表小檗碱(批号J24HB186173)、盐酸小檗碱(批号S01A10K94340)、盐酸黄连碱(批号T21S11C125202)、药根碱(批号D18GB171805)、盐酸巴马汀(批号Z16J10X79792)、非洲防己碱(批号W14J8Z37548)、木兰花碱(批号R21M9F61834)、苦参碱(批号M14GB141405)、氧化苦参碱(批号G14N11KL130769)、槐定碱(批号F18F7S9784),HPLC质量分数均≥98%,均购自上海源叶生物科技有限公司。 1.2.3 试剂 乙腈、甲醇,色谱纯,安徽天地高纯溶剂有限公司;磷酸、盐酸、无水乙醇,分析纯,国药集团化学试剂有限公司;纯净水,屈臣氏集团(香港)有限公司;磷酸二氢钾,分析纯,南京化学试剂股份有限公司。 2 方法与结果 2.1 不同配比黄连-苦参药对指纹图谱的建立 2.1.1 色谱条件 色谱柱为Venusil XBP C18(2)(250 mm×4.6 mm,5 μm);柱温30 ℃;体积流量0.8 mL/min;流动相为乙腈-3 g/L磷酸二氢钾溶液(加入200 μL磷酸调节pH值),梯度洗脱:0~10 min,10%乙腈;10~25 min,10%~24%乙腈;25~35 min,24%乙腈;35~60 min,24%~35%乙腈;60~62 min,35%~60%乙腈;62~65 min,60%~10%乙腈;65~70 min,10%乙腈;分析时间70 min,进样量10 μL;检测波长220 nm。 2.1.2 混合对照品溶液的制备 取非洲防己碱、药根碱、表小檗碱、盐酸小檗碱、盐酸巴马汀、盐酸黄连碱、木兰花碱、苦参碱、氧化苦参碱、槐定碱对照品各适量,分别置于10 mL量瓶中,加甲醇溶解并定容,即得各对照品储备液。分别取适量上述11种对照品储备液,置于同一10 mL量瓶中,加甲醇稀释并定容,制得上述成分质量浓度分别为0.20、0.16、0.24、0.25、0.25、0.44、0.26、0.21、0.80、0.36 mg/mL的混合对照品溶液。 2.1.3 供试品溶液的制备 制备黄连药材粉末(过二号筛)及苦参药材粉末(过三号筛),将上述黄连及苦参依照5∶1、4∶1、3∶1、2∶1、1∶1、1∶2、1∶3、1∶4、1∶5共9个质量比例,进行称取后分别充分混合,并称取单一黄连药材粉末及单一苦参药材粉末作为对照药材,各比例药对总质量及单一药材质量均为12 g。每个比例平行称取各药对3份,将药对以10倍量水浸泡0.5 h后,煎煮1.5 h,取1次滤液;将滤渣加入8倍量水煎煮1.5 h,取2次滤液。将2次滤液混合后抽滤,12 000 r/min离心(离心半径10.4 cm)10 min,取上清液,取1 mL上清液加入4 mL甲醇,以0.45 μm微孔滤膜滤过,即得供试品溶液。 2.1.4 精密度试验 依照黄连与苦参比例1∶3,精密称取黄连药材粉末3 g及苦参药材粉末9 g,按照“2.1.3”项下方法制备供试品溶液,再按“2.1.1”项下色谱条件进样测定6次,考察特征峰的保留时间和峰面积一致性。以盐酸小檗碱的保留时间和峰面积为参照分别计算相对保留时间及相对峰面积。计算得各共有峰相对保留时间的RSD<0.20%,相对峰面积的RSD<2.13%,结果表明仪器精密度良好。 2.1.5 稳定性试验 依照黄连与苦参比例1∶3,精密称取黄连药材粉末3 g及苦参药材粉末9 g,按照“2.1.3”项下方法制备供试品溶液,再按“2.1.1”项下色谱条件每隔4 h进样1次,共测定24 h,考察特征峰保留时间和峰面积的一致性。以盐酸小檗碱的保留时间和峰面积为参照分别计算相对保留时间及相对峰面积。计算得各共有峰相对保留时间的RSD<0.21%、相对峰面积的RSD<2.46%,结果表明该供试品溶液在室温放置24 h内稳定性良好。 2.1.6 重复性试验 依照黄连与苦参比例1∶3,精密称取黄连药材粉末3 g及苦参药材粉末9 g,平行制6份,按照“2.1.3”项下方法制备供试品溶液,分别按“2.1.1”项下色谱条件进样分析,考察特征峰保留时间和峰面积的一致性。以盐酸小檗碱的保留时间和峰面积为参照分别计算相对保留时间及相对峰面积。计算得各共有峰相对保留时间的RSD<0.18%、相对峰面积的RSD<1.57%,表明该方法重复性较好。 2.1.7 黄连-苦参药对指纹图谱的建立及相似度评价分析 将黄连及苦参药材依照“2.1.3”项下方法制备成供试品溶液(S1~S9依次为黄连-苦参比例为5∶1、4∶1、3∶1、2∶1、1∶1、1∶2、1∶3、1∶4、1∶5),再按“2.1.1”项下色谱条件进样分析,记录色谱图。将图谱输入《中药色谱指纹图谱相似度评价系统(2012版)》,设置编号S7的样品(黄连-苦参为1∶3)图谱为参照,采取中位数法[19],将时间窗宽度设置为0.1 s,进行多点校正,建立黄连-苦参药对的HPLC指纹图谱和对照指纹图谱(R,图1),指认9批黄连-苦参药对的16个共有峰。采用《中药色谱指纹图谱相似度评价系统(2012版)》对9批黄连-苦参药对进行相似度评价[20]。结果显示,9批黄连-苦参药对和R之间的相似度均大于0.95,这表明各批次黄连-苦参药对的相似性较好,整体质量稳定,可以用于考察黄连-苦参药对水煎液。以分离度较好、峰面积较大的小檗碱(峰16)为参照峰(S),得到9批黄连-苦参药对16个共有峰相对保留时间的RSD为0.175%~0.894%,提示各批次黄连-苦参药对共有峰的保留时间稳定 2.1.8 黄连-苦参药对指纹图谱色谱峰归属认定 通过比对单味药的色谱峰[21],不同比例配伍黄连-苦参药对HPLC指纹图谱16个共有峰中峰2~6号共5个峰均来源于单味药苦参,峰1、7~16号共11个峰来源于单味药黄连(图1)。通过对比混合对照品溶液色谱图(图2)及黄连、苦参及样品HPLC叠加图(图2)对各样品指纹图谱的各峰进行定性认证[22],得到2、3、6号峰分别为苦参碱、槐定碱、氧化苦参碱,属于单味药苦参;8、11~16号峰分别为木兰花碱、非洲防己碱、表小檗碱、药根碱、黄连碱、巴马汀、小檗碱,属于单味药黄连。 2.1.9 黄连-苦参药对各共有峰相对峰面积差异分析 将各比例药对中黄连-苦参药对生药量以黄连、苦参单煎样品的生药量为标准,换算成一致的量,并以黄连及苦参单煎样品峰面积作为参比,比较不同配比黄连-苦参药对的共有峰相对峰面积,结果见表2。可知在不同程度配比下,各共有峰相对峰面积均有不同程度的变化,绝大部分表现出显著性差异。除属黄连药材的10、13号峰各相对峰面积相比药材单提均有所下降外,其余峰均表现为升高,表明配比后成分的溶出对苦参总体表现为促进作用,而对黄连的不同成分表现为促进和抑制的不同作用。1、5、7号峰在黄连-苦参为2∶1时相对峰面积最大;2~4、8、10号峰在黄连-苦参为5∶1时相对峰面积最大;11~16号峰在黄连-苦参为4∶1时相对峰面积最大;6号峰在黄连-苦参为1∶3时相对峰面积最大;9号峰在黄连-苦参为3∶1时相对峰面积最大,提示在方剂中使用不同配比黄连-苦参药对治疗疾病,可能与不同配比下药对中成分的溶出变化有关[23]。 2.2 不同配比黄连-苦参药对中差异性成分含量测定 2.2.1 色谱条件 按照“2.1.1”项下色谱条件进行测定。设定在波长为205 nm时,对苦参碱、槐定碱、氧化苦参碱进行测定;在波长为220 nm时,对木兰花碱进行测定;345 nm时,对非洲防己碱、表小檗碱、药根碱、黄连碱、巴马汀、小檗碱进行测定。此时各指标性成分均为最大吸收波长。 2.2.2 混合对照品溶液的制备 依照“2.1.2”项下方法制备混合对照品溶液。 2.2.3 供试品溶液的制备 依照“2.1.3”项下方法制备9个比例的黄连-苦参药对供试品溶液,每个比例制备3个供试品溶液作为平行对照。 2.2.4 线性关系考察及检测限、定量限 对照品母液的配制:取苦参碱、槐定碱、氧化槐果碱、木兰花碱、非洲防己碱、表小檗碱、药根碱、黄连碱、巴马汀、小檗碱对照品各适量,分别置于10 mL量瓶中,加甲醇溶解并定容,制得上述成分质量浓度分别为0.98、0.40、0.85、0.35、0.31、0.35、0.36、0.36、0.35、0.81 mg/mL的对照品溶液。 取各对照品母液,逐级稀释0、2、4、8、16、32、64倍,按照“2.1.1”项下色谱条件进行测定。以各差异性成分的质量浓度为横坐标(X)、峰面积为纵坐标(Y)绘制标准曲线,进行线性回归,得回归方程,结果见表3,表明各成分线性关系良好。 依照信噪比,即S/N为3∶1及S/N为10∶1对各成分的检测限及定量限进行检测,结果见表3。 2.2.5 精密度试验 依照黄连与苦参比例1∶3,精密称取黄连药材粉末3 g及苦参药材粉末9 g,按照“2.1.3”项下方法制备供试品溶液,按“2.1.1”项下色谱条件连续进样6次,记录各差异性成分的峰面积。结果显示,苦参碱、槐定碱、氧化苦参碱、木兰花碱、非洲防己碱、表小檗碱、药根碱、黄连碱、巴马汀、小檗碱峰面积的RSD分别为1.40%、2.13%、1.37%、2.11%、0.91%、0.69%、1.25%、1.19%、0.17%、0.14%,结果表明仪器精密度良好。 2.2.6 稳定性试验 依照黄连与苦参比例1∶3,精密称取黄连药材粉末3 g及苦参药材粉末9 g,按照“2.1.3”项下方法制备供试品溶液,于室温放置0、4、8、12、16、20、24 h,按“2.1.1”项下色谱条件进样分析,记录各差异性成分的峰面积。结果显示,苦参碱、槐定碱、氧化苦参碱、木兰花碱、非洲防己碱、表小檗碱、药根碱、黄连碱、巴马汀、小檗碱峰面积的RSD分别为1.57%、2.24%、2.22%、2.46%、0.22%、0.16%、0.65%、0.05%、0.14%、0.20%,表明各差异性成分在室温放置24 h内稳定性较好。 2.2.7 重复性试验 依照黄连与苦参比例1∶3,精密称取黄连药材粉末3 g及苦参药材粉末9 g,按照“2.1.3”项下方法平行制备供试品溶液6份,再按“2.1.1”项下色谱条件进样分析,记录各差异性成分的峰面积,并根据标准曲线计算含量。结果显示,苦参碱、槐定碱、氧化苦参碱、木兰花碱、非洲防己碱、表小檗碱、药根碱、黄连碱、巴马汀、小檗碱质量分数的RSD分别为1.16%、1.24%、1.33%、1.57%、1.05%、1.19%、1.42%、1.30%、1.21%、1.22%,表明该方法重复性良好。 2.2.8 加样回收率试验 依照黄连与苦参比例1∶3,精密称取黄连药材粉末3 g及苦参药材粉末9 g,平行称取6份,分别加入含有苦参碱0.31 mg、槐定碱0.20 mg、氧化苦参碱1.52 mg、木兰花碱0.07 mg、非洲防己碱0.08 mg、表小檗碱0.27 mg、药根碱0.06 mg、黄连碱0.22 mg、巴马汀0.21 mg、小檗碱0.79 mg的对照品溶液5 mL,按照“2.1.3”项下方法制备供试品溶液,再按“2.1.1”项下色谱条件进样分析,记录各标志性成分的峰面积,并计算平均加样回收率。结果显示,苦参碱、槐定碱、氧化苦参碱、木兰花碱、非洲防己碱、表小檗碱、药根碱、黄连碱、巴马汀、小檗碱的平均加样回收率分别为100.2%、100.1%、100.3%、100.2%、101.2%、100.7%、99.8%、101.1%、100.60%、101.0%,RSD分别为0.67%、0.97%、0.89%、0.97%、0.56%、0.70%、0.57%、0.71%、0.99%、0.85%,表明该方法准确度良好。 2.2.9 不同配比黄连-苦参药对水煎液成分含量测定及比较 取9个不同比例的黄连-苦参药对药材粉末,精密称定,按照“2.1.3”项下方法制备供试品溶液,再按“2.1.1”项下色谱条件进样分析,记录各差异性成分的峰面积,并根据标准曲线计算苦参碱、槐定碱、氧化苦参碱、木兰花碱、非洲防己碱、表小檗碱、药根碱、黄连碱、巴马汀、小檗碱的含量。将各比例药对中黄连-苦参药对生药量以黄连、苦参单煎样品的生药量为标准,换算成一致的量,计算各特征性成分的含量。通过SPSS 27.0软件,对数据进行单因子方差分析和显著性检验[24],结果见表4。 对含量测定结果进行系统分析。黄连-苦参比例为4∶1时,所得非洲防己碱、表小檗碱、巴马汀、小檗碱含量为各比例最高,且黄连总生物碱含量最高,与单药材提取具有显著性差异(P<0.05);黄连-苦参比例为5∶1时,所得苦参碱、槐定碱、木兰花碱含量为各比例最高,与单药材提取具有显著性差异(P<0.05);黄连-苦参比例为1∶3时,氧化苦参碱含量为各比例最高,与单药材提取具有显著性差异(P<0.05);黄连-苦参比例为1∶1时,苦参总生物碱含量为各比例最高。与黄连、苦参各药材单提相比,各比例下苦参中总生物碱类成分的溶出均有不同程度的提升,黄连中总生物碱类成分在黄连-苦参5∶1及4∶1比例下溶出表现为提升,其他比例表现为降低。随着药对中黄连比例的降低,黄连中整体生物碱类成分呈现下降趋势。对苦参中差异性成分进行比较,随着药对中黄连比例的降低,苦参碱、槐定碱在药液中的溶出降低,而氧化苦参碱的溶出提升,3种成分呈现“U”型分布,提示3者之间的相互影响关系。 3 讨论 本研究考虑与临床应用一致,黄连-苦参药对选择水回流提取法,选择分离效果最佳的乙腈-磷酸二氢钾溶液体系,对黄连及黄连-苦参药对的色谱条件进行优化,并在190~440 nm进行全波长扫描,于220 nm下进行指纹图谱建立以求全面对待测样品的差异性成分进行测定。结果表明,本研究建立的黄连-苦参药对指纹图谱稳定有效,可全面的测定黄连-苦参药对中的标志性成分。 大量文献研究发现,黄连-苦参药对在方剂中多采用1∶5至5∶1区间配比,故选择典型的9个配比进行量-质传递对比性研究。生物碱类成分作为黄连-苦参药对的主要药效成分,研究生物碱类成分在传统方剂煎煮过程中的溶出差异,可以为临床用药提供参考。故采用建立指纹图谱方式进行定性验证,确定稳定可测的生物碱类成分,并根据“2.1.8”项下结果,选择苦参中苦参碱、槐定碱、氧化苦参碱及黄连中木兰花碱、非洲防己碱、表小檗碱、药根碱、黄连碱、巴马汀、小檗碱进行研究[25-26]。 本研究在最佳吸收波长下,对黄连-苦参药对不同配比中10个差异性成分进行含量测定,分析差异性成分在不同配比下的溶出变化。苦参中3种差异性成分的溶出量随黄连比例的降低呈现“U”型分布,而黄连中7种差异性成分溶出量随黄连比例的降低整体呈现降低趋势。在黄连-苦参药对中,高黄连比例更容易促进药对中差异性成分的溶出。初步分析,当黄连-苦参药对中黄连占比的降低,可能会通过改变溶液中pH值、酸碱度等性质,对二者差异性成分的溶出产生影响,也可能对其中成分的相互转化产生促进作用,其具体产生机制有待深入研究。黄连-苦参药对被应用与各类中医经典方及现代经验方剂中[27-28],但其配伍面对临床不同疾病的合理应用仍需深入研究。 本研究首次将黄连-苦参相须药对与中医传统经验方剂药效相结合,探究差异性成分药理作用与临床疾病治疗的联系。黄连-苦参比例为5∶1时,所得苦参碱、槐定碱、木兰花碱含量为各比例最高;非洲防己碱、表小檗碱、药根碱、黄连碱、巴马汀、小檗碱含量较高,相比各药材单提含量有所提升,与单药材提取均具有显著性差异(P<0.05),与《普济方》中“相须为用,其效益彰”的方解一致,发挥各成分共同药效,达到“清热燥湿”效果。氧化苦参碱具有抗肿瘤作用,当黄连-苦参比例为1∶3时,其溶出量达到最大并与单药材提取具有显著性差异(P<0.05),与临床上使用参白解毒方进行抗结直肠道腺瘤[29]的治疗方式一致。药根碱可发挥降糖作用,在黄连-苦参比例为1∶1时含量最高,与国医大师李玉奇治疗消渴症时采用方剂中黄连-苦参药对[30]的配比一致,证明了方剂中黄连-苦参使用该比例配比的合理性。 综上所述,本研究成果预期可为开展黄连-苦参药对的量-效关系研究提供数据支撑,为临床不同疾病采用药对适宜配比用量、开发黄连-苦参药对新方剂提供借鉴。
[size=3]以下是2010年版药典二部中关于盐酸小檗碱中有关物质检查的描述:[检查]有关物质 取本品适量,精密称定,加流动相溶解并定量稀释制成每1ml中含1mg的溶液,作为供试品溶液;另取盐酸药根碱对照品和盐酸巴马汀对照品适量,精密称定,加流动相溶解并定量稀释制成每1ml中含0.1mg的溶液,分别作为对照品溶液(1)和(2);精密量取供试品溶液2ml和对照品溶液(1)和(2)各10ml,置100ml量瓶中,用流动相稀释至刻度,摇匀,作为对照溶液;取对照品溶液(2)1ml,用供试品溶液稀释至10ml,摇匀,作为系统适用性试验溶液。照高效液相色谱法(附录V D)试验,用十八烷基硅烷键合硅烷为填充剂;以0.01mol/L磷酸二氢铵溶液(用磷酸调节pH值至2.8)-乙腈(75:25)为流动相;检测波长为345nm。取系统适用性试验溶液10ul,注入液相色谱仪,巴马汀峰与小檗碱峰间的分离度应符合要求。另取对照溶液10ul,注入液相色谱仪,调节检测灵敏度,使小檗碱色谱峰的峰高约为满量程的25%。精密量取对照溶液与供试品溶液各10ul,分别注入液相色谱仪,记录色谱图至主成分色谱峰保留时间的2倍。供试品溶液的色谱图中,如有与药根碱峰和巴马汀峰保留时间一致的色谱峰,按外标法以峰面积计算,均不得过1.0%;[color=#fe2419]其他杂质峰面积的和不得大于对照溶液中小檗碱峰的峰面积(2.0%)。[/color]我的问题是:最后一句是什么意思?为什么供试品溶液色谱图中的其他杂质峰与对照溶液色谱图中的小檗碱峰面积进行比较?而不在一张色谱图内比较?如果按上面说,我们的结果是50%以上;而最后的括号中的2.0%是什么意思?[/size]
所用对照品批号为110713-200911使用前无需任何处理,按含C20H18ClNO4为86.8%计,而我所检测的产品是以C20H18ClNO4.2H2O计,是不是最后结果得以1.097来折算??? http://bbs.sdatc.com/image/post/smile/26.gif
请教各位大神,新配的对照品溶液,峰面积减小很快是什么原因呢?谢谢。
对照品:用于鉴别、检查、含量测定和校正检定仪器性能的标准物质;对照品由国家药品检定机构审查认可,其标准应不低于制品的质量标准。 标准品:用于生物检定、抗生素或生物药品中含量或效价测定的标准物质,以效价单位(U)表示。 对照品与标准品概念不清?对照品与标准品是2个不同的概念,中国药典凡例中已有明确的定义:对照品系指用于鉴别、检查、含量测定和校正检定仪器性能的标准物质。标准品:用于生物检定、抗生素或生物药品中含量或效价测定的标准物质,以效价单位(U)表示。文献中常将2种概念混淆,认为对照品就是标准品,是1种物质2种提法而已,造成错误的原因,可能是有的药品既有对照品,又有标准品。 例如:当用微生物法测定头孢克罗效价时,用头孢克罗标准品,用HPLC或UV法测定时,则用对照品;非那西丁当用作熔点校准物质时,用熔点标准品,测定含量时,用对照品。即使是同一种物质的标准品和对照品,它们的规格、标定方法以及用途都可能是不同的。
最近在写sop,特别留意了这个方面的内容。我查到的答案跟大家不一样。请高手说明解释一下:[b]标准品系指用于生物检定、抗生素或生物药品中含量或效价测定的标准物质,以效价单位(U)表示。对照品系指用于鉴别、检查、含量测定和校正检定仪器性能的标准物质。[/b]就我看到的说法,标准品与对照品并无级别上的差异,而是用途的不同。这个说法跟本版面以前的这个帖子差异很大,不明真相的群众求解答。[url]http://bbs.instrument.com.cn/shtml/20081021/1541297/[/url]
[color=#333333]对照品与标准品概念[/color][color=#333333]对照品与标准品是2个不同的概念,中国药典凡例中已有明确的定义:对照品系指用于鉴别、检查、含量测定和校正检定仪器性能的标准物质,而标准品系指用于生物检定、抗生素或生物药品中含量或效价测定的标准物质,以效价单位(U)表示.文献中常将2种概念混淆,认为对照品就是标准品,是1种物质2种提法而已[1,2],造成错误的原因,可能是有的药品既有对照品,又有标准品.例如,当用微生物法测定头孢克罗效价时,用头孢克罗标准品,用HPLC或UV法测定时,则用对照品;非那西丁当用作熔点校准物质时,用熔点标准品,测定含量时,用对照品.即使是同一种物质的标准品和对照品,它们的规格、标定方法以及用途都可能是不同的.[/color]
在抗生素类的标准物质使用时,经常会遇到标准品和对照品的概念。关于这二者的区别,现在比较流行的说法是在做HPLC时使用的标准物质应为对照品。摘录典型观点如下:[B]“标准品都是按效价单位(或μg)计,以国际标准品进行标定。标准品的标示量是按生物活性来计算的,不是按纯度来标示,此种标示法对单组分或多组分物质均适用,尤适用于多组分物质,如乙酰螺旋霉素标准品,是由4种有效成分组成,若欲于一个纯度来标示其含量是不可能的,但用效价(即生物活性)来标示是可行的;对照品的标示量则必定是某单一组分的纯度指标。所以日常工作中,标准品和对照品在定量时是不可相互替代的。以罗红霉素为例,现今是国家标准品与对照品并存,以抗生素微生物检定法测其含量时,必须使用罗红霉素标准品;但以HPLC法测定其含量时,又必须使用罗红霉素对照品,不可混淆。”[/B]但是我见过一些行业标准,比方说HPLC测土霉素残留中,在说到标准液的配制时,写得就是“土霉素标准品”。难道这里面的“标准品”是“对照品”的错误用法?[em0716] 请大家发表一下看法
其余的色谱条件相同,如何确定混合对照品的最佳检测波长?我拿甲醇溶解的对照品,因为在低紫外有吸收,结果出现一个倒峰,那是不是一定要换溶剂呢?换成流动相的话,流动相的比例现在还不确定,那又怎么溶解呢?
有没有薄层(TLC)检测专用对照品
做一个甾体皂苷的含量测定,已经做第4次了,问题始终没解决,遂请教。具体要求如下:色谱条件要求:C8的柱子;流动相是乙腈-水(30:70);检测波长210nm。样品处理:取一定量,加75%乙醇超声10分钟,放冷,补足减失重量,滤过,即可。 对照品也是用75%乙醇溶解。测试经过如下:有同一厂家同一品种不同批号两批样品,用C8的柱子做,计算了下,含量合格,但峰实在太难看。怕峰形太差影响结果的准确性,但只有一根这C8的柱子,只好换根C18的柱子,调整流动相试试;峰形很好,进了一针其中一批的样品,计算了也合格,就编个序列继续做下去了。第四天才发现另一批不合格,只好第五天返工。第二次,问题出现了。返工时,同样的仪器,色谱柱,色谱条件,用原来配的对照品溶液,重新处理样品,不合格的那一批还是不合格,结果比上次测的还低一点,合格的那批也不合格了。又另配了对照品溶液,与之前配的浓度相差不大,但峰面积小多了。但是之前配的对照品峰面积反而变大了,估计是因为流动相微调,把之前没分开的小峰分开了,不至于斜着积分的原因吧。怀疑过对照品峰面积小,是不是冷冻(对照品要求冷冻保存哈)后没放至室温,直接称,导致称不准。样品怀疑过的原因 A:是不是不合格那一批取样量大了,浓度高了,导致过饱和,未全溶 B:样品是不是未混匀,取样代表性不够 C:超声时间,功率是不是有问题,是不是超声发热导致样品降解(对照品要冷冻保存嘛)E:是不是第二次配的溶样溶剂有问题第三次做,更蹊跷了。之前怀疑的样品的原因都排除了。减少,增加取样量;混匀样品;超声不同时间,用不同超声设备(不同功率);超声中换水,防止温度升高,水浴上回流处理样品;重配75%乙醇几次,用无水乙醇配也试了,还换用甲醇,样品溶液居然都没有目标峰。但之前两次配的对照品溶液峰高变化不大,高的还是高,低的还是低。第四次,就是今天。又重配了对照品溶液,换甲醇(色谱纯)溶解,居然对照品也没峰了,用75%乙醇溶解也是。之前配的对照品峰高还是老样子。将第一次配的对照品溶液跟这次提取的测不到目标峰的样品溶液混合,也能出目标峰。这根柱子换走另一个对照品测试,峰好得很。再换另一根柱,走样品和对照品,还是老样子,之前能出的还出,出不了的还是还是出不了。总结:一共配了三次对照品,浓度差异不大,但峰面积在变小,第三次就没了。样品也是,第一次合格的,第二次提取也不合格了,以后干脆目标峰也不出了。 分析:应该不是目标物不稳定的原因,因为第一次配的对照品,隔了一周,峰高依然变化不大,更何况新配的对照品溶液。 几次都是我操作,对照品溶液都超声助溶过,应该也是溶了的。而且用紫外扫过,稀释后吸收值会降低,虽然最大吸收波长在约205nm。其实用甲醇比用75%乙醇溶解要好些,几乎振摇就能溶,用75%乙醇必须稍超声一下。[/fo
[size=3]今天要干燥对照品,对照品是在安瓿中储存的,所以得先把安瓿打开。而这个安瓿没有预划痕,我只好先用小瓷片在一边划了几下,然后一掰,结果用边太大,把安瓿一边整个给捏碎了,玻璃碎屑还掉到了对照品里。这可怎么办?大家遇到过这种情况吗?是怎么处理的?[/size]
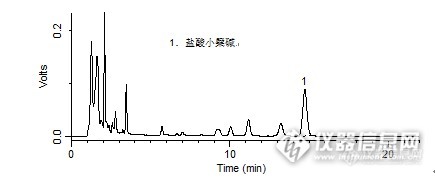
问题:导赤丸中盐酸小檗碱的检测中:对照品中盐酸小檗碱的理论塔板数是多少?答案:13707.960幸运奖获得者:莫名其妙(ID:moyueqiu)大川之子,纵横四海(ID:chuangu120)999youran(ID:999youran)【活动奖励】幸运奖(2钻石币):抽奖软件,当天随机抽取3个回答正确的版友ID号(最后一个ID号,截止至下午3:00),每人奖励2个钻石币积分奖励:所有回答正确的版友奖励10个积分(幸运奖获得者除外)。【注意事项】同样的答案,每人只能发一次PS:该贴浏览权限为“回贴仅作者和自己可见”,回复的版友仅能看到版主的题目及自己的回答内容,无法看到其他版友的回复内容。下午3点之后解除,即可看到正确答案、获奖情况及所有版友的回复内容。导赤丸中盐酸小檗碱的检测http://ng1.17img.cn/bbsfiles/images/2015/11/201511271513_575290_1987954_3.jpghttp://ng1.17img.cn/bbsfiles/images/2015/11/201511271514_575291_1987954_3.jpghttp://ng1.17img.cn/bbsfiles/images/2015/11/201511271514_575292_1987954_3.jpghttp://ng1.17img.cn/bbsfiles/images/2015/11/201511271514_575293_1987954_3.jpg样品制备 制备方法1. 对照品:取盐酸小檗碱对照品适量,精密称定,加甲醇制成每1 mL含80 μg的溶液,即得。2. 供试品:取重量差异项下的本品,水蜜丸研细或大蜜丸剪碎,混匀,取约1.0 g,精密称定,置具塞锥形瓶中,精密加入盐酸-甲醇(1:100)混合溶液25 mL,称定重量,85 ℃水浴中加热回流40 分钟,放冷,再称定重量,用盐酸-甲醇(1:100)混合溶液补足减失的重量,摇匀,离心,取上清液,滤过,取续滤液,即得。分析条件 色谱柱Diamonsil C18(2) 150 x 4.6 mm,5 μm (Cat#:99601)流动相乙腈:0.05 mol/L磷酸二氢钾溶液=50:50(每100 mL中加十二烷基硫酸钠0.4 g,再以磷酸调节pH值至4.0)流速1.0 mL/min柱温30 ℃检测器UV 345 nm进样量5 μL色谱图对照品http://ng1.17img.cn/bbsfiles/images/2015/11/201511271000_575208_1987954_3.png 峰号 保留时间 min 峰面积 μV*s 峰高 μV 理论塔板数* N USP拖尾因子 分离度 1 14.732 962307 50994 13707.960 1.023 -- *药典要求理论板数按盐酸小檗碱峰计算应不低于5000供试品http://ng1.17img.cn/bbsfiles/images/2015/11/201511271000_575209_1987954_3.png 峰号 保留时间 min 峰面积 μV*s 峰高 μV 理论塔板数* N USP拖尾因子 分离度 1 14.728 1653581 87613 13559.527 1.032 -- *药典要求理论板数按盐酸小檗碱峰计算应不低于5000本品种同时使用了Platisil ODS色谱柱,在药典规定条件下进行盐酸小檗碱的检测,满足药典要求。
[size=3]不知大家注意没有,在2010年版药典中,特别是UV-Vis测含量,在“对照品溶液的制备”中,往往是准确指出精密称取的对照品的量,例如,2010年版药典一部第5页,人工牛黄中胆酸的含量测定项下,胆酸对照品溶液的制备:取胆酸对照品12.5mg,精密称定,置25ml量瓶中,加60%冰醋酸溶液使溶解,并稀释至刻度,摇匀,即得(每1ml中含胆酸0.5mg)。而,HPLC或GC等测含量,大多的表述是,如同是第5页,八角茴香中反式茴香脑的含量测定,对照品溶液的制备:取反式茴香脑对照品适量,精密称定,加乙醇制成每1ml含0.4mg的溶液,即得。这两种表述有何不同?[/size]
有一个产品客户要求含量检项,没有对照品,需要用自己做的纯品来标化出对照品。根据合成人员提供的信息,用的乙醇作为溶剂,没有无机盐,含有水分。我标化时检测了纯度、水分,乙醇的残留,炽灼残渣也测了。最终得出一个含量,为99.4%。于是我拿这个含量试着测了一下样品,发现样品只有91%的含量……我又拿买的纯品,按照上面给的数值作为含量,测了一下样品,样品的含量达到了100%以上……很是蒙圈了- -今天把标化的对照品、买的纯品、样品,分别称取了10.00mg的样品,稀释至250ml,进样10ul,对比了一下三者峰面积。如果按照称样量与峰面积的比值来看的话,买来的纯品峰面积最高,是不是说买来的纯品含量应该也是最高的- -造成这种差异的原因是什么,能怎么处理呢?理论上要两种方式标化,但是这个化合物是吲唑上面挂了一个甲基和溴。还有其他方式可以标化吗?
各位前辈,小妹有个问题请教,我用的是安捷伦的高效液相色谱仪,最近做黄柏中盐酸小檗碱的含量测定,347nm,对照品不出峰,样品也就前四分钟内有点杂质峰。很奇怪,以前做的都很正常,这次流动相,对照品以及样品的处理方法都没有变。怀疑是流动相得原因,所以让同事帮忙复检,结果也一样。怀疑是柱子的原因,换了三根柱子,一根做的都有峰,但是个很奇怪的肩峰,另外两根都无峰。又怀疑是检测器的原因,但用它做其他的品种都很好。很是纳闷。想问一下各位前辈在工作中有没有遇到过这种类似的问题,最后是怎么解决的。感激不尽![em09508]
[font=&][color=#333333]对照品系指用于鉴别、检查、含量测定和校正检定仪器性能的标准物质。[/color][/font][font=&][color=#333333]标准品系指用于生物检定、抗生素或生物药品中含量或效价测定的标准物质,以效价单λ(U)表示。[/color][/font][font=&][color=#333333]如果还是感觉不甚明了,是否标准品只用于生物方面?是否化学方面只能称对照品?标准品有什?要求?对照品有什?要求?[/color][/font][font=&][color=#333333]国家药品标准品、对照品系指国家药品标准中用于鉴别、检查、含量测定、杂质和有关物质检查等标准物质,它是国家药品标准不可分割的组成部分。国家药品标准物质是国家药品标准的物质基础,它是测量药品质量的基准;也是做为校正测试仪器与方法的物质标准;在药品检验中,它是确定药品真α优劣的对照,是控制药品质量必不可少的工具。[/color][/font][font=&][color=#333333]目前,中国药品生物制品检定所已能提供各类国家标准物质1242种,其中中药化学对照品288种,对照药材400种,两者占总数的一半以上。[/color][/font][font=&][color=#333333]国家标准品及生物参考品系指用于鉴别、检查含量或效价测定的标准物质,其制备与标定应符合“生物制品国家标准物质制备和标定规程”要求,并由国务院药品监督管理部门指定的机构分发。企业工作标准品或参考品必须经国家标准品或参考品标化后方能使用。[/color][/font][font=&][color=#333333]对照品系指用于生物制品理化等方面测定的特定物质,由生产单λ采用与制品生产工艺相同的方法制备。对照品应尽可能与制品原液配方一致,稳定性较差的,可加不含对测定有干扰物质的适宜的稳定剂。对照品由国家药品检定机构审查认可,其标准应不低于制品的质量标准。[/color][/font][font=&][color=#333333]标准品、对照品:是指用于鉴别、检查、含量测定的标准物质,均由国务院药品监督管理部门指定的单λ制备、标定和供应。标准品系指用于生物测定、抗生素或生化药品中含量或效价测定的标准物质,一国际标准品进行标定;对照品出另有规定外,按干燥进行计算后使用。[/color][/font][font=&][color=#333333]标准品和对照品均附有使用说明书,质量要求,有效期和装量等。[/color][/font][font=&][color=#333333]生物制品标准物质系指用于生物制品效价、活性或含量测定的或其特性鉴别、检查的生物标准品或生物参考物质。[/color][/font]转自:食品伙伴网
请问各位老师,我们这边进了对照品、空白供试、加标供试品,加标对照品的响应值远远大于对照品溶液里的响应,请问是什么原因呢?已知:空白供试品里的检出非常小不会有这么大的影响,对照品自身线性良好,加标供试品重复性良好(也就是一直都很大,并没有逐针降低),并且排除了对照残留的因素。配制过程的区别就是对照与空白供试未离心,加标离心。后来将对照离心后,响应值第一针也变大,但随后也降低至与未离心的响应值差不多。
http://www.greenherbs.com.cn/bbs/dispbbs.asp?boardid=2&Id=7681612594 七氟醚杂质C Sevoflurane Related Compound C 对照品/标准品1612572 七氟醚杂质 B Sevoflurane Related Compound B 对照品/标准品1612550 七氟醚杂质 A Sevoflurane Related Compound A 对照品/标准品1612540 七氟醚 Sevoflurane 对照品/标准品1612539 盐酸舍曲林 Sertraline Hydrochloride 对照品/标准品1612528 盐酸舍曲林杂质A Sertraline Hydrochloride Related Compound A 对照品/标准品1612517 盐酸舍曲林消旋体混合物 Sertraline Hydrochloride Racemic Mixture 对照品/标准品1612506 L-丝氨酸 L-Serine 对照品/标准品1612426 芝麻油杂质B Sesame Oil Related Compound B 对照品/标准品1612415 芝麻油杂质A Sesame Oil Related Compound A 对照品/标准品1612404 芝麻油 Sesame Oil 对照品/标准品1612029 番泻苷 B Sennoside B 对照品/标准品1612018 番泻苷 A Sennoside A 对照品/标准品1612007 番泻苷 Sennosides 对照品/标准品1611955 硒 蛋氨酸 Selenomethionine 对照品/标准品1611900 盐酸司来吉兰 Selegiline Hydrochloride 对照品/标准品1611004 司可巴比妥 CII Secobarbital CII 对照品/标准品1610090 东莨菪亭 Scopoletin 对照品/标准品1610001 氢溴酸东莨菪碱 Scopolamine Hydrobromide 对照品/标准品1609831 沙奎那韦杂质A Saquinavir Related Compound A 对照品/标准品1609829 甲磺酸沙奎那韦 Saquinavir Mesylate 对照品/标准品1609807 双水杨酯 Salsalate 对照品/标准品1609625 沙美特罗杂质B Salmeterol Related Compound B 对照品/标准品1609614 沙美特罗杂质A Salmeterol Related Compound A 对照品/标准品1609603 昔美酸沙美特罗 Salmeterol Xinafoate 对照品/标准品1609501 水杨酸片 Salicylic Acid Tablets 对照品/标准品1609024 水杨酸杂质B Salicylic Acid Related Compound B 对照品/标准品1609013 水杨酸杂质A Salicylic Acid Related Compound A 对照品/标准品1609002 水杨酸 Salicylic Acid 对照品/标准品1608000 水杨酰胺 Salicylamide 对照品/标准品1607506 连翘粉状贯叶提取物 Powdered St. John's Wort Extract 对照品/标准品1607040 糖精钠 Saccharin Sodium 对照品/标准品1607029 糖精钙 Saccharin Calcium 对照品/标准品1607007 糖精 Saccharin 对照品/标准品1606503 芦丁 Rutin 对照品/标准品1606208 硝酚胂酸 Roxarsone 对照品/标准品1605523 罗哌卡因杂质B Ropivacaine Related Compound B 对照品/标准品1605512 罗哌卡因杂质A Ropivacaine Related Compound A 对照品/标准品1605500 盐酸罗哌卡因 Ropivacaine Hydrochloride 对照品/标准品1604916 罗库溴铵合剂峰的识别 Rocuronium Peak Identification Mixture 对照品/标准品1604905 罗库溴铵 Rocuronium Bromide 对照品/标准品1604870 利凡斯的明杂质B Rivastigmine Related Compound B 对照品/标准品1604869 利凡斯的明杂质A Rivastigmine Related Compound A 对照品/标准品1604814 利托那韦杂质混合物 Ritonavir Related Compounds Mixture 对照品/标准品1604803 利托那韦 Ritonavir 对照品/标准品1604701 盐酸利托君 Ritodrine Hydrochloride 对照品/标准品1604665 利培酮系统适用性试验用混合物 Risperidone System Suitability Mixture 对照品/标准品1604654 利培酮 Risperidone 对照品/标准品1604643 利塞膦酸杂质C Risedronate Related Compound C 对照品/标准品1604632 利塞膦酸杂质B Risedronate Related Compound B 对照品/标准品1604621 利塞膦酸杂质A Risedronate Related Compound A 对照品/标准品1604610 利塞膦酸钠 Risedronate Sodium 对照品/标准品1604600 利美索龙 Rimexolone 对照品/标准品1604508 盐酸金刚乙胺 Rimantadine Hydrochloride 对照品/标准品1604348 利鲁唑杂质A Riluzole Related Compound A 对照品/标准品1604337 利鲁唑 Riluzole 对照品/标准品1604202 醌式利福平 Rifampin Quinone 对照品/标准品1604009 利福平 Rifampin 对照品/标准品1603800 利福布丁 Rifabutin 对照品/标准品1603108 核糖 Ribose 对照品/标准品1603006 维生素B2 Riboflavin (Vitamin B2) 对照品/标准品1602706 利巴韦林 Ribavirin 对照品/标准品1602003 间苯二酚 Resorcinol 对照品/标准品1601849 二类残留溶剂-二甲苯 Residual Solvent Class 2 - Xylenes 对照品/标准品1601827 二类残留溶剂-三氯乙烯 Residual Solvent Class 2 - Trichloroethylene 对照品/标准品1601805 二类残留溶剂-甲苯 Residual Solvent Class 2 - Toluene 对照品/标准品1601780 二类残留溶剂-四氢萘 Residual Solvent Class 2 - Tetralin 对照品/标准品1601770 二类残留溶剂-四氢呋喃 Residual Solvent Class 2 - Tetrahydrofuran 对照品/标准品1601769 二类残留溶剂-二氧噻吩烷 Residual Solvent Class 2 - Sulfolane 对照品/标准品1601747 二类残留溶剂-吡啶 Residual Solvent Class 2 - Pyridine 对照品/标准品1601725 二类残留溶剂-硝基甲烷 Residual Solvent Class 2 - Nitromethane 对照品/标准品1601703 二类残留溶剂-N-甲基吡咯烷酮 Residual Solvent Class 2 - N-Methylpyrrolidone 对照品/标准品1601689 二类残留溶剂-甲基环己烷 Residual Solvent Class 2 - Methylcyclohexane 对照品/标准品1601667 二类残留溶剂-甲基丁基酮 Residual Solvent Class 2 - Methylbutylketone 对照品/标准品1601645 二类残留溶剂- 2-甲氧基乙醇 Residual Solvent Class 2 - 2-Methoxyethanol 对照品/标准品1601623 二类残留溶剂-甲醇 Residual Solvent Class 2 - Methanol 对照品/标准品1601601 二类残留溶剂-己烷 Residual Solvent Class 2 - Hexane 对照品/标准品1601587 二类残留溶剂-甲酰胺 Residual Solvent Class 2 - Formam

问题:妇科止带片中盐酸小檗碱的检测药典要求理论塔板数是多少?答案:*药典要求理论板数按盐酸小檗碱峰计算应不低于3000【活动奖励】幸运奖(2钻石币):dahua1981(ID:dahua1981)大川之子,纵横四海(ID:chuangu120)梧桐(ID:mengzhou)http://ng1.17img.cn/bbsfiles/images/2015/12/201512161514_578319_1610895_3.pnghttp://ng1.17img.cn/bbsfiles/images/2015/12/201512161514_578320_1610895_3.png积分奖励:所有回答正确的版友奖励10个积分(幸运奖获得者除外)。【注意事项】同样的答案,每人只能发一次PS:该贴浏览权限为“回贴仅作者和自己可见”,回复的版友仅能看到版主的题目及自己的回答内容,无法看到其他版友的回复内容。下午3点之后解除,即可看到正确答案、获奖情况及所有版友的回复内容。=======================================================================妇科止带片中盐酸小檗碱的检测样品制备 制备方法1. 对照品:盐酸小檗碱对照品适量,精密称定,加甲醇制成每1 mL含50 ug的溶液。2. 供试品:取本品10片,精密称定,研细,取约0.55 g,精密称定,置100 mL容量瓶中,精密加入流动相80 mL,称定重量,超声处理(功率400W,频率40kHz)45分钟,放冷,再称定重量,加流动相至刻度,混匀,滤过,取续滤液,即得。分析条件色谱柱Platisil ODS 150 x 4.6 mm,5 μm (Cat#:99501)流动相乙腈:0.025mol/L磷酸二氢钾:0.025mol/L十二烷基硫酸钠=46:27:27流速1.0 mL/min柱温30 ℃检测器UV 265 nm 进样量10 μL 色谱图对照品http://ng1.17img.cn/bbsfiles/images/2015/12/201512161005_578249_1610895_3.jpg 峰号 保留时间 min 峰面积 μV*s 峰高 μV 理论塔板数* N USP拖尾因子 分离度 1 29.123 2218896 55261 11863.203 1.014 -- *药典要求理论板数按盐酸小檗碱峰计算应不低于3000供试品 http://ng1.17img.cn/bbsfiles/images/2015/12/201512161005_578250_1610895_3.jpg 峰号 保留时间 min 峰面积 μV*s 峰高 μV 理论塔板数* N USP拖尾因子 分离度 1 29.360 27289 762 13797.819 1.022 -- *药典要求理论板数按盐酸小檗碱峰计算应不低于3000本品种同时使用了Diamonsil C18、Inspire C18两款色谱柱,在药典规定条件下进行盐酸小檗碱的检测,均满足药典要求。
有些化合物买不到对照品,但是需要测含量时,怎么标化。看到最多的是,全标,测纯度,水分, 干燥失重,无机物,溶剂残留等。其他没什么问题,但是这个纯度,本身就不太准确。现在有一个化合物,有一个特定杂质始终除不去,用的HPLC检测,占比仅0.5%,产品纯度达99.5%,而[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]检测这个杂质占比1.2%,用核磁鉴定时,这个杂质都达到了8%。也就是说这个杂质在该化合物中占比的实际含量,根据这些检测不是准确值,都是相对的,而且这个杂质是未知的,没办法完全确认其结构,也难以提纯。这种情况下测纯度,然后全标,我认为是不准确的。是否有其他方式,比如用内标法,用其他化合物来标这个产品是否合理?如果用内标法,那么就无需再做全标了。化合物是某苯甲醛,选用什么内标合适?
HPLC法测定盐酸小檗碱时含量偏高,请教盐酸小檗碱含量对照品的处理方法及纯度!!!
[size=3][color=#ff6600][url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法检薄荷桉油含片中薄荷脑的含量,仪器条件和操作方法如下[/color]:[/size][size=3][b][font=宋体]色谱条件与系统适用性试验[/font][/b][font=宋体] 采用弹性石英毛细管柱(30m×0.32mm×0.25[/font][/size][font=宋体][size=3]um )(PEG)聚乙二醇为固定液,进样口温度220℃,检测器温度为250℃。分流进样。[/size][/font][font=宋体][size=3]程序升温,初始90℃,保持1分钟,每分钟5℃升至170℃。理论塔板数按薄荷脑峰计[/size][/font][font=宋体][size=3]算应不低于10000。薄荷脑与内标物质峰的分离度应大于4。[/size][/font][size=3][b][font=宋体]校正因子测定[/font][/b][font=宋体] [/font][font=宋体]取水杨酸甲酯适量,加丙酮稀释成每1ml含1.0mg的溶液,摇匀,作为内标溶液。另取薄荷脑对照品适量,加丙酮稀释成每1ml含1.0mg的溶液。精密取内标溶液与对照品溶液各5ml,置25ml量瓶中,加丙酮至刻度,摇匀,精密吸取1μl注入[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url],计算效正因子。[/font][/size][size=3][b][font=宋体]测定法[/font][/b][font=宋体] [/font][font=宋体]取本品20片,精密称定,研细,取粉末适量(约10片量),精密称定,置具塞瓶中,精密加入丙酮25ml,振摇30分钟,滤过,精密量取续滤液10ml与内标溶液5ml,置25ml量瓶中,加丙酮至刻度,摇匀,精密吸取1μl注入[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url],测定,计算,即得。我按上述方法操作,用的机子是岛津GC-14C,柱子是0.53×30的PEG-20M,各气体流量也正常,但在做时发现只有水杨酸甲酯的内标峰出来,保留时间大约在7分钟的样子,而进薄荷脑对照品怎么没有薄荷脑的峰呢?进样品同样没有薄荷脑的峰,只有内标峰,请大家帮帮忙分析一下原因出在哪里?[/font][/size]
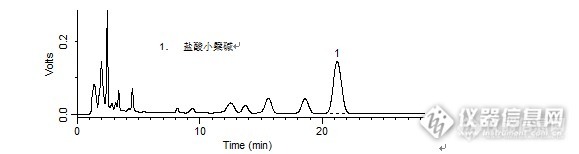
问题:万氏牛黄清心丸中盐酸小檗碱的检测:对照品中盐酸小檗碱峰的拖尾因子是多少呢?答案:1.033获奖公布:zengzhengce163(ID:zengzhengce163)梧桐(ID:mengzhou)馨语(ID:huangdm)http://ng1.17img.cn/bbsfiles/images/2016/03/201603091601_586411_708_3.jpghttp://ng1.17img.cn/bbsfiles/images/2016/03/201603091601_586412_708_3.jpg【活动奖励】幸运奖(2钻石币):抽奖软件,当天随机抽取3个回答正确的版友ID号(最后一个ID号,截止至下午3:00),每人奖励2个钻石币积分奖励:所有回答正确的版友奖励10个积分(幸运奖获得者除外)。【注意事项】同样的答案,每人只能发一次PS:该贴浏览权限为“回贴仅作者和自己可见”,回复的版友仅能看到版主的题目及自己的回答内容,无法看到其他版友的回复内容。下午3点之后解除,即可看到正确答案、获奖情况及所有版友的回复内容。万氏牛黄清心丸中盐酸小檗碱的检测样品制备 制备方法1. 对照品:取盐酸小檗碱对照品适量,精密称定,加甲醇制成每1 mL含80 μg的溶液,即得。2. 供试品:取重量差异项下的本品,剪碎,混匀,取约0.3 g,精密称定,置具塞锥形瓶中,精密加入盐酸-甲醇(1:100)混合溶液25 mL,称定重量,85℃水浴中加热回流40分钟,放冷,再称定重量,用盐酸-甲醇(1:100)混合溶液补足减失的重量,摇匀,离心,上清液滤过,取续滤液,即得。分析条件 色谱柱Diamonsil C18 150 x 4.6 mm,5 μm (Cat#:99901)流动相乙腈:0.05 mol/L 磷酸二氢钾溶液=50:50 (每100 mL中加十二烷基硫酸钠0.4 g,再以磷酸调节pH值为4.0)流速1 mL/min柱温30 ℃检测器UV 345 nm 进样量5 μL 色谱图对照品http://ng1.17img.cn/bbsfiles/images/2016/03/201603091123_586373_708_3.png 峰号 保留时间 min 峰面积 μV*s 峰高 μV 理论塔板数* N USP拖尾因子 分离度 1 21.438 4076937 111683 7718.883 1.033 -- *药典要求理论板数按盐酸小檗碱峰计算应不低于5000供试品http://ng1.17img.cn/bbsfiles/images/2016/03/201603091123_586374_708_3.png 峰号 保留时间 min 峰面积 μV*s 峰高 μV 理论塔板数* N USP拖尾因子 分离度 1 21.193 6691441 142627 4594.908 1.052 -- *药典要求理论板数按盐酸小檗碱峰计算应不低于5000
最近公司要采购中检所出的对照品,不知道从哪里购买?公司要求如果是从经销商购买需要经销商提供药检所的授权书,和检验报告,但是经询问经销商证书和报告都没有,那我怎么证明对照品就是药检所出的, 如果是我们做认证用,我们怎么证明这东西的来源?没有报告没有授权书似乎这东西没有说服力。 如果直接从药检所购买,似乎也很困难,服务差不说,还要托熟人帮忙购买,否则钱打过去都没人理,经销商说药检所都不乐意做生意,遇到这种情况我该怎么办呢?希望大家给出出主意!







 我要推广仪器
我要推广仪器
 下载APP
下载APP