甲拌磷砜GB23200.8-2016,甲拌磷亚砜有国标或农业部的检测方法么,求标准号
问题:各位老师 峰分叉是怎么回事(如图)? 这是甲拌磷标准液回复:可能是甲拌磷砜或甲拌磷亚砜http://ng1.17img.cn/bbsfiles/images/2017/01/201701191702_673981_3114888_3.jpg
本人系一名在校研究生,现需要采购多种农药标准品,用于科研。标准品浓度需在1000PPM以上或者固体,有意者请把标准品价格、浓度、含量,发到myou@xmu.edu.cn 或者mh_you@126.com氯氰菊酯、醚菌酯腈菌唑杀螟腈二甲戊乐灵氟虫腈乙草胺异菌脲氟丙菊酯丙溴磷异丙甲草胺丙环唑三氯杀螨醇ddv乙拌磷四氯间二甲苯氟乐灵甲拌磷乐果二嗪哝百菌清甲基毒死蜱七氯杀螟松马拉硫磷环氧七氯硫丹1丁草胺稻瘟灵异狄氏剂环氟菌胺硫丹2乙硫磷联苯菊酯甲氰菊酯三氯杀螨砜三氟氯氰菊酯氯菊酯氟氯氰菊酯氰戊菊酯溴氰菊酯三唑磷丙线磷敌敌畏甲胺磷异吸硫磷甲拌磷治螟磷内吸磷二嗪哝乙拌磷稻瘟净久效磷乐果甲基毒死蜱甲基对硫磷倍硫磷马拉硫磷杀螟松对硫磷甲基异柳磷喹硫磷稻丰散丙溴磷乙硫磷苯硫磷
Talin?索马甜提取自非洲竹竽(Thaumatococcus Daniellii),现已正式被批准列入中国食品国家标准,继Talin?索马甜成功进入欧洲、日本和韩国等多个市场后,我们将Talin?索马甜进而带入了庞大的中国市场。Talin?为Naturex索马甜产品之品牌凭借30余载之经验,Naturex可谓索马甜的全球引领者。Talin?为一种多功能食品添加剂,于食品饮料具有口感改良作用,同时可掩盖不良口感,此外亦可增强风味从而改善糖和盐的口感。索马甜采自于西非雨林地带,其果实称为西非竹竽(Thaumatococcus Daniellii),此果通过水提取可确保索马甜的百分百天然特质。纯度标准及分析方法的建立Naturex于2010年启动Talin?索马甜在中国的食品添加剂法规项目的申请工作,此次得到中国卫生部官方批准并公布,着实是对我们团队做出努力的最佳回报。中国法规中的纯度标准以及严格的分析检测方法皆基于Naturex所开发和使用的独有的测试方法。使用范围及市场机遇此次荣获批准,为我们中国食品饮料行业带来了绝佳的机遇。Talin?索马甜目前可使用范围为绝大多数饮料类,加工坚果与籽类,焙烤食品以及餐桌甜味剂等。Naturex预见在“低热量”饮料类中索马甜将大显神通,因其可使口感更圆润,甜味更丰满;对于餐桌甜味剂,由于Talin?索马甜为一天然甜蛋白,加之其热量极低,可谓是更健康的选择。
限量标准上说的是合计以甲拌磷、涕灭威……计,那么最后数据处理计算时该怎么计算,峰面积加和还是分子量换算?不是很理解,请各位大佬指点。还有根据标准上保留时间及相对离子丰度该如何判定样品是否检出,是否在仪器离群值那里设定后,离群就可以判断其未检出还是需要自己根据实际情况再做判断?
[sup]我最近做甲胺磷标准曲线,用丙酮配制标准储备液,然后用乙酸乙酯稀释到1、5、10、20、50ppm,结果都没出峰,怎么回事啊,很郁闷。以下是我的配置(6890N):8位自动进样器,DB-17的柱子,不分流进样口,FPD检测器。另外跟工程师沟通过,他说让把衬管里的玻璃棉拿掉,我拿掉以后还是不出峰,请各位大侠帮我分析分析。 昨天看到大家的回帖很感动!我再把我的情况说一下. 程序升温: 第一阶段:60℃(保持2min),升温速度:10℃/min 第二阶段:200℃(保持0.2min),升温速度2℃/min 第三阶段:250℃ 进样口温度:270℃ 检测器温度:250℃(方法要求270℃,但仪器只能升到250℃) 另外,在4min左右会出现一溶剂峰.
食品包装用纸的轻工业标准出炉,几大纸制品巨头联合打造!《食品包装用淋膜纸和纸板》国家标准征求意见
甲拌磷、甲拌磷砜、甲拌磷亚砜在气相出峰如何,三个峰会重合吗?检测条件如何,有图谱发上来,会有积分奖励。
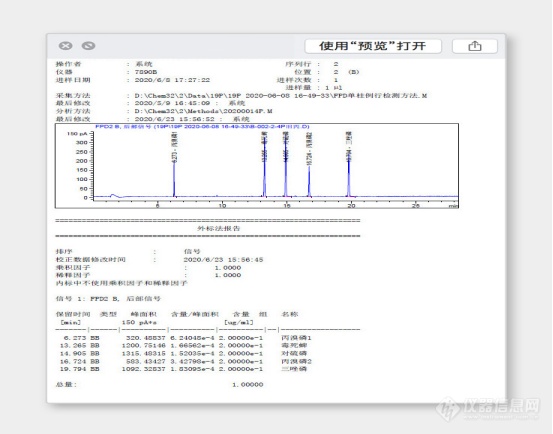
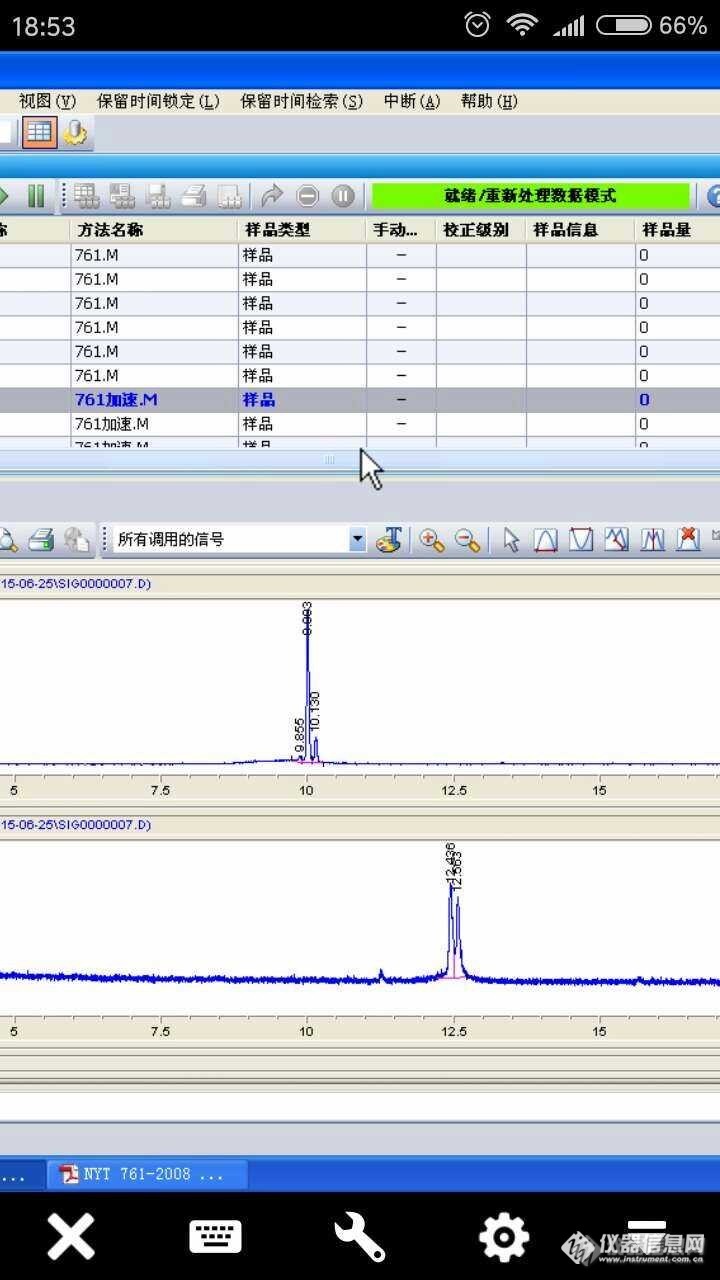
[font=宋体][size=14px] 〖标准品专题〗[/size][/font][font=宋体][size=14px]奇怪的丙溴磷[/size][/font][font=宋体][size=14px] 单位因为检验检测任务,每年都会根据需要购买新的标液,多年来我单位一直使用的是同一国产厂家的标准品,且都是正式批准过的有证标准物质。新的标准物质按照单位标准物质的管理办法进行入库出库手续后,检测室就会对标准品的量值按规定的方法进行测试、核定、比对确定,以便能溯源到国家基准、国家测量基准或国家标准物质基准,经过分析、比对验证、符[/size][/font][font=宋体][size=14px]合要求方能使用。[/size][/font][font=宋体][size=14px][font=宋体] 10月10日,在对购进的标准品测试过程中,发现有机磷色谱图中色谱峰出峰个数比加标液种数多一个,查阅NY/T 761-2008《蔬菜和水果中有机磷、有机氯、拟除虫菊酯和氨基甲酸酯类农药多残留的测定》标准和色谱图第[/font][font=宋体]Ⅰ—Ⅳ组有机磷农药标准溶液色谱图[/font][font=宋体]附件,按照该方法条件下测定,有机磷都应该是单峰,没有组峰情况,多出现的这个峰是什么呢[/font][font=宋体]?[/font][font=宋体]鬼峰[/font][font=宋体]?未知峰?杂峰?[/font][font=Arial]……[/font][/size][/font][font=宋体][size=14px] [img=,553,434]https://ng1.17img.cn/bbsfiles/images/2020/07/202007090822325118_4989_3389022_3.png!w553x434.jpg[/img][/size][/font][font=宋体][size=14px] [img=,480,480]https://ng1.17img.cn/bbsfiles/images/2020/07/202007090823520016_850_3389022_3.png!w480x480.jpg[/img][/size][/font][font=宋体][size=14px] 一、排查问题[/size][/font][font=宋体][size=14px] 1 实验部分[/size][/font][font=宋体][size=14px] 1.1 仪器与试剂[/size][/font][font=宋体][size=14px] [url=https://insevent.instrument.com.cn/t/Mp]gc[/url]7890B[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url](安捷伦公司)FPD检测器、高速分散均质机、离心机、混匀器、十分之一天平。乙腈(色谱纯)、丙酮(色谱纯)。[/size][/font][font=宋体][size=14px] 1.2实验方法[/size][/font][font=宋体][size=14px] 采用[/size][/font][font=宋体][size=14px]NY/T 761-2008[font=宋体]《蔬菜和水果中有机磷、有机氯、拟除虫菊酯和氨基甲酸酯类农药多残留的测定》[/font][/size][/font][font=宋体][size=14px]方法进行检测。[/size][/font][font=宋体][size=14px] 1.3 标准溶液的配置[/size][/font][font=宋体][size=14px][font=宋体] 新购进的某厂家标准物质,质量浓度均为[/font]100ug/mL,有机磷的标液用丙酮稀释至10ug/mL。再根据检测需要配置需要浓度的混标或单标溶液。[/size][/font][font=宋体][size=14px] 1.4实验条件[/size][/font][font=宋体][size=14px][font=宋体] 有机磷的检测条件[/font] [font=宋体]色谱柱:[/font]DB-1701(30m×0.25mm×0.25um);温度:80℃保持1min,以20℃速度上升到180℃,不保持,再以5℃升温到230℃,再以15℃升温到250℃保持11.00min。进样体积:1uL;进样口:250℃,不分流进样;检测器300℃;进样口温度:250℃,氮气流速:20mL/min;柱流速:2mL/min。[/size][/font][font=宋体][size=14px]1.5实验排查[/size][/font][font=宋体][size=14px] 1.5.1色谱柱污染的问题可以基本排除:9月底[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url]检定时新换的色谱柱,且色谱柱在使用前才刚刚进行了老化。[/size][/font][font=宋体][size=14px] 1.5.2先进一针丙酮溶剂,看看是否有大峰出现,排除溶剂污染的可能。[/size][/font][font=宋体][size=14px] 1.5.3鬼峰的排除:重新进2针该标液,该峰保留时间,峰的形状从显性特别好,且衬管、进样垫、色谱柱、检测器、载气都处于最佳状态,排除鬼峰的可能。[/size][/font][font=宋体][size=14px] 1.5.4分别进单标,丙溴磷出了两个峰,且保留时间同混标色谱图一致,确定该峰为丙溴磷2。[/size][/font] [img=,578,732]https://ng1.17img.cn/bbsfiles/images/2020/07/202007090828164259_7355_3389022_3.png!w578x732.jpg[/img][font=宋体][size=14px][font=宋体] 查阅单位保存的前期检测的图谱,在色谱柱不同时丙溴磷确实存在有时一个峰有时两个峰的情况。[/font]DB-5出一个峰,[/size][/font][font=宋体][size=14px]DB-1701[/size][/font][font=宋体][size=14px]、[/size][/font][font=宋体][size=14px]VF-1701出现过2个峰。[/size][/font] [img=,591,484]https://ng1.17img.cn/bbsfiles/images/2020/07/202007090831478975_4933_3389022_3.png!w591x484.jpg[/img][font=宋体][size=14px] 1.6进一步确定[/size][/font][font=宋体][size=14px] 针对上述情况,需要具体排除是色谱柱选择问题还是丙溴磷标液的问题。[/size][/font][font=宋体][size=14px] 1.6.1分别进17年、18年、19年[/size][/font][font=宋体][size=14px]购进的[/size][/font][font=宋体][size=14px]该[/size][/font][font=宋体][size=14px]厂家[/size][/font][font=宋体][size=14px]丙溴磷[/size][/font][font=宋体][size=14px]标准物质[/size][/font][font=宋体][size=14px]。[/size][/font][font=宋体][size=14px] 仪器试剂、实验方法、[/size][/font][font=宋体][size=14px]标准溶液的配置[/size][/font][font=宋体][size=14px]同上。[/size][/font][font=宋体][size=14px][font=宋体] 检测条件[/font] [font=宋体]色谱柱:[/font]VF-1701(30m×0.25mm×0.25um);温度:80℃保持1min,以20℃速度上升到180℃,不保持,再以5℃升温到230℃,再以15℃升温到250℃保持11.00min。进样体积:1uL;进样口:250℃,不分流进样;检测器300℃;进样口温度:250℃,氮气流速:20mL/min;柱流速:2mL/min。[/size][/font][font=宋体][size=14px][font=宋体] 实验结果如下:[/font]17年、18年标准品丙溴磷出2个峰[/size][/font][font=宋体][size=14px] 19年标准品丙溴磷出1个峰[/size][/font] [img=,576,632]https://ng1.17img.cn/bbsfiles/images/2020/07/202007090829339292_1022_3389022_3.png!w576x632.jpg[/img][font=宋体][size=14px] 结论:同一厂家生产的丙溴磷标准品同实验条件下的出峰情况不仅仅受色谱柱的影响,还受标液不同生产年份不同批次的影响。[/size][/font][font=宋体][size=14px] 建议:标准物质是检测实验室用于保证检测数据的准确性和精密度,实现量值传递的重要工具,国内试剂质量不够稳定,造成了检测人员时间和人力资源的浪费,也是导致市场认可度较低的主要原因。[/size][/font][font=宋体][size=14px] 随着我国科研投入的加大,以及人们对食品、健康、环境等民生问题的重视,对试剂的需求也越来越大,加快国产试剂的发展,整体提升国产试剂的质量势在必行。[/size][/font]
各位老师,请教一个GCMS检测邻苯二甲酸酯的问题,检测标准品时,每个峰前都会有一个小峰,采购的100ppm原标是用甲醇配制的,校准曲线是用丙酮稀释的,大家有遇到过这个现象吗?是甲醇丙酮溶剂汽化体积不同造成的吗?谢谢。
按照NY/T 761-2008里面,硫环磷和甲基硫环磷是用FPD检测器检测的,但是用标准品0.1ug/mL和1.0ug/mL进样,均未发现出峰,DB-1701柱。
有吃韭菜中毒的,质谱检出甲拌磷砜,色谱想定量,但无其标准,色谱中检出有少量的甲拌磷。甲拌磷代谢产物是甲拌磷砜,但不知道二者关系。望高手解答
要CNAS认可了, 标准 SNT 1924-2007 进出口动物源性食品中克伦特罗,莱克多巴胺,沙丁胺醇 要换版成 标准SNT 1924-2011进出口动物源食品中克伦特罗、莱克多巴胺、沙丁胺醇和特布他林残留量的测定 液相色谱-质谱质谱法 需不需要重新对方法进行验证呢?遇到这种情况大家都是怎么做的?
配制甲拌磷亚砜1.0ug/mL,出现两个峰,前一个峰很小,而且与甲拌磷出峰时间重合,那么甲拌磷亚砜出几个峰,与甲拌磷重合的小峰也是甲拌磷亚砜的吗?
最近有幸参加了“吉林省人社厅2013年专业技术人才知识更新工程高级研修班项目-生物资源保护与食品安全评价”的讲课。我的讲课题目是“食品企业管理相关国家标准介绍 ”。将我所讲的有关食品行业管理的国家及行业标准部分介绍给大家,请大家参考,讨论,并留下宝贵意见,作者期待中。
请问哪里能买到拌种灵和苯醚甲环唑的标准品呢?
甲拌磷亚砜哪有卖?要标液不要标品,标品需要配制,麻烦。
我用的是Rtx-1701的柱子检测有机磷类农药,但是进纯标准品岀峰的时间与加在样品中标准品岀峰的时间相差太大了,就像是两个不同的峰,而且甲胺磷现在根本都不出峰了,这种情况是我的柱子寿命已尽了吗?
用气相可以做甲拌磷砜与甲拌磷亚砜吗?上级下的任务是检测有机磷农药残留,其中有甲拌磷、甲拌磷砜与甲拌磷亚砜,甲拌磷出峰很好,但这两种能出峰吗?标液还在购买中,希望做过的说说出峰情况。
[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质联用仪[/color][/url]三重四级杆跑了甲拌磷和甲拌磷亚砜 为什么出峰时间是重叠的 甲拌磷和甲拌磷亚砜应该离的挺远啊 求帮助
最近在做塑料制品中的18种多环芳烃,5ug/mL浓度(为了确定保留时间所以浓度较高)的峰形都很好。配制了20ng/mL~400ng/mL共6个浓度点的标准溶液,出来的谱图无一例外表现出:除了前面3个化合物外,其余15种都拖尾严重,挨着的2个或3个化合物以一个峰宽很大的峰出来,而且保留时间都比5ug/mL时延迟。奇怪的是,我做了中间浓度点的加标实验,样液中的一系列化合物峰形都很正常。标准物质用甲苯配制,塑料制品用甲苯提取。 也就是说,标准物质在溶剂中拖尾严重,在样液中峰形正常。这是怎么回事? 我老化了柱子还是如此。望高人指点。
SPME测定果实香气,进行半定量,选了环己醇或者3-辛醇做内标,内标跟标准品的溶剂要尽量一致对吧?那用什么溶剂来溶解内标呢?正己烷?SPME测定果实香气是气体进样,做内标曲线的时候,配制不同梯度的标准品再加内标,这一步的话是液体进样还是气体进样?另外,如何根据特征离子峰来确定该化合物的相对浓度?望大神不吝赐教!感谢!!!
《食品安全国家标准 食品中农药最大残留限量》2016版正式颁布实施,这一农药残留的新国标,在标准数量和覆盖率上都有了较大突破,规定了433种农药在13大类农产品中4140个残留限量,较2014版增加490项,基本涵盖了我国已批准使用的常用农药和居民日常消费的主要农产品。 食品伙伴网标法中心结合2016版标准前言部分内容与2014版进行相应的对比分析,供参考: 1、对原标准中氟唑磺隆、甲咪唑烟酸、氟吡菌胺、三唑酮和三唑醇等5种农药残留物定义,敌草快等5种农药每日允许摄入量等信息进行了核实,修订了敌草快、三环锡等5种农药的ADI值。 2、增加了2,4-滴异辛酯等46种农药;增加了490项农药最大残留限量标准 2014版规定了食品中2,4-滴等387种农药3650项最大残留限量,2016版规定了433种2,4-滴等农药4140项最大残留限量。增加了46种农药:2,4-滴异辛酯、2甲4氯异辛酯、苯嘧磺草胺、苯嗪草酮、吡唑草胺、丙硫多菌灵、除虫菊素、毒草胺、多抗霉素、呋虫胺、氟吡菌酰胺、复硝酚钠、甲磺草胺、井冈霉素、抗倒酯、苦参碱、醚苯磺隆、嘧啶肟草醚、扑草净、嗪草酸甲酯、氰氟虫腙、氰烯菌酯、炔苯酰草胺、噻虫胺、三苯基乙酸锡、三氯吡氧乙酸、杀螺胺乙醇胺盐、莎稗磷、虱螨脲、特丁津、调环酸钙、五氟磺草胺、烯丙苯噻唑、烯肟菌酯、烯效唑、辛菌胺、辛酰溴苯腈、溴氰虫酰胺、唑胺菌酯、唑啉草酯、啶菌噁唑、丁吡吗啉、噁唑酰草胺、甲哌鎓、丁酰肼、唑嘧菌胺。 3、增加 12 项检测方法标准,删除1项检测方法标准 增加了SN/T 0162、SN 0198、SN/T 0931、SN/T 1624、SN/T 1989、SN/T 2229、SN/T 2231、SN/T 2237、SN/T 2323、SN/T 2387、SN/T 2795、SN/T 2807,删除了SN/T 0711,其中SN 0198标准已于2015年12月31日被认监委废止,废止依据为《国家认监委办公室关于公布2015年检验检疫行业标准复审结论的通知》。 4、修改了丙环唑等8种农药的英文通用名 修改了丙环唑、六六六、烯肟菌胺、氯啶菌酯、杀虫双、四氯苯酞、氯氟吡氧乙酸和氯氟吡氧乙酸异辛酯 5、将苯噻酰草胺和灭锈胺的限量值由临时限量修改为正式限量;对资料性附录 A 进行了修订,增加了干制蔬菜等3种食品名称,修改1项作物名 食品伙伴网对附录A部分内容的对比发现如下变化: 1)水果(核果类)的类别说明增加了青梅,枣修改为枣(鲜)。 2)水果(浆果和其他小型水果)的类别说明中露莓增加了备注:包括波森莓和罗甘莓。 3)水果(热带和亚热带水果)的类别说明中将大型果的木瓜修改为番木瓜。 4)干制水果的类别说明中增加了枣(干)等。 5)食品类别名称修改:将饮料修改为饮料类。 6、食品伙伴网在对比过程中发现,2016版标准除了以上所列变化外,还修正了其他一些内容: 1)引用的标准名称的修正,如GB/T 19648、GB/T 19469等部分标准的名称中 “兽”字已删除。 2)引用的作废标准的修正,如2014版标准中引用的是2006版GB/T 20770的标准名称,2016版标准已经修正为2008版GB/T 20770的标准名称。 3)农药中文名称修改:2014版标准中的2甲4氯(钠)修改为2甲4氯钠。 4)附录A中动物源食品部分类别的测定部位描述进行了修正。
蔬菜中乙拌磷的国家标准限值是多少?
消费品的一些标准品,邻苯类增塑剂,多溴阻燃剂,多环芳香烃,偶氮,多氯联苯这一类的标准品,都有在哪买,要比较有口碑的,可信任的?我们一般在百灵威买,还有其他家?
采用液液萃取的方法测三卤甲烷,做出了标准品的曲线,然后用去离子水做了一个加标的,测了发现加标的峰面积比标准品的峰面积大很多,是不是很不正常?这怎么计算回收率?而且萃取不是会有损失,怎么峰面积还变大了?前处理步骤,就是20ml水样加入比色管中,然后加100ppb的标准品,加4ml的甲基叔丁基醚萃取,再加入8g的无水硫酸钠。也做了个空白,发现空白没有这些出峰,峰面积大小可以忽略不计。加标和测标准品的方法是一样的。请高手解答一下
[b][font=宋体]问题描述:按照[/font]HJ 786-2014[font=宋体]测定土壤和沉积物中多环芳烃,色谱条件:岛津[/font]LC-20AT[font=宋体]四元低压液相系统,二极管阵列检测器([/font]SPD-M20A[font=宋体])和荧光检测器([/font]RF-20A[font=宋体]),流动相为乙腈水溶液,流速为[/font]1mL/min[font=宋体]。结果出峰数量少两个并且与标准中多环芳烃的出峰顺序不同,这是为什么?[/font][font=宋体]解答:[/font][/b][font=宋体]([/font]1[font=宋体])现有流动相洗脱条件无法使所有目标物完全分离,可设置不同的梯度洗脱程序,观察单峰是否有分离成两个峰的迹象,并且验证峰纯度,如条件允许,也可使用质谱或购买单标进行定性。[/font][font=宋体]([/font]2[font=宋体])借助单标或者质谱验证,确认是否混标中本身就缺少某种组分,或是某种组分已经分解或是损失。如确认是标准品本身的原因,应重新购置合格的标准品。[/font][font=宋体]([/font]3[font=宋体])尽量采用推荐的色谱柱,液相色谱法测试[/font]16[font=宋体]种多环芳烃有专门的色谱柱,可以使用的安捷伦公司和岛津公司的[/font]PAH[font=宋体]专用柱,规格是[/font]4.6mm×250mm[font=宋体],[/font]5μm[font=宋体]。[/font][font=宋体]([/font]4[font=宋体])分离效果与仪器的延迟体积也有一定关系,比如岛津的[/font]LC-20AT[font=宋体]带四元比例阀加混合器,延迟体积可以大到[/font]3mL[font=宋体]左右,安捷伦的[/font]1260[font=宋体]带四元比例阀延迟体积一般是[/font]900μL[font=宋体]左右;因此同样的梯度、同样的色谱柱分离也不一定完全一样,也会出现某些峰不能完全分离的情况。[/font][font=宋体]([/font]5[font=宋体])出峰顺序与柱子、流动相、柱温等很多因素有关。如果条件不能做到,无法与标准中出峰顺序完全一致,应对色谱峰进行定性确认。如能定性,即便有个别峰顺序不对,结果也是可以接受的,这并不会影响定量分析。[/font][font='微软雅黑','sans-serif'][color=black][back=white]领取更多《实战宝典》请进:[url]http://instrument-vip.mikecrm.com/2bbmrpI[/url][/back][/color][/font][font='微软雅黑','sans-serif'][color=black][back=white] [/back][/color][/font]

气相色谱法检测食品中甲拌磷的残留摘要:农药残留是引起食物中毒的主要原因之一。甲拌磷是一种高效内吸性杀虫剂,广泛应用于防治植物虫害,其过度残留会污染食物,严重者导致误食者中毒。中国(GB2763-2014)规定粮食中甲拌磷农药的允许标准最高值是0.1mg/kg(食品)。目前,气相色谱-火焰光度法(Flame Photometric Detector,FPD)是检测农药残留最重要的手段。本文针对特定模拟情况,分析中食品中毒物来源,利用GC-FPD对放置在被甲拌磷污染的木架上包装完好的薯片进行检测,以确定薯片是否被甲拌磷污染。1. 实验部分1.1 试剂丙酮为色谱纯(美国Fisher公司)。1.2 样品前处理1.2.1木板中甲拌磷的提取将木板样品浸泡在盛放有10 mL丙酮的试管中,36 KHz超声,振荡提取10 min。离心,取上清液,GC-FPD检测。1.2.2薯片中甲拌磷的提取对薯片进行了研磨粉碎,称取薯片粉末1.000g加入盛放有10 mL丙酮的试管中,36 KHz超声,振荡提取10 min。离心,取上清液,GC-FPD检测。1.2.3情况模拟将甲拌磷农药2 mL滴加在木架表面,室温放置到表面干燥。将包装完整薯片码放在木架上面,室内阴凉处放置7天,取样。1.3 仪器条件气相色谱条件:7890A型气相色谱仪配有FPD检测器(美国,Agilent)、DB-1701型毛细管色谱柱(30 m×0.25 mm×0.25 μm)。2. 结果与讨论按照1.2.2中方法对薯片样品中甲拌磷含量进行检测。取出一包薯片进行隔离防止甲拌磷造成污染,对其他薯片进行模拟实验。取甲拌磷农药2mL滴加在木板表面,图1、2为甲拌磷在木板表面的滴加情况。将薯片放置在滴加了甲拌磷的木板表面,室温静置7天,图6为薯片在污染木板上的放置情况。http://ng1.17img.cn/bbsfiles/images/2015/07/201507311858_558390_2648817_3.jpg图1. 未滴加3911(甲拌磷)的木板照片http://ng1.17img.cn/bbsfiles/images/2015/07/201507311858_558391_2648817_3.jpg图2. 滴加3911(甲拌磷)后的木板照片 分别对放置7天的薯片样品(以下简称为放置7天薯片样品)和隔离薯片样品(以下简称为隔离污染薯片样品)进行粉碎。向隔离薯片粉末中添加已知量的甲拌磷,静置1小时。按照1.2.2中的方法对放置7天薯片样品和隔离样品进行甲拌磷提取。经过气相色谱仪检测,有明显污染痕迹区域的木板样品中甲拌磷含量为130.968μg/g。检测结果如下:http://ng1.17img.cn/bbsfiles/images/2015/07/201507311859_558393_2648817_3.jpghttp://ng1.17img.cn/bbsfiles/images/2015/07/201507311859_558394_2648817_3.jpg样品谱图信息 名称 保留时间 min 峰面积 浓度 μg/mL 浓度平均值 μg/mL 放置7天的薯片样品 10.415 207778 50.161 49.855 10.414 205591 49.549 隔离污染薯片样品 10.419 641557.0 171.552 171.205 10.424 639082.5 170.859 3. 结论与展望甲拌磷在20-1000μg/mL浓度范围内呈现较好线性,回归方程为Y=2989.6X+48312,R2=0.9976,平均回收率为87.5%(n=3)。经过分析得到包装完整的薯片中甲拌磷含量为130.97mg/kg,明显超过GB2763-2014中0.1mg/kg的限量值,说明木板中的甲拌磷已经污染到包装完整的薯片。通过模拟事发过程,利用GC-FPD对放置在被甲拌磷污染的木架上包装完好的薯片进行再次检测,也确认了木板上的甲拌磷通过接触进入了包装完整的薯片中。利用该法对包装完好的薯片进行甲拌磷检测,符合农药残留测定的技术要求,可以准确的对甲拌磷进行定量分析。参考文献 1、 GB/T 5009.20-2003食品中有机磷农药残留量的测定 2、 余萍中,戴荣彩,贺敏,陈莉,贾春虹.气相色谱法测定土壤中甲拌磷及其代谢物甲拌磷砜.湖南农业大学学报(自然科学版).2008(06):732-734. 3、 张以春,雍宗峰.固相萃取-毛细管柱色谱柱气相色谱法测定韭菜中甲拌磷及其代谢产物甲拌磷砜.中国食品卫生杂志.2012(01):34-37. 4、 张杰,许家胜,刘连生.气相色谱法快速测定蔬菜水果中甲拌磷、甲基对硫磷残留量.科学技术与工程.2011(13):3049-3051.
FPD检测器,DB1701(30*0.25*0.25),以前做甲拌磷砜与甲拌磷亚砜,可以分离,两峰相差0.5min,买了新DB1701柱,老化后进样,砜与亚砜两峰完全重合,是标液标错了,还是新柱有问题,但其它20种农药出峰都正常,有人遇见过这种情况吗?
如何才能买到多西环素(强力霉素)的两个代谢产物:4-epidoxycycline和6-epidoxycycline的标准品?







 我要推广仪器
我要推广仪器
 下载APP
下载APP