http://ng1.17img.cn/bbsfiles/images/2012/09/201209272130_393425_2255248_3.gifHPLC测定木犀草苷对照品为什么峰前面有个小峰????
草乌薄层喷稀碘化铋钾试液后三种对照品都显什么颜色
目前我们实验室用的维生素A醋酸酯的对照品的供应商断货了,求问一下大家都用的是哪些供应商的对照品?我们也可以去买。我们试用过Sigma的和USP的发现都不行。Sigma的是实际含量和COA上的含量出入较大。USP的是一个混合物有全反式的和CIS的,由于我们不是用的中国药典附录上测定维生素A的方法,所以我们的液相分不开这2种物质,所以也不能用。
对照品甘草苷 后面出杂峰 是怎么回事[img]https://ng1.17img.cn/bbsfiles/images/2018/08/201808221432219383_6429_3461163_3.jpeg[/img]
药典 金钱草含量测定以甲醇一O.4%磷酸溶液(50:50)为流动相;检测波长为360nm,温度30℃。 对照品溶液的制备 取槲皮素对照品、山柰素对照品适量,精密称定,加80%甲醇制成每1ml各含槲皮素4μg、山柰素20μg的溶液,即得 供试品溶液的制备 取本品粉末(过三号筛)约1.5g,精密称定,置具塞锥形瓶中,精密加入80%甲醇50ml,密塞,称定重量,加热回流1小时,放冷,再称定重量,用80%甲醇补足减失的重量,摇匀,滤过。精密量取续滤液25ml,精密加入盐酸5ml,置90℃水浴中加热水解1小时,取出,迅速冷却,转移至50ml量瓶中,用80%甲醇稀释至刻度,摇匀,滤过,取续滤液,即得。 本品按干燥品计算,含槲皮素和山柰素的总量不得少于O.10%。
高效液相色普法测定甘草酸苷含量,甘草酸苷对照品峰面积与之前相比较偏高,导致含量偏高,请问有可能是哪些原因?
[color=#444444]请问大神们,麻烦大家帮我分析一下,我实在没法了:我用浓度为30微克/ml的山奈素对照品溶液跑高液,色谱条件是甲醇:0.4%磷酸水比例50:50,,流速1ml/min,柱温30℃,检测波长为360nm.进样量为20微升.为什么跑不出来任何峰形啊。。用异鼠李素对照品都能跑出来的,山奈素的出峰位置应该是在异鼠李素之前的。。。[/color][color=#444444][/color]
问题1:内毒素日常检查中:在加入鲎试剂前的供试品对照溶液(0.1ml)药典规定是含内毒素浓度2λ的。假如MVD=2,C=10mg/ml,λ=0.25EU/ml。那么应该用1EU/ml(也就是4λ)的内毒素溶液对半稀释浓度为10mg/ml的供试品溶液,终浓度才是含量为0.5EU/ml(2λ)内毒素、供试品为MVD浓度的供试液的供试品对照液!!! 问题2:还有如果供试品为注射用水,假如L=0.25,那么鲎试剂的灵敏度最少要用λ=0.125的;如果用λ=0.25的,因为供试品液(0.1ml)加入鲎试剂(0.1ml)后,稀释了1倍,假如结果为+,那么说明供试品的内毒素限制大于0.5EU?而不是大于0.25EU吧?
样品为复方维生素,其中一项是核黄素磷酸钠,没找到核黄素磷酸钠的对照品,故用的核黄素对照品样品制备:先用水溶,然后用流动相稀释,流动相弱酸性做出的结果比标示量低了很多啊用核黄素对照品代替核黄素磷酸钠对照品,请问结果可信吗?
如题请问用芦丁对照品绘制标准曲线是否能用槲皮素或其他的黄酮对照品替代?由于实验室的芦丁标准品不见了,老师建议先用槲皮素替代,请问是否可行?
做青霉素发酵液的HPLC检测时,所用的对照品到底应该是什么?我买的是青霉素G钾盐,到底能不能用来做青霉素G的对照品,另外我看到网上卖的青霉素G的标准品是Benzathine penicillin tetrahydrate,难道是苄青霉素?青霉素G和钾盐在反相色谱上的保留行为是一样的还是有差异的?
请教:注射用水溶性维生素中关于烟酰胺、等5项的液相检测方法其标准为烟酰胺、盐酸吡哆辛、硝酸硫胺、泛酸钠、维生素C钠和核黄素磷酸钠 照高效液相色谱法(中国药典1995年版二部附录Ⅴ D)测定。 色谱条件与系统适用性试验 用氨基键合多孔硅胶为填料,以(0.02mol/L)磷酸二氢钾溶液-乙腈(27:73),用10%盐酸溶液调节pH为5.3的溶液为流动相,流速为1.5ml/min,检测波长:烟酰胺、盐酸吡哆辛、硝酸硫胺、泛酸钠、维生素C钠为214nm;核黄素磷酸钠用萤光检测λEX=445nm、λEM=520nm。各组分的分离度应符合要求。 对照品溶液的制备 (1)取烟酰胺对照品约150mg、硝酸硫胺对照品约12mg、盐酸吡哆辛对照品约18mg、泛酸钠对照品约62mg,分别精密称量置50ml量瓶中,加水溶解并稀释至刻度摇匀,精密量取2ml置50ml量瓶中,用流动相稀释至刻度,摇匀,即为对照品溶液(Ⅰ),此溶液置暗处充氮气于零下20℃可保存1个月。(2)取维生素C钠对照品约425mg、核黄素磷酸钠对照品约19mg,精密称定,置50ml量瓶中加水溶解并稀释至刻度摇匀,精密量取2ml置50ml量瓶中,用流动相稀释至刻度,摇匀即为对照品溶液(Ⅱ),此溶液必须临用新鲜配制,并于零下20℃保存,用前放置至室温。 等容混合对照品溶液(Ⅰ)和对照品溶液(Ⅱ)即为对照品溶液。 供试品溶液的制备 取装量差异项下的内容物约2瓶重量,精密称定,置100ml量瓶中,加水溶解并稀释至刻度,摇匀,精密量取15ml置200ml量瓶中,用流动相稀释至刻度。 测定法 取对照品溶液和供试品溶液各10μl,交替注入液相色谱仪,测定,用外标法计算各组分含量,即得。目前存在问题用紫外检测的分不开5种组分,大家有什么好办法,谢谢
中国药典2010年版新增很多对照品,但大部分时间中检所断货。SIGMA、及USP对照品供应商能提供部分,但价格很贵。如何解决?
中检所对照品常见问题与答复1、标准物质的用途和应用范围药品标准物质不能作为药物或医疗器械而施用于人或动物。药品标准物质主要用于法定药品质量标准中的相关项目的检测用,详细内容请见使用说明书。2、有效期除了说明书上注明有效期的品种外,药品标准物质一般没有像药品一样设置有效期。在规定的储存和使用条件下,定期进行特性量值的稳定性核查,若发现影响使用将及时处理。3、储存标准物质一般应密闭、避光保存,对有特殊储存要求(如低温、避光等)的标准物质,说明书上均有说明,今后标签上也将注明。建议不要一次购买大量的标准物质,以免储存不当出现问题。需要冷藏或冷冻保存的品种,短时间短距离的冰盒运输对特性不会造成影响。4、纯度目前含量测定用的化学对照品的标签及说明书均赋有量值,以前的部分中药化学含量测定用未赋值的品种,按 100.0%计。5、是否能用于说明书用途范围外的检验、科研需要用户进行分析与验证。6、使用前是否需要干燥标准物质说明书上对使用前是否需要干燥等情况,均有相关说明。除另有规定外,对照药材不需要特殊处理。7、标准物质证书或测试报告暂时还不能提供证书或者测试报告。8、新批号标准品出来后旧批号能否继续使用 新旧批号更换过程中,部分品种将设置3-6个月以上的缓冲期。9、用五氧化二磷干燥的标准物质是否要在相同条件下保存不需要。按说明书的条件保存即可。10、从哪里可以查到标准物质的结构、物理化学特性等中药化学对照品的说明书大多附有结构,化学药品的可以通过中国药典二部查阅。另外,分发的标准物质都提供了英文名,可以通过文献查阅有关详情。11、内毒素标准品是否有10EU一支的 标准品均为100EU/支,10EU的工作品是鲎试剂生产企业生产的,低效价的内毒素稳定性差,我们不建议使用这种工作品进行检验工作。12、为什么对照品在色谱上不出峰? 请按国家标准中提供的条件考察自己的色谱条件因素。色谱不出峰一般来讲有如下原因:一是色谱条件不合适;二是信号采集时衰减过高,建议减小衰减;三是采集时间过短,建议增加采集时间。13、为什么对照品出2个或多个峰 标准物质除多组分的以外,均只有一个主峰,杂质峰不会超过赋值的范围。如果杂质峰超过赋值的范围,可能属于以下原因:① 配置对照品溶液的容器或溶剂被污染;② 盛放流动相的容器或配置溶剂被污染;③ 进样器被污染;④ 高效液相进样阀被污染;⑤ 色谱柱填充物出现断裂等。
[em09509]最近一直在做茶皂素的检测,从某试剂公司买来茶皂素想作为对照品,可是用液相怎么也做不出有紫外吸收的峰来,自己这边的样品就会出一些峰,用蒸发光做也一样。我是用乙腈跟PH3.0的冰醋酸水溶液做的流动相,走梯度,有哪位好心人来指点一下[em09509]
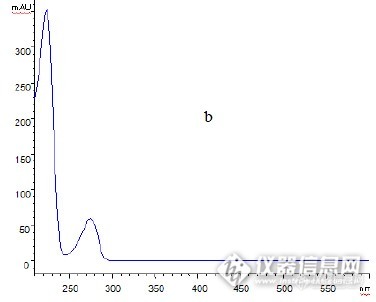
HPLC-DAD分析酸浆中木犀草素及木犀草素-7-β-D-葡萄糖甙成分酸浆(拉丁文名:Physali alkekengi L.)又名红菇娘、挂金灯、戈力、灯笼草、灯笼果、洛神珠、泡泡草、鬼灯等北方称为菇蔫儿、姑娘儿,以果实供食用。化学成分含酸浆苦素A(Physalin A)、酸浆苦素B、酸浆苦素C、木犀草素(Luteolin)及木犀草素-7-β-D-葡萄糖甙。果实含枸橼酸、草酸、维生素C、酸浆红色素(physalien)、酸浆醇(physanol)A,B。花萼含α胡萝卜素、酸浆黄质(physoxanthin)及叶黄素等,种子油的不皂化物中分得多种4α-甲基甾醇,主要为禾本甾醇(gramisterol)和钝叶醇(obtusifoliol)及4种新甾体。此外尚含多种4-脱甲基甾醇,如胆甾醇和24-乙基胆甾醇等。还含有多种三萜3β-一元醇,其中环木菠萝烷醇(cycloartanol)35%,环木菠萝烯醇(cycloartenol)27%、羊毛脂-8-烯-3β-醇(lanost-8-en-3β-ol)。木犀草素(luteolin)是一种天然黄酮类化合物,存在于多种植物中,具有抗炎、抗肿瘤、抗过敏等方面的作用。化学是如下:http://ng1.17img.cn/bbsfiles/images/2016/08/201608311303_607620_2217446_3.jpg目前,国内传统中药有效成分的提取方法普遍存在提取率低、杂质清除率不高、生产周期过长、能耗高、溶剂用量大等缺点。随着中药现代化进程的不断深入,许多现代高新技术不断地被应用到中药有效成分的提取和分离,使得中药有效成分的提取更高效和简便。超声-微波协同萃取技术直接将超声振动与开放式微波两种作用方式相结合,充分利用超声波振动的空化作用以及微波的高能作用,实现了低温常压条件环境下,对固体样品进行快速、高效、可靠的预处理,与常规提取方法相比,超声-微波协同萃取技术具有快速、节能、节省溶剂、污染小等优点。本实验应用超声-微波协同萃取法提取酸浆中的木犀草素及木犀草素-7-β-D-葡萄糖甙,采用高效液相-二极管阵列检测法(HPLC-DAD)测定提取物中木犀草素及木犀草素-7-β-D-葡萄糖甙的含量,药材中二者成分的含量分别为:1.200mg/g 和0.43mg/g,二个峰,木犀草素-7-β-D-葡萄糖甙峰位置分别为:221nm,270nm,木犀草素峰位置分别为:226nm,276nm,由于木犀草素-7-β-D-葡萄糖甙比木犀草素多了一个 β-D-吡喃葡萄糖基团,天麻素二个峰位置都发生了蓝移,样品中二个峰的光谱图与标准品二个峰的光谱图相同,可以进一步确定酸浆中含有木犀草素及木犀草素-7-β-D-葡萄糖甙。主要仪器与试剂主要仪器Agilent1100型四元梯度高效液相色谱仪(美国 Agilent 公司)Agilent TC-C18(ODS)色谱柱(5μm,4.6×250mm,美国 Agilent 公司)CW-2000 超声-微波协同萃取仪(新拓微波溶样测试技术有限公司)DJ-10A 型倾倒式粉碎机(上海隆拓仪器设备有限公司)RE-52AA 型旋转蒸发仪(河南巩义仪器厂)LXJ-IIB 型低速大容量多管离心机(上海安亭科学仪器厂)试剂木犀草素(中检所,含量98%;)木犀草素-7-β-D-葡萄糖甙(中检所,含量98%;)酸浆全草(采于黑龙江)除甲醇、乙腈为色谱纯(国药集团化学试剂有限公司),其余试剂除专门提到外,均为分析醇,实验用水为二次蒸馏水。实验方法供试品溶液的制备 精密称取酸浆粉末1.0g,置于超声-微波萃取仪玻璃容器中,加入50mL70%甲醇,开启超声微波,控制在恒温50℃下提取40min,萃取3次,合并提取液,浓缩至近干,残渣加入甲醇溶解,转移至10mL 量瓶中,加甲醇稀释至刻度,摇匀,过0.45μm 的微孔滤膜,取续滤液,即得。提取条件的考察溶剂的选择:精密称取酸浆粉末1.0g,置于超声-微波萃取仪玻璃容器中,分别用水、70%甲醇、70%乙醇溶液超声-微波协同萃取40min(n=3),萃取3次,合并提取液,浓缩至近干,残渣加入甲醇溶解,转移至10mL 量瓶中,加甲醇稀释至刻度,摇匀,过0.45μm的微孔滤膜,取续滤液,HPLC 测定萃取率。溶剂体积分数的选择:分别用体积分数为40%、50%、60%、70%、80%、90%和纯甲醇溶液超声-微波协同萃取30min(n=3),方法同上。溶剂用量的选择:分别用10mL、20mL、50mL、80mL、100mL70%甲醇提取,方法同上。提取时间的选择:分别用70%甲醇超声-微波协同萃取20min、30min、40min、50min、60min(n=3),方法同上。提取温度的选择:分别在40、45、50、55、60℃下用70%甲醇超声-微波协同萃取40min,方法同上。对照品溶液的制备 分别精密称取常温减压干燥12h 的木犀草素及木犀草素-7-β-D-葡萄糖甙对照品适量,加甲醇配制成木犀草素-7-β-D-葡萄糖甙为200μg/mL、木犀草素为100μg/mL 的混合对照品溶液,冷藏备用。色谱条件 色谱柱:Agilent TC-C18柱(5μm,4.6×250mm);流动相:A-0.1%乙酸水溶液;B-甲醇,线性梯度洗脱:0~30 min,3%~5% B;30~35 min,5%~20%B;35~40min,20%~20%B;检测波长:270nm;流速:1mL/min;柱温:30℃;进样量:20μL。结果与讨论提取条件的优化结果溶剂的优化结果:分别用水、70%甲醇、70%乙醇溶液超声-微波协同萃取30min(n=3),结果表明70%甲醇提取木犀草素-7-β-D-葡萄糖甙的量较高,而木犀草素的量差异不明显,因此选择70%甲醇提取。溶剂体积分数的优化结果:分别用体积分数为40%、50%、60%、70%、80%、90%和纯甲醇溶液超声-微波协同萃取30min(n=3),结果表明,在甲醇体积分数70%时,木犀草素-7-β-D-葡萄糖甙和木犀草素的提取率随着甲醇浓度的增加而增加;但当甲醇体积分数在70%以上时,木犀草素葡萄糖甙的提取率呈现下降趋势,木犀草素没有明显的变化。木犀草素葡萄糖甙属于一种苷,分子量小,极性较大,当甲醇体积分数过高时,溶液极性降低,使得极性较强的木犀草素葡萄糖甙不易溶出,而木犀草素极性相对木犀草素葡萄糖甙小,影响不明显,因此实验选择70%甲醇作为提取溶剂。溶剂用量的优化结果:分别用10mL、20mL、50mL、80mL、100mL70%甲醇提取,结果表明溶剂体积在50mL时木犀草素葡萄糖甙和木犀草素的提取率最高,之后随着溶剂用量的增加,木犀草素葡萄糖甙和木犀草素的提取率趋于稳定,因此溶剂用量选用50mL 进行提取 。提取时间的优化结果:分别用70%甲醇超声-微波协同萃取20min、30min、40min、50min、60min(n=3),结果表明超声-微波协同萃取时间从20~40min的过程中木犀草素葡萄糖甙和木犀草素的提取率逐渐增加;而提取时间超过40min之后,提取率反而逐渐下降。超声-微波协同萃取时间太长,植物中大量细胞细胞破碎,使得大量粘性物质等进入提取液,溶剂杂质增多、粘度增大,影响了有效成分的溶出,有效成分含量反而减少,因此选择提取时间为40min。提取温度的优化结果:分别在40、45、50、55、60℃下用70%甲醇超声-微波协同萃取40min,实验表明,提取温度在50~60℃的范围内,木犀草素葡萄糖甙和木犀草素的提取率没有明显差异,考虑到温度太高容易破坏活性成分,因此选择提取温度为50℃。流动相的考察在实验过程中,流动相首先考察了甲醇-水、乙腈-水等度洗脱对酸浆超声-微波协同萃取样品溶液进行分离,乙腈-水作为流动相时,出峰较快,不能较好地把木犀草素葡萄糖甙和木犀草素与其他杂质成分分离;甲醇-水作为流动相时,出现峰形拖尾现象,分离效果不理想。为改善上述现象,改用0.1%乙酸代替水并采用梯度洗脱,经过反复筛选之后,最终确定流动相组成为 A -0.1%乙酸水溶液, B -甲醇,洗脱程序为0~30 min , 3%~5% B;30~35 min ,5%~20% B ;35~40 min 20%~3% B,木犀草素葡萄糖甙和木犀草素和其他杂质成分能够很好的分离,得到较理想的色谱图。对照品溶液和酸浆萃取样品的HPLC-DAD 分析下图分别显示了在上述的色谱条件下,采用 DAD 进行检测得到的两种混合对照品及酸浆萃取样品的 HPLC 分离色谱图。图1色谱图中木犀草素葡萄糖甙和木犀草素的保留时间分别为18.74min, 26.87min,根据保留时间判断,图2中的 a、b 色谱峰分别初步鉴定为木犀草素葡萄糖甙和木犀草素。图3、4分别显示了混合对照品和酸浆萃取物中保留时间18.74min, 26.87min 的色谱峰进行 DAD 检测后得到的光谱图,木犀草素葡萄糖甙和木犀草素 UV 光谱图形状相似,出现 二个峰,木犀草素葡萄糖甙峰位置分别为:221nm,270nm,木犀草素峰位置分别为:226nm,276nm,由于木犀草素葡萄糖甙比木犀草素多了一个 β-D-吡喃葡萄糖基团,木犀草素葡萄糖甙二个峰位置都发生了蓝移,样品中二个峰的光谱图与
http://www.greenherbs.com.cn/bbs/dispbbs.asp?boardid=2&Id=7651724918 唑吡坦杂质A CIV Zolpidem Related Compound A CIV 对照品/标准品1724907 酒石酸唑吡坦 CIV Zolpidem Tartrate CIV 对照品/标准品1724893 唑吡坦 CIV Zolpidem CIV 对照品/标准品1724805 盐酸唑拉西泮 Zolazepam Hydrochloride 对照品/标准品1724769 硫酸锌 Zinc Sulfate 对照品/标准品1724747 氧化锌 Zinc Oxide 对照品/标准品1724689 齐留通杂质C Zileuton Related Compound C 对照品/标准品1724678 齐留通杂质B Zileuton Related Compound B 对照品/标准品1724667 齐留通杂质 A Zileuton Related Compound A 对照品/标准品1724656 齐留通 Zileuton 对照品/标准品1724532 齐多夫定杂质C(胸腺嘧啶) Zidovudine Related Compound C (thymine) 对照品/标准品1724521 齐多夫定杂质B Zidovudine Related Compound B 对照品/标准品1724500 齐多夫定 Zidovudine 对照品/标准品1724317 扎西他滨杂质A Zalcitabine Related Compound A 对照品/标准品1724306 扎西他滨 Zalcitabine 对照品/标准品1724000 盐酸育亨宾 Yohimbine Hydrochloride 对照品/标准品1722005 木糖 Xylose 对照品/标准品1721002 盐酸赛洛唑啉 Xylometazoline Hydrochloride 对照品/标准品1720600 木糖醇 Xylitol 对照品/标准品1720429 盐酸赛拉嗪 Xylazine Hydrochloride 对照品/标准品1720407 赛拉嗪 Xylazine 对照品/标准品1720203 呫吨酮 Xanthone 对照品/标准品1720000 呫吨酸 Xanthanoic Acid 对照品/标准品1719102 华法林杂质 A Warfarin Related Compound A 对照品/标准品1719000 华法林 Warfarin 对照品/标准品1717708 牡荆素(牡荆甙) Vitexin 对照品/标准品1717504 含量测定系统适用性用维生素D Vitamin D Assay System Suitability 对照品/标准品1716002 维生素A Vitamin A 对照品/标准品1715000 硫酸紫霉素 Viomycin Sulfate 对照品/标准品1714528 长春瑞滨杂质A Vinorelbine Related Compound A 对照品/标准品1714506 酒石酸长春瑞滨 Vinorelbine Tartrate 对照品/标准品1714007 硫酸长春新碱 Vincristine Sulfate 对照品/标准品1713004 硫酸长春碱 Vinblastine Sulfate 对照品/标准品1711508 阿糖腺苷 Vidarabine 对照品/标准品1711472 维替泊芬杂质A Verteporfin Related Compound A 对照品/标准品1711461 维替泊芬 Verteporfin 对照品/标准品1711440 维拉帕米杂质F Verapamil Related Compound F 对照品/标准品1711439 维拉帕米杂质E Verapamil Related Compound E 对照品/标准品1711428 维拉帕米杂质D Verapamil Related Compound D 对照品/标准品1711406 维拉帕米杂质B Verapamil Related Compound B 对照品/标准品1711304 维拉帕米杂质A Verapamil Related Compound A 对照品/标准品1711224 维库溴铵杂质F Vecuronium Bromide Related Compound F 对照品/标准品1711202 盐酸维拉帕米 Verapamil Hydrochloride 对照品/标准品1711188 维库溴铵杂质C Vecuronium Bromide Related Compound C 对照品/标准品1711177 维库溴铵杂质B Vecuronium Bromide Related Compound B 对照品/标准品1711166 维库溴铵杂质A Vecuronium Bromide Related Compound A 对照品/标准品1711155 维库溴铵 Vecuronium Bromide 对照品/标准品1711133 赖氨加压素 Lypressin 对照品/标准品1711100 加压素 Vasopressin 对照品/标准品1711009 香草醛熔点标准品 Vanillin Melting Point Standard 对照品/标准品1710006 香草醛 Vanillin 对照品/标准品1709018 Vancomycin B with Monodechlorovancomycin 对照品/标准品1709007 盐酸万古霉素 Vancomycin Hydrochloride 对照品/标准品1708795 缬沙坦杂质 C Valsartan Related Compound C 对照品/标准品1708784 缬沙坦杂质 B Valsartan Related Compound B 对照品/标准品1708773 缬沙坦杂质 A Valsartan Related Compound A 对照品/标准品1708762 缬沙坦 Valsartan 对照品/标准品1708751 戊柔比星分离度用混合物 Valrubicin Resolution Mixture 对照品/标准品1708730 戊柔比星 Valrubicin 对照品/标准品1708729 丙戊酸杂质A Valproic Acid Related Compound A 对照品/标准品1708718 丙戊酸杂质 B Valproic Acid Related Compound B 对照品/标准品1708707 丙戊酸 Valproic Acid 对照品/标准品1708503 L- 缬氨酸 L-Valine 对照品/标准品1708015 D-缬更昔洛韦 D-Valganciclovir 对照品/标准品1708004 缬更昔洛韦盐酸盐 Valganciclovir Hydrochloride 对照品/标准品1707908 缬草烯酸 Valerenic Acid 对照品/标准品1707894 万乃洛韦杂质G Valacyclovir Related Compound G 对照品/标准品1707883 万乃洛韦杂质F Valacyclovir Related Compound F 对照品/标准品1707872 万乃洛韦杂质E Valacyclovir Related Compound E 对照品/标准品1707861 万乃洛韦杂质D Valacyclovir Related Compound D 对照品/标准品1707855 万乃洛韦杂质C Valacyclovir Related Compound C 对照品/标准品1707839 盐酸万乃洛韦 Valacyclovir Hydrochloride 对照品/标准品1707806 熊去氧胆酸 Ursodiol 对照品/标准品1706701 C13尿素 Urea C 13 对照品/标准品1706698 尿素 Urea 对照品/标准品1706009 乌拉莫司汀 Uracil Mustard 对照品/标准品1705800 阿糖尿苷 Uracil Arabinoside 对照品/标准品1705505 十一烯酸 Undecylenic Acid 对照品/标准品1705323 泛癸利酮杂质A Ubidecarenone Related Compound A 对照品/标准品1705312 系统适用性试验用泛癸利酮 Ubidecarenone for System Suitability 对照品/标准品1705301 泛癸利酮 Ubidecarenone 对照品/标准品1705006 L- 酪氨酸 L-Tyrosine 对照品/标准品1704003 泰洛沙泊 Tyloxapol 对照品/标准品1703850 酒石酸泰洛星 Tylosin Tartrate 对照品/标准品1703805 泰洛星 Tylosin 对照品/标准品1702008 氯筒箭毒碱 Tuboc
请问各位大神,高效液相测得给一个峰出峰时间与对照品[color=#333333]HPLC[/color][color=#333333]法在做欧前胡素的阴性对照,然后在和欧前胡素对照品溶液出峰时间差半分钟地方出现一个峰,请问各位老师这算有干扰么?[/color]
木犀草素(luteolin),别称草木犀、黄示灵等,大多以糖苷的形式广泛存在于多种中药材、天然药用植物[1]及蔬菜[2]中的一种黄酮类化合物,是一种天然色素成分,可以作为食用色素添加于食品中。木犀草素的化学名为3′,4′,5,7-四羟基黄酮(3′,4′,5,7-tetrahydroxyflavone),物理状态为淡黄色结晶状粉末,熔点为330 ℃,包含4个酚羟基,具有弱酸性,可溶于碱性溶液中,因脂溶性高而难溶于水,从而阻碍了其在体内的吸收与利用[3]。木犀草素具有抗炎和抗菌[4-5]、抗氧化[6]、抗肿瘤[7]、神经保护[8]、抑制肺纤维化[9]及肺癌[10-11]和心血管疾病[12]等多种药理作用。由于水溶性差(仅为6.0 mg/L)、生物利用度率低等原因限制了其成药性和临床应用。针对这一问题,近年来许多学者开展了增加木犀草素溶解度的研究,如微球[13]、纳米胶束[14]、金属配合物[15]、自微乳[16]、脂质体[17]等,并明显提高了其生物利用度,这表明木犀草素的肠道渗透性不是限制其生物利用度的关键因素,其属于生物药剂学系统II类药物。因此,采用制剂技术提高木犀草素的溶解性是可以改善其成药性和生物利用度的,将有利于推广其临床应用。然而上述开发的剂型仍存在诸多的缺点,如工艺复杂、载药量低、生物安全性差、成本高等,难以大范围推广应用。近年来,逐步发展成熟的纳米混悬剂[18]作为一种新剂型,与传统纳米制剂相比,它具有载药量高、溶出度高、添加剂用量少、易于放大生产等优点。因此,本实验尝试将难溶性木犀草素制备成纳米混悬剂以提高其水溶性和生物利用度,改善其成药性和临床优势。 为此,本实验首先采用微沉淀-高压匀质法制备口服木犀草素纳米混悬剂(luteolin nano-suspension,LNS),并以纳米粒的粒径、稳定性、多分散性指数(polydispersity index,PDI)、ζ电位等为考察指标,采用单因素考察法筛选LNS的稳定剂和最优药物-稳定剂比;接着,对LNS的理化性质进行考察,并分析其物理状态和体外溶出行为;最后通过大鼠外翻肠模型考察药物在肠道不同部位的吸收转运情况,探索药物在肠道内的吸收速率和最佳部位,预测纳米混悬剂可能存在的体内吸收行为,既可以用于木犀草素口服给药的潜在剂型,也为其进一步加工成其他剂型研究提供基础。 1 仪器与材料 1.1 仪器 ZNCL-BS180型恒温磁力搅拌器,北京市永光明医疗仪器有限公司;AL104-1C型精密分析天平,上海鼎科科学仪器有限公司;NS1001L型高压匀质机,意大利GEA [url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]NIR[/color][/url]o Soavi公司;Nanotrac wave II型激光粒度仪型激光粒度仪,美国麦奇克有限公司;LC3100型高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱仪[/color][/url],安徽皖仪科技股份有限公司;ZWY-103D型恒温振荡仪,上海智诚分析仪器制造有限公司;H1650-W型医用离心机,湖南湘仪实验室仪器开发公司;DZF-6030型真空干燥箱,上海精宏实验设备有限公司。JEOL 2010型透射电子显微镜(TEM),日本JEOL公司。 1.2 试剂 木犀草素原料药,批号JZ19021403,质量分数97.0%,南京狄格尔医药科技有限公司;木犀草素对照品,批号ps1032-0025,HPLC质量分数≥98%,成都普思生物科技有限公司;十二烷基磺酸钠(sodium dodecyl sulfonate,SDS),医药级,河南圣拓实业有限公司;泊洛沙姆188(Poloxamer 188,Pluronic,F68),医药级,西安天正药用辅料有限公司;维生素E聚乙二醇琥珀酸酯(D-α-tocopherol polyethylene glycol 1000 succinate,TPGS),医药级,上海惠诚生物科技有限公司;二甲基亚砜(dimethyl sulfoxide,DMSO),分析纯,天津市德恩试剂有限公司。 1.3 动物 SD大鼠购买于河南省实验动物中心,体质量(200±20)g,合格证号:SCXK(豫)2017-0001。所有动物实验均经过河南大学动物伦理委员会审核批准(HUSOM2019-216)。 2 方法与结果 2.1 LNS的制备 2.1.1 LNS中稳定剂的选择 将40 mg木犀草素原料药超声溶解于1 mL的DMSO中作为有机相,再取等量的稳定剂(SDS、F68、TPGS)溶解于纯水中(作为水相,或称反溶剂相);在室温下,将有机相通过注射器快速注入转速为1 800 r/min的反溶剂相中,继续搅拌10 min,得到预混悬剂;将预混悬剂转移至高压匀质机中,分别以20.0、50.0、80.0 MPa的压力循环匀质5、5、25次,得到LNS。 利用动态光散射仪分别考察LNS的粒径、多分散系数(polydispersity index,PDI)、表面电荷(ζ电位)和稳定性。本实验以不同稳定剂(SDS、F68、TPGS)制备的LNS粒径大小、PDI、ζ电位结果如表1所示。3种稳定剂所制备的粒径均在100~500 nm。以SDS为稳定剂制备的纳米混悬剂粒径最大,以F68为稳定剂制备的纳米混悬剂PDI最大,以TPGS为稳定剂制备的纳米混悬剂ζ电位最大,但是3者没有较大的差异,因此对于预测稳定性来说,上述结果难以判断哪个稳定剂制备的LNS会有良好的贮存稳定性。 因此,本实验又对各种条件的贮存稳定性进行了研究,结果见图1。以SDS、F68为稳定剂制备的纳米混悬剂在1周内粒径呈现持续增长的趋势,而以TPGS为稳定剂制备的LNS粒径未出现明显变动,由此可知,本实验中以TPGS为稳定剂制备的LNS具有较好的物理稳定性。 2.1.2 LNS中药物-稳定剂质量比的筛选 将40 mg的木犀草素原料药超声溶解于1 mL的DMSO中作为有机相,再分别按照木犀草素与TPGS的质量比为1∶2、1∶1、2∶1称取TPGS,溶解于水中,得到反溶剂相;再按上述工艺制备LNS,得到不同药物-稳定剂质量比的LNS。利用动态光散射仪分别考察纳米混悬剂的粒径、分布、ζ电位和稳定性。不同药物-稳定剂比制备的LNS的理化性质研究结果见表2和图2。如表2所示,3种不同药物-稳定剂比制备的LNS的粒径分别为(289.3±6.6)、(210.7±2.0)、(34.6±3.7)nm,3种LNS的PDI接近,1∶2时ζ电位最大,2∶1时ζ电位没测到。虽然药物与稳定剂的质量比为2∶1时,其粒径与1∶2、1∶1时相差较大,但是粒径难以反映稳定性情况。因此,接下来考察了1∶2、1∶1、2∶1 3种不同比例下制备的LNS的稳定性,结果如图2所示。当药物-稳定剂比为2∶1和1∶2时,在2周内粒径变化幅度都较为明显,说明其稳定性表现均极差;而当药物-稳定剂比为1∶1时,制备的纳米混悬剂的粒径基本保持稳定,表明其稳定性较好。因此,本实验最终选用药物-稳定剂比为1∶1。 2.1.3 最优制备处方和方法的确定 依照LNS的稳定剂及药物-稳定剂比的筛选结果,初步确定LNS的最优制备处方与方法如下:将精密称取40 mg的木犀草素原料药超声溶解于1 mL的DMSO中作为有机相;将40 mg TPGS搅动溶解于40 mL纯水中作为水相,将有机相快速注入转速为1 800 r/min的水相中,搅动10 min,得到预混悬剂;将制备的预混悬剂倒入高压匀质机的导入槽中,分别以20.0、50.0、80.0 MPa的压力,分别循环匀质5、5、25次,得到LNS。重复制备3批,以粒径、PDI和ζ电位考察制剂处方和制备工艺的稳定性。 2.2 LNS的表征 2.2.1 粒径、ζ电位及形态分析 将最优处方制备的3批LNS分别通过激光粒度分析仪测定其粒径、PDI、ζ电位,结果LNS的粒径为(209.00±3.24)nm(n=3),PDI都低于0.228±0.013(n=3),粒径分布图见图3;ζ电位值为(?16.80±0.27)mV (n=3),较小的PDI和绝对值较大的ζ电位,意味着LNS可能具有较好的长期稳定性[19]。 再取适量的LNS加蒸馏水稀释到适当倍数后,滴在覆有支持膜的铜网上,自然环境下干燥后,通过TEM观察其形态特征及大小,并成像,结果见图4。LNS呈现均匀分散的球形或椭圆形颗粒,粒径约为180 nm,比动态光散射测定结果较小,这可能是由于TEM样品为干燥品,导致粒子外层亲水部分失水而收缩[20]。 2.2.2 储存稳定性 将制备的LNS分别放在4 ℃和室温环境中,在预定的时间点取样,通过激光粒度分析仪测定其粒径和PDI,连续考察14 d,每个样品平行操作3份,结果见表3。LNS在4 ℃和室温下储存2周后,粒径和PDI稍有增加,但变化范围都较小,说明该LNS的储存稳定性较好。 2.2.3 体外胃肠环境中的稳定性 以pH 1.2和pH 6.8的缓冲溶液模拟胃液和肠液,将制备的LNS分别以1∶1与上述2种缓冲溶液混合,并于37 ℃水浴中放置,在预定的时间点0、2、4、6、8、12、24 h时取样,通过激光粒度分析仪测定其粒径,连续考察24 h,每个样品平行操作3份,结果见表4。在2种37 ℃的缓冲溶液中孵育24 h内,LNS的粒径和PDI几乎无变化,表明LNS在2种环境中能保持稳定,这表示LNS口服给药后,在经胃肠道给药时能保持良好的稳定性,这有利于木犀草素到达肠道后仍以纳米晶存在,从而有利于木犀草素的快速释放而获得较高的生物利用度。 2.2.4 纳米混悬剂的物理状态研究 本实验选用DSC来确定LNS中的木犀草素晶型是否发生了改变,测试样品有木犀草素、TPGS、木犀草素与TPGS的物理混合物和LNS。以空铝盘作为空白对照,分别精密称取3~5 mg的木犀草素、TPGS、物理混合物(木犀草素+TPGS)、LNS干粉放于差式扫描量热分析(differential scanning calorimetry,DSC)仪中,N2流(40 mL/min)保护下,以10 ℃/min升温速度持续升温,升温范围设置为40~600 ℃,记录差式扫描量热分析图谱,所有测试样品重复分析3批,结果见图5。木犀草素和LNS、物理混合物均是结晶,其熔融温度为339.38 ℃,稳定剂对木犀草素的熔融温度基本无影响。这表明LNS中的木犀草素仍处于结晶状态,稳定剂的存在不会改变木犀草素的晶型。在木犀草素和LNS中,在50~150 ℃出现了1个宽峰,这可能是由于药物吸收了水分造成的。 再分别称取适量的木犀草素、TPGS、物理混合物(木犀草素+TPGS)、LNS置于X射线粉末衍射(X-ray powder diffraction,XRPD)仪中,以步进测定方式,散射角扫描范围设为5°~60°,电压设为40 kV,电流为30 mA,结果见图6。由图6可知,木犀草素在19.12、23.20、26.32 ℃有3个衍射峰,衍射峰的峰形较为尖锐,峰值较高,表明木犀草素的晶型为结晶型。稳定剂TPGS在15.72、17.48、22.86、25.60、29.26 ℃有衍射特征峰。制备成纳米混悬剂后,虽然LNS图谱中木犀草素的特征峰有所减弱,但与木犀草素相比,在相应位置特征峰均存在,进一步证实制备成LNS后木犀草素并未显著改变晶型,说明稳定剂的加入不会影响木犀草素的晶型,这与DSC分析的结果一致。 2.3 平衡溶解度与过饱和溶出度测试 为了测定木犀草素的平衡溶解度与木犀草素纳米混悬剂的过饱和溶出度,本实验参考文献方法[21]建立了HPLC法。 2.3.1色谱条件 色谱柱为Sino Chrom ODS-BP色谱柱(250 mm×4.6 mm,5 μm);流动相为甲醇-0.3%磷酸水溶液(60∶40);柱温30 ℃;检测波长350 nm;体积流量1 mL/min;进样量10 μL。 2.3.2对照品溶液的配制 精密称取木犀草素对照品2.50 mg,放入100 mL棕色量瓶中,以适量色谱甲醇使之完全溶解,并定容至刻度线,摇匀得到质量浓度为25 μg/mL的木犀草素对照品储备液。 2.3.3 线性关系考察 采用色谱甲醇稀释成质量浓度分别为0.5、1.0、2.0、5.0、7.0、10.0 μg/mL系列的木犀草素对照品溶液,按“2.3.1”项下色谱条件进行分析,以对照品质量浓度为横坐标(X)、峰面积为纵坐标(Y)进行线性回归,得线性回归方程为Y=44 670 X-2 498.3,R2=0.999 8,结果表明木犀草素在0.5~10.0 μg/mL线性关系良好。 2.3.4 专属性、精密度和准确度考察 在建立的HPLC色谱条件下,木犀草素色谱峰不会受pH 1.2和pH 6.8的溶出介质、稳定剂TPGS、Tyrode液以及肠吸收液中所有成分的干扰(图7),表明本实验所建立的含量测定方法具有较好的专属性,能够满足体外溶出和肠吸收试验中木犀草素的含量测定要求。另外,其精密度实验的RSD为1.2%,高、中、低3个质量浓度的样品加样回收率在99.67%~101.47%,RSD均小2%,符合《中国药典》2020年版的规定。 2.3.5 平衡溶解度的测定 为了测定木犀草素在pH值为1.2、6.8缓冲溶液中的平衡溶解度,取5 mL 2种缓冲溶液各3份于西林瓶中,加入过量的木犀草素,将西林瓶置于恒温振荡箱中,在温度为37℃,转速为75 r/min条件下振荡24 h。取出各样品,3 000 r/min下离心10 min后取上清液,然后用0.2 μm滤膜滤过,取续滤液于进样瓶中,按照“2.3.1”项下色谱条件进样测定,并计算木犀草素的平衡溶解度,结果可知,木犀草素在pH值为1.2、6.8的缓冲溶液中的平衡溶解度分别为(3.83±0.23)、(7.81±0.13)μg/mL。 2.3.6 过饱和溶出度的测定 为了考察LNS体外溶出行为,参照《中国药典》2020年版中桨法进行。具体操作如下:在智能溶出仪中,以500 mL模拟胃液为溶出介质,温度为37℃,桨旋转速度为75 r/min,将30 mL LNS加入溶出介质中,以相同质量浓度的木犀草素乙醇溶液作为对照,二者均平行操作3份。以药物刚接触溶出介质开始计时,分别于5、15、30、60、120、130、150、180、240、360、480 min时取样4 mL,取完样后立即补充4 mL相应的新鲜溶出介质。另外,于120 min取样后,每个溶出杯中分别加入适量的Na3PO4溶液,调节pH值为6.8,以模拟肠液。将所取样品溶液经0.2 μm微孔滤膜滤过,取续滤液置于进样瓶中,照“2.3.1”项下色谱条件测定,计算累积溶出度,结果见图8。为了测定过饱和溶出水平,在整个实验过程中,介质中药物的质量浓度都应保持远远大于药物的饱和溶解度[22]。结果如图8所示,在pH 1.2和pH 6.8时,木犀草素-原料药的过饱和溶出始终低于对应的平衡溶解度,LNS的过饱和溶出始终高于对应的平衡溶解度,说明制剂的过饱和度高;在溶出介质的pH值调为6.8后,过饱和溶出水平明显下降,在150 min后过饱和溶出水平逐渐稳定,说明LNS能维持较高的过饱和溶出水平。 结果表明,LNS较木犀草素原料药具有明显优势,其饱和溶出度约是木犀草素原料药的15倍,过饱和度高并能维持较长时间,可以延缓药物在体内因析出晶体而沉淀的过程,从而使稳定剂在较小用量下也能保证药物分子成溶解态,提高了原料药的溶解度,有利于增加其生物利用度[23]。 2.4 小肠吸收实验 为了探索LNS对木犀草素在胃肠道的吸收部位和吸收速率的影响,采用外翻肠囊法[24]研究LNS在肠道不同肠段的吸收特征,以探究药物在肠道内的最佳吸收部位。 2.4.1 对照品溶液的制备 精密量取“2.3.1”项下相应体积的储备液,置于50 mL棕色量瓶中,用Tyrode液定容至刻度,摇匀,配制出质量浓度为1、2、4、8、16、32、40 μg/mL木犀草素对照品溶液。 2.4.2 线性关系考察 按照“2.3.1”项下色谱条件测定,以木犀草素对照品质量浓度为横坐标(X),峰面积为纵坐标(Y)进行线性回归,得到回归方程为Y=45 475 X-19 575,R2=0.999 6,结果表明木犀草素在1~40 μg/mL线性关系良好。 2.4.3 供试品溶液的制备 大鼠按实验质量浓度随机分为3组,每组4只,实验前12 h禁食,自由饮水。颈椎脱臼处死,打开腹腔,小心分离出小肠,分别截取十二指肠、空肠、回肠、结肠相应肠段各10 cm,用生理盐水冲洗至无内容物流出。将肠段放入37 ℃ Tyrode液中,冲洗,在不损伤肠管的情况下,小心剥离肠表面的脂肪及血管,取出,用滤纸吸干表面水分。 将肠管一端结扎,用光滑的玻璃棒外翻,用Tyrode液冲洗过后,向不同肠段中注入3 mL的空白Tyrode液后将另一端也进行结扎形成囊状的肠管。将肠管放入盛有Tyrode液的烧杯中,实验中始终保持37 ℃的恒温,并不断通入95% O2/5% CO2的混合气体。平衡5 min后,将烧杯中的液体倒出,分别加入不同质量浓度(0.15、0.30、0.60 mg/mL)的木犀草素及LNS药液。以肠囊和药液接触时开始计时,取样时间点分别为15、30、45、60、75、90、105、120 min,每个时间点从肠囊内取样500 μL,同时补充同温同体积的空白Tyrode液。待试验结束后,将各段肠囊置于空白Tyrode液中孵育1 h,以清除掉肠囊及肠组织中残留的药物;随后将上述用于木犀草素和LNS吸收实验的各肠段互换,再按上述操作同法重复试验,以进行自身对照交叉试验的后段实验。取上述肠吸收液,加入甲醇500 μL,超声混匀,15 898×g离心(离心半径6.32 cm)2次,每次15 min,取上清液用0.2 μm滤膜滤过,取续滤液适量即得。 按照“2.3.1”项下色谱条件测定,并计算药物在各时间点的累积吸收量(Q,μg)和药物吸收速率常数[Ka,μg/(mincm2)],结果见图9。 由公式计算不同质量浓度下木犀草素在各个时间点的累积吸收量(Q)。 Q是每个时间点木犀草素的累积吸收量,Ci是每个时间点的实际检测质量浓度,V1是加入肠囊内的空白Tyrode液,V2是每次取样的体积 由图9可知,通过对比2种制剂在各肠段中不同质量浓度的药物吸收情况,可以发现药物的同一时间点的吸收量表现出质量浓度相关性。相同质量浓度下,在各肠段中2制剂组吸收量相比,LNS组的药物累积吸收量显著大于木犀草素溶液组,表明LNS相比于木犀草素溶液能够促进药物在肠道的吸收。 根据小肠内(4个肠段)的Q值,通过线性拟合,由公式Ka=L(斜率)/A(肠管平铺面积)求得吸收速率常数(Ka)和相关系数(R2),结果见表5。2种制剂中木犀草素在肠道的不同部位中的吸收速率大小顺序均为十二指肠>空肠>回肠>结肠,这可能归因于十二指肠和空肠肠段的吸收面积较大;这一结果还表明LNS并没有改变木犀草素在肠道内的主要吸收部位和机制。对比相同质量浓度、相同肠段中2种制剂的吸收情况可以发现,LNS中木犀草素的吸收速率显著高于木犀草素溶液的情况,尤其是十二指肠和空肠中LNS和木犀草素溶液的木犀草素吸收速率差异更加明显,这表明LNS可以增加木犀草素的肠吸收,且十二指肠和空肠是主要吸收部位。 另外,还可以发现2种制剂在每一肠段中的吸收速率都存在显著的质量浓度相关性(P<0.01),但是2种制剂在同一肠段中的吸收速率随质量浓度增加而提高的程度有明显差异,即木犀草素溶液随质量浓度的增加,各肠段中吸收速率增幅增大,而LNS随质量浓度的增加,各肠段中吸收速率增幅减小,这些结果表明2种制剂在各肠段中的吸收均有质量浓度相关性,但其吸收速率与质量浓度之间均存在非线性关系,且仅在Ka<0.052时,木犀草素的肠吸收过程可能只受木犀草素溶解度限制,而不受吸收速度限制。然而,木犀草素的实际口服吸收情况是否符合上述规律以及其具体吸收机制如何,将有待于后期开展体内外吸收途径探索和体内药动学研究来进一步证实。 3 讨论 3.1 稳定剂的选择及药物-稳定剂比的确定 由于不同的稳定剂中化学基团的差异,导致稳定剂与药物微粒之间的分子间作用力以及胶粒间的作用力都有明显差异,所以稳定剂种类会影响到纳米混悬剂的稳定性[25]。因此,本实验首先以粒径和稳定性为考察指标,通过单因素筛选法优化了LNS的稳定剂种类,并确定了以TPGS作为稳定剂能达到较好的预期效果;考虑到稳定剂用量对稳定效果的影响[26],随后本实验又考察了药物-稳定剂比对纳米混悬剂的粒径、稳定性、PDI、ζ电位的影响,最终确定最佳药物-稳定剂比为1∶1。 3.2 LNS体外分析方法的建立及研究 3.2.1 波长的选择 木犀草素对照品与稳定剂TPGS在紫外波长200~800 nm扫描,结果显示木犀草素在207、254、350 nm 3处波长处有强吸收;而TPGS在219、286 nm显示出强吸收,350 nm处没有显示出强吸收。为了排除稳定剂TPGS对木犀草素测定的干扰,选用350 nm作为木犀草素的测定波长。 3.2.2 Tyrode溶液的配制 在木犀草素的肠吸收情况研究中,虽有文献报道了外翻肠囊模型和在体单向肠灌流模型[27-29],但关于木犀草素及其制剂在大鼠不同肠段中的吸收情况鲜有报道,且大多数文献对其吸收情况所提甚少。 本实验采用离体外翻肠囊法,可操作性强、重复性好;能够保留较为完整的肠道组织和黏膜特性,其实验结果与机体药物吸收水平比较接近,具有说服力;但肠外翻肠囊法也存在缺点,如长时间暴露在体外,肠管没有血管和神经的控制,肠黏膜功能和形态会失去作用。因此,本研究为解决这一问题,利用Tyrode培养液改善肠管的存在环境,具体配制方法如下:将NaCl(8.0 g/L)、KCl(0.2 g/L)、CaCl2(0.2 g/L)、NaHCO3(1 g/L)、NaH2PO4(0.05 g/L)、MgCl2(0.1 g/L)、葡萄糖(1.0 g/L),用蒸馏水定容至1 000 mL,稀盐酸调pH值为7.2~7.4,由于CaCl2不好溶解,应在其他无机盐溶解完全后再加入,葡萄糖于临用前再加入。并且在实验过程中连续通入95% O2/5% CO2,保证了在实验期间肠管上肠黏膜的活性。实验证明用该模型了解药物的离体吸收,其结果可靠。 3.3 LNS的过饱和溶出 药物在纳米混悬剂中所处的物理状态关系着其粒径和溶出稳定性,通常无定形药物微粒具有较高的饱和溶出度,但其属于热力学不稳定状态,因此物理稳定性差,容易引起纳米混悬剂粒径分布发生变化,同时溶出速率和溶出度下降;而结晶型药物具有较好的热力学稳定性,随着其粒径的减小,其饱和溶出度会明显提高[30]。根据本实验对LNS中木犀草素物理状态的研究结果可知,本实验制备的LNS中木犀草素以结晶形式存在,这表明LNS可能存在稳定的粒径和溶出度。 在过饱和溶出实验中发现,相比于木犀草素原料药,LNS具有显著的长期高过饱和溶出水平,这可归因于LNS中药物以粒径远小于原料药的状态存在,正如开尔文定律所描述的小粒径药物具有高溶解度一样[31]。药物的长期高过饱和溶出水平将有助于避免或减少口服给药后因胃肠道pH变化而引起的析晶沉淀现象,从而增加药物的吸收速度和时间,提高药物的口服生物利用度。 综上所述,本实验制备的LNS,分散性和储存稳定性良好,方法也简单易行,本实验建立的木犀草素体外分析方法,经方法学验证可知,该方法快速、可靠、准确度高,适合LNS的体外溶出和外翻肠囊吸收实验研究。 同时,外翻肠实验表面,LNS能促进药物在肠道的吸收,可作为木犀草素口服给药的潜在剂型,也为其进一步加工成其他剂型研究提供坚实基础。同时,在木犀草素肠道吸收的具体机制方面还有很大的研究空间。
我是做中药检验的,以前用的对照品溶液都经0.45滤膜过滤后使用,还做过某两种对照品溶液的测试,峰面积差异不大。但最近我使用黄芩苷对照品时发现检品含量总是很高,反复测试才找到原因,过滤后对照品的峰面积为:3249725.000,没过滤的对照品峰面积为:4933873.000,差异很大,不知道各位朋友使用对照品溶液时是否也经过0.45滤膜过滤?有没有文件规定对照品溶液能否过滤?
在抗生素类的标准物质使用时,经常会遇到标准品和对照品的概念。关于这二者的区别,现在比较流行的说法是在做HPLC时使用的标准物质应为对照品。摘录典型观点如下:[B]“标准品都是按效价单位(或μg)计,以国际标准品进行标定。标准品的标示量是按生物活性来计算的,不是按纯度来标示,此种标示法对单组分或多组分物质均适用,尤适用于多组分物质,如乙酰螺旋霉素标准品,是由4种有效成分组成,若欲于一个纯度来标示其含量是不可能的,但用效价(即生物活性)来标示是可行的;对照品的标示量则必定是某单一组分的纯度指标。所以日常工作中,标准品和对照品在定量时是不可相互替代的。以罗红霉素为例,现今是国家标准品与对照品并存,以抗生素微生物检定法测其含量时,必须使用罗红霉素标准品;但以HPLC法测定其含量时,又必须使用罗红霉素对照品,不可混淆。”[/B]但是我见过一些行业标准,比方说HPLC测土霉素残留中,在说到标准液的配制时,写得就是“土霉素标准品”。难道这里面的“标准品”是“对照品”的错误用法?[em0716] 请大家发表一下看法
[color=#333333]HPLC法在做欧前胡素的阴性对照,然后在和欧前胡素对照品溶液出峰时间差半分钟地方出现一个峰,请问各位老师这算有干扰么?[/color]
AAbrine 相思豆碱O-Acetyl-3,6-di-O-β-D-xylopy-rano- astragaloside O-乙烯 3,6-双氧-β-D- 吡喃木糖基绵毛黄芪甙Acetylastragaloside 乙酰黄芪甙N-Acetyl-D-Glucosamine N-乙酰氨基葡萄糖糖6''-acetylhyperoside 6''-乙酰氧基金丝桃甙Acetylshikonin 乙酰紫草素14-Acetyltalatisamine Achyranthan 牛膝多糖Acobreting D 短距乌头碱丁Acobreting E 短距乌头碱戊Aconine 乌头原碱Aconitine 乌头碱Aconosine 爱康诺辛Actein 黄肉楠碱Actinodephnine Acuminatin Acuminatoside Adenanthin 腺华素Adenine 腺嘌呤Adenoside 腺苷Adynerin 欧夹竹桃甙乙Aescin 七叶皂甙Aesculetin 七叶素(七叶内脂,秦皮乙素)马栗树皮素Aesculin 七叶甙 马栗树皮甙Agaricic acid 落叶松覃酸Agaricus blazei murrill P.E 姬松茸提取物Agrimophol 仙鹤草酚Ajmalicine(δ-Yohimbine) 阿吗碱,δ-育亨宾碱,阿吗里新Ajmaline 阿马林Akebia saponin D 木通皂甙 DAlantolactone 土木香内酯Albopilosin A Aleuritic acid 苏式-紫胶桐酸Alfalfa P.E 紫苜蓿提取物Alizarin 茜素Alkaloids 罗勒生物碱Allantoin 尿囊素Allasecurinine 别一叶秋碱Allantolin Allicin 大蒜素α-Allocryptopine α-别隐品碱Alloisoimperatorin 别异欧前胡素Alloxanthoxyletin Allose 阿罗糖Aloeemodin 芦荟大黄素Aloe-saponol Aloin 芦荟甙Aloesin 芦荟苦素Aloperin 苦豆碱Alpinetin 山姜素Amentoflavone 1- Amino-cyclopropane-1-acid hydrochloride 1-氨基环丙烷-1-羧酸盐酸盐Amethystoidin A 香茶菜甲素Ampelopstin 福建茶素Amygdalase 苦杏仁酶Amygdalin 苦杏仁甙Aamylase 淀粉酶β-Amyrin β-香树脂醇α-Amyrin acetate α-香树脂醇乙酸乙酯β-Amyrin acetate β-香树脂醇乙酸乙酯β-Amyrin palmitate 棕榈酰β-香树酯Anabasine 毒藜碱Anagyrine 臭豆碱,安那吉碱Andrographis Paniculata P.E 穿心莲提取物Andrographolide 穿心莲内酯 穿心莲乙素Anemonin 白头翁素Angelica Dongquai P.E 当归提取物Anisodamine 山莨菪碱Anisodine 樟柳碱Anthocyanosides 花色素Anthraglycoside A 大黄素-6-甲醚-8-D-葡萄糖甙Anthraglycoside B 大黄素-8-D-葡萄糖甙Apigenin 芹菜甙元 芹菜素Apigenin-7-glucoside 芹菜甙元-7- 葡萄糖甙Apigenin-7-O-glucoside 芹菜甙元-7- O-葡萄糖甙Apigenin-8-O-glucoside 芹菜甙元-8- O-葡萄糖甙Apigenin-7-O-α-L-rhamnoside 芹黄素-7-O-α-L-鼠李糖甙Apiin 芹菜甙Apple Extract 苹果提取物Araloside A 楤木皂甙 AArbutin 熊果甙Arctigenin 牛蒡甙元Arctiin 牛蒡子甙Arctinal 牛蒡子醛Arctinol b 牛蒡子醇-bAristolactone 马兜铃内酯Aristolochic acid A, D 马兜铃酸A,DAristolochic acid B, C 马兜铃酸B,CArmillarisin A 亮菌甲素,假蜜环菌甲素Arteannuic acid 青蒿酸Arteannuin 青蒿素Artemisinin 青蒿提取物α-Artemether α-蒿甲醚β-Artemether β-蒿甲醚Artemetin 青蒿亭Artemisinic acid 春活力 青蒿酸Arundoin 芦竹素Asarinin 细辛脂素α-Asaron α-细辛醚β-Asarone β-细辛醚Asiatic acid 积雪草酸Asiaticoside 积雪草甙Astilbin 落新妇甙Astragaloside 黄芪甙Astragalin Astragaloside II 黄芪皂甙 IIAstragaloside IV 黄芪甲甙Astragalus Root P.E 黄芪提取物Atisine chloride Atractydin 苍术素Atractylon 苍术酮Atraotydin 苍术甙Atropine 阿托品Aucubin 桃叶珊瑚甙Avacularin 扁蓄甙
今天我把昨天的两份对照品储备液分别稀释成对照品溶液,顶空进样,但是第一份对照品溶液待测组分未出峰,第2份对照品溶液出峰正常,我想问下隔24小时以后再用待测组分都会挥发完全吗?为什么第二份正常出峰呀?对照品溶液里面的组分分别是乙醇,乙酸乙酯,丙酮,二氯甲烷,水为溶剂

[align=center][b]中风Ⅰ号合剂[/b][/align][align=center][b]医院制剂用药品的原料(药材)和成品的质量标准草案[/b][/align][b]一. 医院制剂用药品的原料(药材)的质量标准草案:[/b](1)大黄:本品为蓼科植物掌叶大黄Rheumpalmatuml.、唐古特大黄Rheumtanguticum.Maxim.exBalf. 或药用大黄RheumoffcihaleBaill. 的干燥根和根茎。秋末茎叶枯萎或次春发芽前采挖,除去细根,刮去外皮,切瓣或段,绳穿成串干燥或直接干燥。应符合中华人民共和国药典2015年版一部23页大黄项下的有关规定。(2)钩藤:为茜草科植物钩藤 [i]Unacaria rhynchophylla[/i](Miq.)Miq.ex Havil.、大叶钩藤[i] Uncaria macrophylla[/i] Wall.、毛钩藤[i]Uncaria hirsuta[/i] Havil.、华钩藤 [i]Uncaria sinensis[/i](Oliv.)Havil.或无柄果钩藤[i]Uncaria sessilifructus[/i] Roxb.的干燥带钩茎枝。秋、冬二季采收,去叶,切段,晒干。主产于[color=#333333]浙江、福建、广东、广西[/color][color=#333333]等省。[/color]应符合中华人民共和国药典2015年版一部257页钩藤项下的有关规定。(3)白芍:为毛莨科植物芍药Paeonia lactiflora PalL.的干燥根。夏、秋二季采挖,洗净,除去头尾和细根,置沸水中煮后除去外皮或去皮后再煮,晒干。主产于[color=#333333]浙江、安徽、四川等省。[/color]应符合中华人民共和国药典2015年版一部105页白芍项下的有关规定。 (4)夏枯草:为唇形科植物夏枯草Prunella vulgarisL.的干燥果穗。夏季果穗呈棕红色时采收,除去杂质,晒干。主产于[color=#333333]江苏、安徽、浙江、河南[/color]等省。应符合中华人民共和国药典2015年版一部280页夏枯草项下的有关规定。(5)浙贝母:为百合科植物浙贝母Fritillariathunbergii Miq.的干燥鱗茎。初夏植株枯萎时采挖,洗净。大小分开,大者除去芯芽,习称“大贝”;小者不去芯芽,习称“珠贝”。分别撞擦,除去外皮,拌以锻过的贝壳粉,吸去擦出的浆汁,干燥;或取鱗茎,大小分开,洗净,除去芯芽,趁鲜切成厚片,洗净,干燥,习称“浙贝片”。主产于[color=#333333]浙江、江苏、湖南[/color]等省。应符合中华人民共和国药典2015年版一部292页浙贝母项下的有关规定。(6)地龙:为钜蚓科动物参环毛蚓Pheretima aspergillum(E.Perrier)、通俗环毛蚓Pheretima vu1garis Chen、威廉环毛蚓Pheretima guillelmi(Michaelsen)或栉盲环毛蚓Pheretima pectinifera Michaelsen的干燥体。前一种习称“广地龙”,后三种习称“沪地龙”。广地龙春季至秋季捕捉,沪地龙夏季捕捉,及时剖开腹部,除去内脏和泥沙,洗净,晒干或低温干燥。[color=#333333]广地龙[/color][i]主产于[/i]广东、海南、广西、福建等省。沪[i]地龙主产于[/i]上海、浙江等省。应符合中华人民共和国药典2015年版一部122页地龙项下的有关规定。(7)石决明:为鲍科动物杂色鲍Haliotis diversicolor Reeve、皱纹盘鲍Haliotis discus hannai Ino、羊鲍Haliotis ovinaGmelin、澳洲鲍Haliotis ruber(Leach)、耳鲍Haliotis asinina Linnaeus或白鲍Haliotislaevigata(Donovan)的贝壳。夏、秋二季捕捞,去肉,洗净,干燥。主产于[color=#333333]浙江、福建、台湾、广东、海南、广西[/color]等省。应符合中华人民共和国药典2015年版一部91页石决明项下的有关规定。(8)鲜竹沥:为禾木科植物粉绿竹Phyllostachys glaucaMcClure、净竹Phyllostachysnuda McClure及同属数种植物的鲜杆经加热后自然沥出的液体,煮沸后,加适量防腐剂制得。主产于四川、江西等省。应符合中华人民共和国卫生部药品标准中药材第一册99页鲜竹沥项下的有关规定。[b]二.医院制剂用药品的成品的质量标准草案:[/b][align=center][b]中风Ⅰ号合剂[/b][/align][align=center][/align][b]【处方】 [/b]大黄60g 钩藤120g 白芍100g 夏枯草150g浙贝母90g 地龙100g 石决明240g 鲜竹沥100ml[b]【制法】 [/b]以上八味药材,除鲜竹沥,其余七味用水浸渍30分钟,煎煮两次,第一次1.5小时,第二次1小时,合并煎液,滤过,滤液静置24小时,取上清液浓缩至约800ml,加入鲜竹沥、甜菊苷、对羟基苯甲酸乙酯和苯甲酸,搅匀,过滤,滤液加水使成1000ml,灌装,灭菌,即得。[b]【性状】 [/b]本品为棕褐色液体,味微苦、甜。[b]【鉴别】 [/b](1)取本品20ml,加盐酸2ml,水浴加热30分钟,放冷,用乙醚振摇提取3次,每次25ml,合并乙醚液,挥干,残渣加甲醇1ml使溶解,作为供试品溶液。取大黄素对照品、大黄酚对照品及大黄酸对照品,加甲醇分别制成每1ml含0.2mg的溶液,作为对照品溶液。照薄层色谱法(中华人民共和国药典2015年版四部 通则0502)试验,吸取上述两种溶液各5μl,分别点于同一硅胶G薄层板上,以石油醚(30〜 60°C)-甲酸乙酯-甲酸(15:5:1)的上层溶液为展开剂,展开,取出,晾干,置紫外光灯(365nm)下检视。供试品色谱中,在与对照品色谱相应的位置上,显相同颜色的荧光斑点。(2)取本品20ml,用乙醚振摇提取2次,每次20ml,弃去乙醚液,水液用水饱和的正丁醇振摇提取2 次,每次20ml,合并正丁醇液,用水20ml洗涤1次,取正丁醇液,蒸干,残渣加甲醇2ml使溶解,作为供试品溶液。另取芍药苷对照品,加甲醇制成每1ml含2mg的溶液,作为对照品溶液。照薄层色谱法(中华人民共和国药典2015年版四部 通则0502)试验,吸取上述两种溶液各5μl,分别点于同一硅胶G薄层板上,以三氯甲烷-乙酸乙酯-甲醇-甲酸(8:1:2:0.1)为展开剂,展开,取出,晾干,喷以5%香草醛硫酸溶液,在105℃加热至斑点显色清晰。供试品色谱中,在与对照品色谱相应的位置上,显相同颜色的斑点。[b]【检查】 相对密度[/b] 应不低于1.02(中华人民共和国药典2015年版四部通则0601)。[b] pH值[/b] 应为4.0~6.0(中华人民共和国药典2015年版四部 通则0631)[b] 其他 [/b]应符合合剂项下有关的各项规定(中华人民共和国药典2015年版四部 通则0181)。[b]【含量测定】 [/b]照高效液相色谱法(中华人民共和国药典2015年版四部通则0512)测定[b]色谱条件与系统适应性试验[/b] 以十八烷基硅烷键合硅胶为填充剂,以乙腈-0.1%磷酸(14:86)为流动相,检测波长为230nm。理论塔板数按芍药苷峰计算应不低于3000。[b]对照品溶液的制备 [/b]精密称取芍药苷对照品适量,精密称定,加流动相制成每1ml含芍药苷80μg的对照品溶液,即得。[b]供试品溶液的制备[/b] 精密吸取样品1ml,置25ml量瓶中,用流动相稀释并定容至刻度,即为供试品溶液。[b]测定法[/b] 分别精密吸取对照品溶液与供试品溶液各10μl,注入液相色谱仪,测定,即得。本品含芍药苷(C[sub]23[/sub]H[sub]28[/sub]O[sub]11[/sub])不得少于0.35mg/ml。[b]【功能与主治】 [/b]平肝熄风、化痰通腑。用于各类急性期中风,半身不遂,肢体麻木,口眼喎斜,舌强语蹇。[b]【用法与用量】 [/b]口服,一日2次,一次50ml。或遵守医嘱。[b]【规 格】 [/b] 100ml/瓶。[b]【贮 藏】 [/b] 密封。[b]【有效期】 [/b]2年。[align=center][b]中风Ⅰ号合剂[/b][/align][align=center][b]医院制剂用药品的原料(药材)和成品的[/b][/align][align=center][b]质量标准草案起草说明[/b][/align][b]一.医院制剂用药品的原料(药材)的质量标准草案起草说明:[/b](1)大黄:同正文。 (2)钩藤:同正文。 (3)白芍:同正文。(4)夏枯草:同正文。 (5)浙贝母:同正文。 (6)地龙:同正文。(7)石决明:同正文。 (8)鲜竹沥:同正文。[b]二.临床用药品成品的质量标准草案起草说明:【名称】 [/b]中风Ⅰ号合剂 ZhongfengYihao Heji[b]【处方】[/b] 同正文。[b]【制法】 [/b]同正文。[b]【性状】 [/b]同正文。[b]【鉴别】 [/b]处方由8味中药材组成。本标准建立2项薄层色谱鉴别方中2味药材:大黄、白芍。【鉴别】(1)、(2)均试验了三批样品,并分别与对应的阴性样品进行了比较,均无干扰,且薄层色谱斑点清晰,表明方法可行。[b](1)系方中大黄的定性鉴别。[/b]以大黄素、大黄酚、大黄酸对照品鉴别方中大黄,通过阴性对照试验及三批样品的实验观察,阴性无干扰,专属性强,故选大黄素、大黄酚及大黄酸对照品作为鉴别指标,列入正文(见图1)。[img=,596,504]https://ng1.17img.cn/bbsfiles/images/2019/07/201907010912419216_3978_2166779_3.png!w596x504.jpg[/img][b](2)系方中白芍的定性鉴别。[/b]以芍药苷对照品鉴别方中白芍,通过阴性对照试验及三批样品的实验观察,阴性无干扰,专属性强,故选芍药苷对照品作为鉴别指标,列入正文(见图2)。[img=,690,649]https://ng1.17img.cn/bbsfiles/images/2019/07/201907010913524007_844_2166779_3.png!w690x649.jpg[/img][b]【含量测定】[/b]白芍为方中主药,据《本草拾遗》记载,具有[color=#333333]养血柔肝,缓中止痛,敛阴收汗[/color]的作用。本文采用HPLC法测定中风I号合剂中白芍所含有的芍药苷,在测定波长下,阴性无干扰,方法快捷,简便。因此,本文采用HPLC法测定芍药苷的含量,以达到控制中风I号合剂质量的目的。[b](一)方法[/b]照高效液相色谱法(中华人民共和国药典2015年版四部 0512)测定[b]色谱条件与系统适应性试验[/b] 以十八烷基硅烷键合硅胶为填充剂,以乙腈-0.1%磷酸(14:86)为流动相,检测波长为230nm。理论塔板数按芍药苷峰计算应不低于3000。[b]对照品溶液的制备[/b] 精密称取芍药苷对照品适量,精密称定,加流动相制成每1ml含芍药苷80μg的对照品溶液,即得。[b]供试品溶液的制备[/b] 精密吸取样品1ml,置25 ml量瓶中,用流动相稀释并定容至刻度,摇匀,即为供试品溶液。[b]测定法[/b] 分别精密吸取对照品溶液与供试品溶液各10μl,注入液相色谱仪,测定,即得。本品含芍药苷([color=#333333]C[sub]23[/sub]H[sub]28[/sub]O[sub]11[/sub][/color])的量不得少于0.35mg/ml。[b](二)方法学考察1 仪器与试药[/b]戴安U3000高效液相色谱仪;梅特勒XS205DU电子天平;艾科浦超纯水器。中风I号合剂由福建省南平市人民医院制剂室提供。芍药苷对照品(批号110736-201842,含量97.4%)购自中国食品药品生物检定研究院。乙腈为色谱纯;水为超纯水。[b]2 方法与结果2.1 色谱条件[/b]色谱柱:Welch Ultimate XB-C18(4.6mm×250mm,5μm);流动相:乙腈-0.1%磷酸(14:86)检测波长:230nm;流速:1.0mlmin[sup]-1[/sup];柱温:30 ℃;进样量:10μl理论塔板数:按芍药苷峰计算应不低于3000。[b]2.2 提取方法的选择 [/b]在供试品溶液的制备中,进行了直接稀释法、超声法的对比研究,结果两者无显著性差别,从操作简便快捷的角度选择直接稀释法,结果见表2。 表2 芍药苷不同提取方法含量测定结果比较 [table=594][tr][td] [align=center]提取方法[/align] [/td][td] [align=center]芍药苷含量(mg/ml)[/align] [/td][td] [align=center]平均含量(mg/ml)[/align] [/td][/tr][tr][td=1,2] [align=center]稀释法[/align] [/td][td] [align=center]0.6890[/align] [/td][td=1,2] [align=center]0.69[/align] [/td][/tr][tr][td] [align=center]0.6888[/align] [/td][/tr][tr][td=1,2] [align=center]超声法[/align] [/td][td] [align=center]0.6892[/align] [/td][td=1,2] [align=center]0.69[/align] [/td][/tr][tr][td] [align=center]0.6890[/align] [/td][/tr][/table][b]2.3 溶液的制备[/b]2.3.1对照品储备液的制备 精密称取芍药苷对照品10mg,置10ml量瓶中,用甲醇溶解并稀释至刻度,制得对照品储备液(0.974g/L芍药苷)。2.3.2 供试品溶液的制备 精密吸取样品1ml,置25ml量瓶中,用流动相稀释并定容至刻度,摇匀,即为供试品溶液。2.3.3 阴性对照溶液的制备 按处方比例制备不含芍药苷的阴性样品,同2.3.2制备方法制备阴性对照溶液。[b]2.4 线性关系考察[/b]将芍药苷对照品储备液逐步稀释,得到浓度分别为4.87,9.74,24.35,48.70,73.05,97.40μg/ml六个浓度的系列标准溶液,进样测定,结果见表3 [table][tr][td=3,1] [align=center]表3 芍药苷线性关系测定结果[/align] [/td][/tr][tr][td] [align=center]进样体积(μl)[/align] [/td][td] [align=center]芍药苷浓度(μg/ml)[/align] [/td][td] [align=center]峰面积(mAU*min)[/align] [/td][/tr][tr][td] [align=center]10[/align] [/td][td] [align=center]4.87[/align] [/td][td] [align=center]1.105[/align] [/td][/tr][tr][td] [align=center]10[/align] [/td][td] [align=center]9.74[/align] [/td][td] [align=center]2.252[/align] [/td][/tr][tr][td] [align=center]10[/align] [/td][td] [align=center]24.35[/align] [/td][td] [align=center]5.853[/align] [/td][/tr][tr][td] [align=center]10[/align] [/td][td] [align=center]48.70[/align] [/td][td] [align=center]11.909[/align] [/td][/tr][tr][td] [align=center]10[/align] [/td][td] [align=center]73.05[/align] [/td][td] [align=center]17.511[/align] [/td][/tr][tr][td] [align=center]10[/align] [/td][td] [align=center]97.40[/align] [/td][td] [align=center]23.240[/align] [/td][/tr][/table]以峰面积(Y)为纵坐标,以芍药苷浓度(X)为横坐标绘制标准曲线。结果表明,芍药苷在4.87~97.40μg/ml的范围内线性关系良好(见图3)[img=,611,350]https://ng1.17img.cn/bbsfiles/images/2019/07/201907010916394318_5586_2166779_3.png!w611x350.jpg[/img][img=,650,539]https://ng1.17img.cn/bbsfiles/images/2019/07/201907010916445229_4881_2166779_3.png!w650x539.jpg[/img][img=,631,383]https://ng1.17img.cn/bbsfiles/images/2019/07/201907010916515496_9756_2166779_3.png!w631x383.jpg[/img][img=,618,717]https://ng1.17img.cn/bbsfiles/images/2019/07/201907010916572812_7861_2166779_3.png!w618x717.jpg[/img][img=,646,703]https://ng1.17img.cn/bbsfiles/images/2019/07/201907010917032958_603_2166779_3.png!w646x703.jpg[/img][b]【功能与主治】 [/b] 同正文。[b]【用法与用量】 [/b] 同正文。[b]【规 格】 [/b] 同正文。[b] 【贮 藏】 [/b]同正文。[b]【有效期】 [/b]同正文。
大家好,中检所的维生素A对照品才开始提供,我们买来做实验后发现维生素A对照品出现三个峰,但是省所提供的数据是只有两个峰。咨询省所,问我们是否有光照破坏峰,对于这个我们不是很明白。药典附录里说如果对照品里有顺式的维生素A醋酸酯就不必做光照破坏,中检所的维生素A对照品说明书里说明其中反式维生素A醋酸酯占96.9%,那么就是有顺式的了吗?有没有做过的同行们,给予我们指导啦,谢谢了。
哪里可以买到符合USP标准的维生素C对照品?有USP证书的。
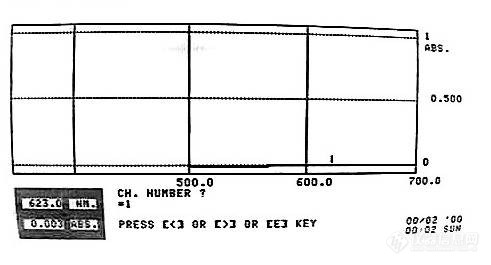
[align=center][b]鱼腥草注射液中增溶剂吐温80的限量测定[/b][/align][b]1 [/b][font=黑体]溶液的制备[/font][font=黑体]对照品溶液的制备[/font][font=宋体]取吐温[/font]80[font=宋体]对照品适量至[/font]100mL[font=宋体]量瓶中,精密称定,加重蒸水制成[/font]9.83 mgmL[sup]-1[/sup][font=宋体]的溶液。[/font][font=黑体]供试品溶液的制备[/font][font=宋体]精密移取鱼腥草注射液[/font]1mL[font=宋体]置[/font]100 mL[font=宋体]离心管中,加铵钴硫氰酸盐溶液[/font]15 mL[font=宋体](取硝酸钴[/font]6g[font=宋体],硫氰酸铵[/font]40 g[font=宋体],加水溶解并稀释至[/font]200 mL[font=宋体],摇匀,即得),精密加入二氯甲烷[/font]10mL[font=宋体],称定重量,置调速多用振荡器振荡[/font]30 min[font=宋体],取出,用二氯甲烷补足减失的重量,移至分液漏斗中,静置[/font]30 min[font=宋体],分取二氯甲烷层溶液,作为供试品溶液。[/font][b]2[/b][font=黑体]测定波长的选择[/font] [font=宋体]对照品溶液与供试品溶液经显色反应后,在[/font]400~ 700 nm[font=宋体]波长范围内分别进行扫描,结果发现吐温[/font]80[font=宋体]对照品溶液与鱼腥草注射液供试品溶液峰形一致,最大吸收波长一致,均为[/font]623 nm[font=宋体]。[/font][b]3[/b][font=黑体]方法学考察[/font] [font=黑体]专属性试验[/font][font=宋体]取空白溶液(水)、鱼腥草重蒸馏液(未加吐温[/font]80[font=宋体],作阴性对照)、对照品吐温[/font]80[font=宋体]、鱼腥草注射液照,按“供试品溶液的制备”项下方法显色后比较吸光度,结果在[/font]623 nm[font=宋体]波长处,空白溶液的吸光度为[/font]0.002[font=宋体],阴性对照溶液的吸光度为[/font]0.001[font=宋体],鱼腥草注射液和对照品均有较好吸收,表明鱼腥草注射液中其它组分对本试验测定结果无干扰,阴性对照溶液与空白溶液在[/font]623nm[font=宋体]波长处无吸收。见[/font]Fig.1[font=宋体]。[/font][align=center][img=,491,265]https://ng1.17img.cn/bbsfiles/images/2022/09/202209011436509666_4892_3527494_3.jpg!w491x265.jpg[/img][/align][align=center][b](A)[/b][/align][font='Times New Roman'][/font][align=center][img=,490,250]https://ng1.17img.cn/bbsfiles/images/2022/09/202209011437018380_6503_3527494_3.jpg!w490x250.jpg[/img][/align][font='Times New Roman'][/font][b](B)[/b][align=center][img=,490,267]https://ng1.17img.cn/bbsfiles/images/2022/09/202209011437115097_5280_3527494_3.jpg!w490x267.jpg[/img][/align][align=center] ([b]C)[/b][/align][align=center] [/align][align=center][img=,491,265]https://ng1.17img.cn/bbsfiles/images/2022/09/202209011437184190_6882_3527494_3.jpg!w491x265.jpg[/img][/align][align=center][b](D)[/b][/align][align=center]Fig.1 The ultraviolet spectra of the blank solution (A)[font=宋体]、[/font]negative control sample (B)[font=宋体]、[/font]tween80 control sample (C) and real sample (D)[/align][b] [/b][font=黑体]线性和范围[/font][font=宋体]精密量取吐温[/font]80[font=宋体]对照品溶液[/font]0.5[font=宋体],[/font]1.0[font=宋体],[/font]2.0[font=宋体],[/font]4.0[font=宋体],[/font]8.0[font=宋体],[/font]10mL,[font=宋体]置[/font]100 mL[font=宋体]离心管中,分别加入硫氰钴铵溶液[/font]15mL[font=宋体],精密加入二氯甲烷[/font] 10 mL[font=宋体],称定重量,置调速多用振荡器上振荡[/font][font=宋体](室温,[/font]88rpm[font=宋体])[/font]30 min[font=宋体],取出,用二氯甲烷补足减失的重量,移至分液漏斗中,静置[/font]30min[font=宋体],分取下层溶液,在[/font] 623 nm[font=宋体]波长处测定吸光度,以二氯甲烷为空白对照,以吸光度([/font][i]A[/i][font=宋体])对吐温[/font]80[font=宋体]对照品浓度([/font][i]C[/i][font=宋体])进行线性回归,绘制标准曲线,回归方程为[/font]:[i]A [/i]= 1.57×10[sup]-1[/sup][i]C[/i]+ 2.5×10[sup]-3[/sup][font=宋体],[/font][i]r[/i]= 0.9999[font=宋体]。结果表明,吐温[/font]80[font=宋体]在[/font]0.49 ~ 9.83 mgmL[sup]- 1[/sup][font=宋体]范围内与吸光度呈良好的线性关系。[/font][b] [/b][font=黑体]精密度试验[/font][font=宋体]取同一“供试品溶液的制备”项下的鱼腥草注射液供试品溶液,在[/font]623nm[font=宋体]波长下进行测定[/font]6[font=宋体]次,测定结果[/font]RSD[font=宋体]为[/font]0.4%[font=宋体],表明仪器精密度良好。[/font][font=黑体]重复性试验[/font][font=宋体]取同一批鱼腥草注射液[/font]6[font=宋体]份,按“供试品溶液的制备”项下制备供试品溶液,在[/font]623 nm[font=宋体]波长处测定,所测吐温[/font]80[font=宋体]含量的[/font]RSD[font=宋体]为[/font]0.5%[font=宋体],表明该方法重复性良好。[/font][font=黑体]稳定性试验[/font][font=宋体]取“供试品溶液的制备”项下的鱼腥草注射液供试品溶液,在[/font]623nm[font=宋体]波长下分别于[/font]0[font=宋体]、[/font]1[font=宋体]、[/font]2[font=宋体]、[/font]4[font=宋体]、[/font]8[font=宋体]、[/font]12h[font=宋体]测定吸光度,测定结果[/font]RSD[font=宋体]为[/font]0.5%[font=宋体],表明供试品溶液在显色后[/font]12 h[font=宋体]内稳定。[/font][font=黑体]回收率试验[/font][font=宋体]精密量取已知含量的鱼腥草注射液[/font]0.5mL [font=宋体]置[/font]100 mL[font=宋体]离心管中,共[/font]9[font=宋体]份,每[/font]3[font=宋体]份[/font]1[font=宋体]组,分别精密加入吐温[/font]80[font=宋体]对照品[/font]sd[sub]2[/sub][font=宋体]溶液[/font]1.1[font=宋体],[/font]1.3[font=宋体],[/font]1.6mL[font=宋体],按“供试品溶液的制备”项下的方法制备供试品溶液,在[/font]623 nm[font=宋体]波长下测定,测定吐温[/font]80[font=宋体]的平均回收率为[/font]99.2%[font=宋体],[/font]RSD[font=宋体]为[/font]0.4%[font=宋体]([/font][i]n[/i]=9[font=宋体])。[/font][b]4.[font=宋体]样品测定[/font][/b][font=宋体]精密移取各批鱼腥草注射液[/font]1 mL[font=宋体],置[/font]100mL[font=宋体]离心管中,按“供试品溶液的制备”项下进行显色反应,在[/font]623 nm[font=宋体]波长下测定吸光度。对鱼腥草注射液([/font]10[font=宋体]批)中的吐温[/font]80[font=宋体]含量进行了测定,其含量在[/font]1.44~ 2.50 mgmL[sup]-1[/sup][font=宋体]。[/font]
[size=4]1.所用对照品(标准品)中检所已经发放提供(可参阅中国药典2005年版二部附录ⅩⅤG),且使用方法相同时,应使用中检所提供的现行批号对照品(标准品),并提供其标签和使用说明书,说明其批号,不应使用其他来源者;如使用方法与说明书使用方法不同(如定性对照品用作定量用、效价测定用标准品用作理化测定法定量、UV法或容量法对照品用作色谱法定量等),应采用适当方法重新标定,并提供标定方法和数据;若色谱法含量测定用对照品用作UV法或容量法,定量用对照品用作定性等,则可直接应用,不必重新标定。 2.申报临床研究时,如中检所尚无供应,为不影响注册进度,可先期与中检所接洽制备和标定,申报时提供标定报告、标签(应标明效价或含量、批号、使用效期)和使用说明书;也可与省所合作标定,申报时提供标准品或对照品研究资料,“说明其来源、理化常数、纯度、含量及其测定方法和数据”;标定有困难时,可使用国外药品管理当局或药典委员会发放的对照品(标准品)或国外制药企业的工作对照品(标准品),进行标准制订和其他基础性研究,但应提供其标签(应标明其含量)和使用说明书,能保证其量值溯源性;也可使用国外试剂公司(如sigma公司等)提供的对照品(标准品),但应提供试剂公司该批对照品(标准品)的检测报告(用作含量测定时,应有确定的含量数据),如为高纯度试剂,提供了国外试剂公司检测报告(用作含量测定时,应有确定的含量数据)时,也可使用,并应能保证其量值溯源性,但申请人应及时与中检所接洽对照品(标准品)的标定事宜,临床研究期间完成此工作。 3.直接申报生产品种,如中检所尚无供应,可参照2中要求进行,并提供相应研究资料,但申请人在标准试行期间应与中检所接洽并完成的标定事宜。 4.对照品(标准品)标定的技术要求: 4.1.创新药物 应说明对照品(标准品)原料的制备路线、精制方法、质检报告,提供理化常数和纯度的测定数据及分析结果(包括相关图谱),提供标定方法的研究和验证资料(如与原料药质量研究项下相同,可不再提供)、含量测定数据及经统计分析得到的对照品(标准品)含量结果,并说明进行临床前药学研究、药理毒理学研究所用样品的含量是否用该批对照品(标准品)确定或可用该批对照品(标准品)进行量值溯源。 ●纯度测定方法应选用色谱法,并采用两种以上不同分离机理或不同色谱条件并经验证的色谱方法相互验证比较,同时采用二极管阵列检测器或其它适宜方法检测HPLC法的色谱峰纯度,而后根据测定结果经统计分析确定对照品(标准品)原料的纯度。 ●对于组份单一、纯度较高的药物,对照品(标准品)标定方法宜首选可进行等当量换算、精密度高、操作简便快速的容量法。可根据药物分子中所具有的官能团及其化学性质,选用不同的容量分析方法,但应符合如下条件:(1)反应按一个方向进行完全;(2)反应迅速,必要时可通过加热或加入催化剂等方法提高反应速度;(3)共存物不得干扰主药反应,或能用适当方法消除;(4)确定等当点的方法要简单、灵敏;(5)标化滴定液所用基准物质易得,并符合纯度高、组成恒定且与化学式符合、性质稳定(标定时不发生副反应)等要求。 标定方法的选择要关注如下事项:(1)供试品的取用量应满足滴定精度的要求(消耗滴定液约20ml);(2)滴定终点的判断要明确,提供滴定曲线。如选用指示剂法,应考虑其变色敏锐,并用电位法校准其终点颜色。(3)为排除因加入其它试剂而混入杂质对测定结果的影响,或便于剩余滴定法的计算,可采用“将滴定的结果用空白试验校正”的办法;(4)要给出滴定度(采用四位有效数字)的推导过程。 标定结果要根据3个以上实验室各不少于15组测定结果经统计分析,去除离群值和可疑值后的结果,并报告可信限。 ●如该药物没有可进行等当量换算并符合要求的容量法时,可采用反复纯化的原料,色谱法确定纯度后扣除有关物质、炽灼残渣、水分和挥发溶剂等后的理论含量确定为标准品含量,以此为基准进行对照品(标准品)的换代和量值传递。 ●用于抗生素微生物检定法的第一代基准标准品可参照上述方法标定,如为多组份抗生素,其组份比例应与拟上市产品组份比例一致或接近,或以其中某一组份纯品为基准标准品,但要注意标准品换代时量值传递的恒定。 ●仅用于鉴别定性的化学对照品,注重其结构确证的研究资料,纯度和含量的要求一般可适当降低。 ●杂质对照品,用作限度要求时,应提供其来源(合成路线)、结构确证的研究资料,应具备较高的纯度和含量,并提供纯度和含量的的测定结果,提供质量控制标准。 4.2其他类别药物,可参照4.1要求进行。 ●用于抗生素微生物检定法的标准品须用上市国的国家标准品或原发厂的工作标准品为基准标准品进行标定。标定时采用的原料药应符合相应要求,并提供原料的制备路线、精制方法、质检报告,提供理化常数和纯度的测定数据及分析结果(包括相关图谱)。标定须用现行版中国药典附录收载的“抗生素微生物检定法”-三剂量法,并提供详细的方法学研究,包括检定菌和培养基的选择、剂量和剂距选择、缓冲液选择(如与质量研究项下相同,可不再提供)。每次标定结果均应照“生物检定统计法-量反应平行线测定法(3.3)”法进行可靠性测验及效价计算。按照《药品注册管理办法》,上市药品质量标准所用标准物质均须由中检所负责标定和管理,药品研发过程中,研制单位应注意及时与中检所联系标定事宜,以保证研发工作的连续性。[/size]
求助大神,我外标法测定含量,前后含量结果差异很大,发现对照品前后的响应差异也很大,同一台仪器,重新配置了流动相,重新配置了对照品溶液,同一个色谱条件下,对照品的响应真的会差很大吗,会是什么原因造成的呢,求助求助







 我要推广仪器
我要推广仪器
 下载APP
下载APP