FID使用时相关问题,我是在乙醇里加入等体积四氢呋喃、甲苯、氟苯浓度大概在百分之一,出峰顺序是四氢呋喃、乙醇、氟苯、甲苯,提高柱温以后,乙醇的含量减少了是怎么回事?相同样品,同样的注射量,恒温程序走的,当提升温度把柱温由60变到80,乙醇的积分面积百分数从90%下降到80%是因为什么,请各位大神解惑
我想做甲苯乙醇混合液定量分析,使用的柱子是VF-5MS,刚刚接触GC-MS,求助各位指点一下进样条件
朋友们谁有苯甲酸甲酯、对甲苯磺酸、TEBAC、甲醇钠、4-二甲胺基吡啶、苯甲酰氯、乙硫醇、巴豆醛、乙酰乙酸甲酯、丙酰氯、DCP的国标、行标或企业标准啊?帮忙找一下啊,谢谢!
版友问题,做乙醇甲苯残留溶剂顶空进样不出峰,做其他的直接进样正常,会有什么原因呢?
最近,接到一个项目,用氨基柱做糖酯的分析,样品里面含有吡啶、甲苯、叔戊醇等溶剂。这些溶剂有没有好的办法除去,而不损失糖醇、糖酯。若不除去,直接进液相色谱,会不会对柱子有损伤。
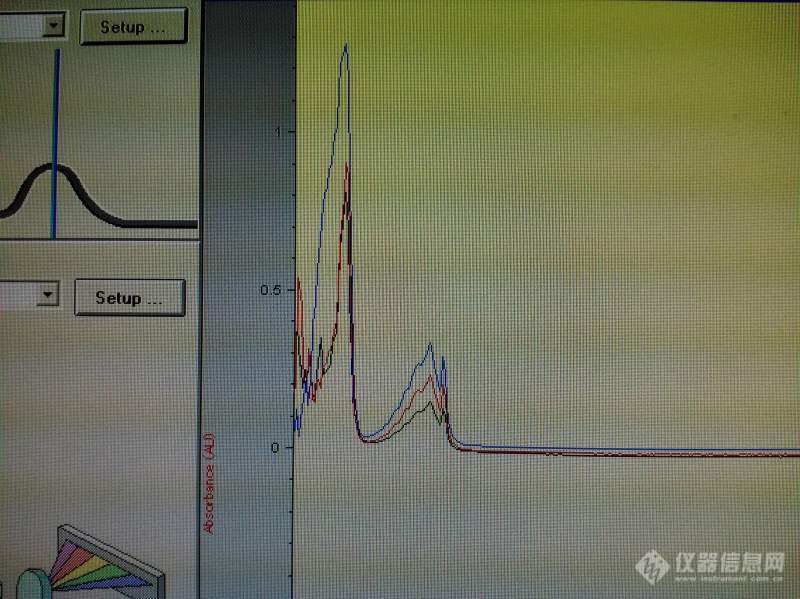
[table=100%][tr][td]我是使用甲苯作为有机溶剂提取某些天然代谢产物的,在做紫外可见光谱分析时,使用乙醇作为参比溶剂及稀释甲苯溶液。这张图是我稀释了2000倍之后的3个不同代谢产物的甲苯提取物光谱图,但是感觉所有的峰都是甲苯,找不到我所提取的产物的峰。很急!要疯了![/td][/tr][/table][img=,690,516]https://ng1.17img.cn/bbsfiles/images/2019/09/201909301440080773_5609_1827556_3.jpg!w690x516.jpg[/img]
急求标准对二氯苯、五氯化磷、乙酰氯、对甲苯磺酰胺、乙二醇甲醚、马来酸国标、化工标准、地方标准都行谢谢!![em0808]
[em06] 四氢呋喃、二氧六环、吡啶、甲苯 照残留溶剂测定法(附录Ⅷ P第三法)试验。精密称取苯适量,加甲醇制成每1 ml中约含60μg的溶液,作为内标溶液。精密称取四氢呋喃、二氧六环、吡啶、甲苯适量,加甲醇制成每1ml中各含720μg、380μg、200μg和890μg的溶液,作为对照贮备溶液;精密量取对照贮备溶液1ml与内标溶液1ml,置10ml量瓶中,加水稀释至刻度,摇匀,作为对照溶液。精密称取本品1.0g,置10ml量瓶中,加内标溶液1ml,加水溶解并稀释至刻度,摇匀,作为供试品溶液。用二乙烯基-乙基乙烯苯型高分子小球作为固定相,柱温190℃,依法测定。残留溶剂含量应符合规定。我让色谱公司按这个要求做了不锈钢柱子(柱填料:401有机载体(二乙烯基苯/乙基乙烯苯共聚体)60-80目),可是不出峰,后来把柱寄回去了,现在又寄给我的柱子(柱填料:10%PEG-20M CHROMOSORB PAW-DMCS 80-100目),峰是有了,可是分不开,我做[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]的氮气4圈,空气4.2圈,氢气4.5圈,后我又把氮气开到3圈,还是这个样子.是怎么回事呢,请高手赐教.谢谢!!!
现在要分离水、乙醇、草酸二乙酯、二甲苯,假设各占1/4的含量,二甲苯不用分离异构体,准备用GDX-401或parapak P/Q作担体,PEG20M作固定液,不知道理论能不能分离?
Materials 材料1.Toluene – 99.8% 甲苯 - 99.8%2.DME (1,2-Dimethoxyethane, ethylene glycol dimethyl ether) – 99+% DME(1,2 -- 二甲氧基乙烷, 乙二醇二甲醚) -- 99+%3.Triethylsilane – 99% 三乙基硅烷 -- 99%4.Tetraethylsilane – 99% 四乙基硅烷 -- 99%标准样品1成分标准样中成分的重量%含量样品中的成分重量甲苯20%10.0gDME75%37.5g三乙基硅烷3%1.5g四乙基硅烷2%1.0g总量100%50g标准样品2成分标准样中成分的重量%含量样品中的成分重量甲苯96%48.0gDME1%0.5g三乙基硅烷2%1.0g四乙基硅烷1%0.5g总量100%50g都没有人做过[em09508][em09508][em09508]
小弟想买根柱子,用来分离甲苯、乙醇和丙酮,但不知道该买什么型号的,求大神指点,小弟跪谢了
一般GC-MS的检出限是多少?例如乙醇中的微量甲苯、苯等?
最近用液相在检测氯羟吡啶和喹乙醇残留量,一个样品要做这两个项目,但是样品处理方法缺不一样。氯羟吡啶:乙腈匀浆后过SPE柱喹乙醇:乙腈匀浆后正己烷萃取杂质后留乙腈层[color=#DC143C]因为样品量大,不知道有哪位大侠做过,看知道有没有好办法同时处理样品同时测定。[/color][color=#00008B](色谱条件不是问题,关键是处理)[/color]
请问:分析一有机药物中甲苯、乙醇、乙腈、丙酮、三乙胺残留量的柱子型号和分析方法谢谢!
刚刚摸索用内标法测定2-氰基吡啶和3-氰基吡啶纯品的含量,不知道选哪种内标物比较好?(纯品中可能还含有甲苯、吡啶、2-甲基吡啶/3-甲/4-甲、4-氰基吡啶)看到一篇文献中以3-氰基吡啶为内标物测定2-氰基吡啶水溶液的含量,但以我们现在的[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]条件,2-氰基吡啶和3-氰基吡啶的样品峰并不能完全分开,还有一小部分互溶,好像达不到内标法的要求。用甲醇或乙醇作内标物不知道合适不?期盼高手解答一下。不胜感激!
要测吡啶\丙酮\乙醇\DMF\乙二醇应选哪种型号色谱柱?
[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]配制5%乙醇5%苯5%甲苯,怎么配制?用分析天平称取质量()g甲醇()g苯()g甲苯,用( )溶液稀释100ml容量瓶中,这种配制溶液可以么?算百分含量用密度么?有公式帮忙写下,非常感谢
有哪位高手知道对甲苯磺酰基甲基异腈检测方法?
请问,1000克含有35%甲苯40%乙酸丁酯15%乙酸乙酯10%乙醇的有机溶剂可以溶解多少克硝酸纤维素酯?如果要减小以上溶液的表面张力在不改变溶液透明有光泽的前提下,应该加哪一种表面活性剂?
请问乙基苯和二甲苯的保留时间是不是很接近? 容易分离吗? 我在定性时,这两个组分是重叠的。 请大家帮下忙!
如何检测哌嗪、乙二胺、三乙烯二胺、二乙烯三胺、氨乙基哌嗪、二甲苯、均三甲苯?
[img]http://ng1.17img.cn/bbsfiles/images/2010/03/201003081849_204529_1621482_3.jpg[/img]我想请教各位一个问题 做[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]分析二甲苯时,导致里面乙基苯含量变高的原因是什么?谢谢!
如何对于机动车尾气(非极性的C1-C8烷烃、烯烃、苯、甲苯等,乙醇、乙醛、乙酸等)的定性与定量分析?这些有机物的含量一般在10ppm左右。用GC-MS仪器定性时,选用什么色谱柱?
你好,我是一家企业,经常碰到在高价的有机溶剂里面添加低价溶剂的行为,特此请教!甲醇与乙醇的鉴别:求助:1,如乙醇里面加入5~10%的甲醇,请问怎么鉴别 2.甲苯,二甲苯,里面添加少量甲醇,乙醇,怎么鉴别 3,甲醇里面加入少量水怎么鉴别:可以用常用的仪器,用物理或者化学的方法鉴别?
新进的批号是201004的吡啶已经是淡黄色的了,配制溶液过夜后,苯酐溶液颜色更深了,请问,吡啶变色原理是什么?对多元醇测定影响是如何产生的,请高人指点。我用的电位滴定。吡啶变黄不能用是针对使用指示剂干扰终点判断还是其他的原因呢?

10,抽取5个版友);中奖名单:dahua1981(注册ID:dahua1981)牛一牛(注册ID:v2700892)莫名其妙(注册ID:moyueqiu)吕梁山(注册ID:shih20j07)翠湖园(注册ID:hhx050)http://ng1.17img.cn/bbsfiles/images/2016/12/201612261506_01_1610895_3.jpghttp://ng1.17img.cn/bbsfiles/images/2016/12/201612261506_02_1610895_3.jpg【注意事项】同样的答案,每人只能发一次PS:该贴浏览权限为“回贴仅作者和自己可见”,回复的版友仅能看到版主的题目及自己的回答内容,无法看到其他版友的回复内容。下午3点之后解除,即可看到正确答案、获奖情况及所有版友的回复内容。=======================================================================黄酒中β-苯乙醇的测定(GBT 13662-2008)方法:GC基质:黄酒应用编号:101863化合物:β-苯乙醇固定相:DM-InertWAX色谱柱/前处理小柱:DM-InertWax 60m x 0.25mm x 0.25um样品前处理:制备方法: β-苯乙醇溶液:做标样用。吸取β-苯乙醇(色谱纯)2 ml,用60%乙醇溶液(体积分数)定容至100 mL 2-乙基正丁酸:做内标用。吸取(色谱纯)2 ml,用60%乙醇溶液(体积分数)定容至100 mL 校正因子(f值)的测定:吸取β-苯乙醇标准溶液1 mL,移入100 mL容量瓶中,加入内标溶液1 mL,用60% 乙醇定容色谱条件:分析条件 色谱柱: DM-Inertwax,60×0.25 mm,0.25 μm (Cat#:8522) 柱温: 50 ℃(2 min)—200 ℃, 5 ℃/min,(10 min) 载气: 氮气 柱前压: 0.6 Mpa 进样方式: 不分流,230℃ 检测器: FID,250 ℃ 进样量: 2.0 μL文章出处:天津迪马实验室关键字:黄酒,β-苯乙醇,GBT 13662-2008,DM-Inertwax,8522摘要:黄酒中β-苯乙醇的测定谱图:http://www.dikma.com.cn/Public/Uploads/images/huangjiu.PNG图例:β-苯乙醇
用岛津的2010plus[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url]做液体,一般做的都是甲醇、乙醇、正丁醇、苯、对二甲苯等纯度较高的产品,怎么样清洗进样用的小瓶,是不是100下烘箱烘干就可以了?
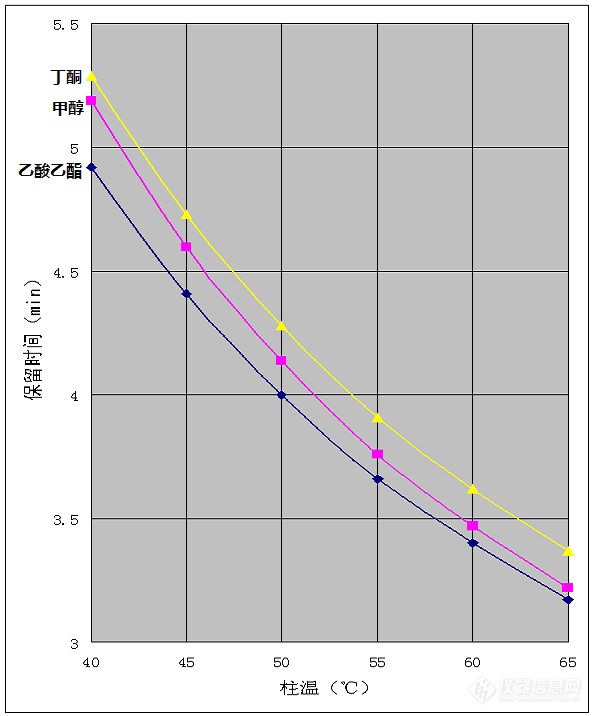
溶剂残留分析是[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]的重要应用之一,在药品、食品、包装等领域都是必测的项目。常见溶剂中涉及到的检测目标物经常有乙酸乙酯、甲醇、丁酮,以及二甲苯异构体这几项。最近看到 @m3091333、@p3109800、@Insm_c1196d2b 等多人发帖子讨论相关问题,我从原理上进行了一些解释,但终究纸上谈兵,于是找别的实验室要了这几种试剂,用实践检验了一下。首先,如果二甲苯异构体不要求分离,用624柱可以很容易的解决问题,这里就不讨论了。如果要求乙苯、对二甲苯、间二甲苯、邻二甲苯四种异构体分离,用624柱是无法完成的。因为二甲苯异构体色散力差异非常小,只能靠诱导力的差异分离,不同异构体在强极性柱上的极化率不同,乙苯极化率最低,其次是对二甲苯、间二甲苯,邻二甲苯极化率最大,出峰时间也随极化率的增加而延长。而624柱的极性比较弱,不能产生足够的极化作用,特别是对二甲苯与间二甲苯的极化差异非常小,无法实现分离。这个问题是由分子结构决定的,无论怎么调节色谱条件都不能解决。要想解决只能换强极性柱,常见的就是聚乙二醇柱,包括各种wax柱和FFAP柱等。三氟丙基柱也是强极性的,可以分离二甲苯异构体,但是这种柱很少使用。在聚乙二醇类的色谱柱上,乙酸乙酯、甲醇、丁酮三种目标物分离困难,各种类型的聚乙二醇柱选择性略有差异,但这三种物质都是较为接近的,想要分离是不太容易的。但是这三种物质与聚乙二醇固定相之间的作用力存在本质上的差异,因此通过调整柱温条件是可以分离的。下面三幅图是用60米*0.53mm*1um的INNOWAX柱分离乙酸乙酯、甲醇、丁酮的效果,柱温分别是40℃、50℃、60℃。[img=,690,796]http://ng1.17img.cn/bbsfiles/images/2018/08/201808022157168864_5041_2204387_3.png!w690x796.jpg[/img][img=,690,796]http://ng1.17img.cn/bbsfiles/images/2018/08/201808022157170984_7926_2204387_3.png!w690x796.jpg[/img][img=,690,796]http://ng1.17img.cn/bbsfiles/images/2018/08/201808022157172914_736_2204387_3.png!w690x796.jpg[/img]图中很明显,柱温低时甲醇与丁酮出峰时间接近分不开,高温时甲醇与乙酸乙酯出峰时间接近分不开,温度适中时三者可以实现分离。虽然未达到基线分离,但分离度都超过1,用来定量是完全可以的。这是找别人借的一根旧柱子,柱效只有4万塔板,如果是新柱子柱效应该能达到七八万塔板,分离度肯定更高,如果是0.32mm口径的柱子分离就更没问题了。要强调的是,能够实现分离的条件并不是完全靠盲目尝试获得的。我们看一看三种目标物的保留时间随柱温的变化就能发现其中的规律,见下图:[img=,594,716]http://ng1.17img.cn/bbsfiles/images/2018/08/201808022156374904_6999_2204387_3.png!w594x716.jpg[/img]图中可以看出,三种目标物的保留时间都是随温度升高而减小的,但是减小的幅度却并不相同。甲醇的保留时间随温度升高而减小的幅度明显大一些。这是因为甲醇具有羟基,与聚乙二醇固定相的相互作用力以氢键为主,氢键的强度随温度升高而迅速减弱。而乙酸乙酯、丁酮与聚乙二醇固定相的作用力都是以诱导力和取向力为主,这种力是由分子偶极矩决定的,受温度的影响要小一些。甲醇峰位置在乙酸乙酯与丁酮之间,温度升高时保留时间都减小,但甲醇减小更多,于是甲醇与乙酸乙酯靠的更近,与丁酮的分离度提高。温度降低时保留时间都增大,但甲醇增大更多,于是甲醇与丁酮靠的更近,与乙酸乙酯的分离度提高。用其他的柱子,如DB-wax或者FFAP时,各组分之间的相对位置会有差别,甚至有时出峰顺序都会变,但是保留时间随温度变化的这种规律仍然是适用的。所以遇到分不开的情况,一定不要盲目的乱试一通,也不用盲目的换柱子,一定要把问题想明白,有针对性的优化条件。最后要强调的是,这里虽然是以溶剂检测为例讨论了如何只用一根柱子就实现分离,但实际样品很复杂,并不是每次都能通过这种优化实现全部分离目的。所以色谱实验室配备多种不同极性的色谱柱是非常重要的。特别是做复杂样品时,即使谱图上看起来分离不错,最好也能用另外一种柱子进行一次验证,以免实际样品中有干扰物共流出,造成假阳性。
有谁做过糖精钠中邻甲苯磺酰胺和对甲苯磺酰胺?按2015版药典设置柱温180度。用fid检测器做,检测器设置为280,进样口250。只有溶剂峰,没见目标峰,有谁做过,请教一下色谱条件。
要检测,乙醇,异丙醇,二氯甲烷,甲苯,四种溶剂。用SH-RTX-5色谱柱,30m*0.32mm*0.25um分离效果不好,用WAX柱,百分百聚乙二醇色谱柱,30*0.53*0.25分离度也不好,分流比30,柱温40℃,流速也几乎调到最低标准了?,RTX-5设置的是1.2ml/min,wax柱设置的是3.0ml/min乙醇,二氯,异丙醇,三个都出的非常近。两种方法都是2.5-2.8分钟全部出峰了。而且有重叠







 我要推广仪器
我要推广仪器
 下载APP
下载APP