瑞士罗氏制药公司昨天向外界简单介绍了抗禽流感药物“达菲”的成分和生产工艺。出人意料的是,“达菲”成分中竟包含一种可爆炸物质、一种中国调味料以及一种无害的大肠杆菌。 “达菲”共有12道生产工序,最初是从八角茴香中提取莽草酸。 由于30公斤八角茴香只能加工出1公斤莽草酸,而八角茴香只在中国4个省份生长,通常在5月和3月收获,因此有人担心一旦收成欠佳或价格过高,罗氏公司将无法获得足够原料。 罗氏公司为此开发了不用八角茴香制造莽草酸的方法,并在去年与德国一家生物技术中心合作。目前,罗氏公司三分之二的莽草酸取自八角茴香,余下三分之一由大肠杆菌提供。 莽草酸经过反应器、过滤器和干燥器这三道工序后,转变为一种中间化学物质环氧化物。随后,环氧化物的结构必须被打开,通过原子转换,变成另外一种中间化学物叠氮化物。接着,叠氮化物被制成活性成分,再与其他添加成分混合加工,最后经过真空干燥制成“达菲”颗粒。 [img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=38952]莽草酸的hplc图谱[/url]

实验室打算用GC-MS检测一下莽草酸,用的衍生剂是MTBSTFA,莽草酸中六元环上有三个相邻的羟基还有一个羧基,不知道衍生剂能否将其完全衍生化,会不会有空间位阻。http://ng1.17img.cn/bbsfiles/images/2012/12/201212021521_408726_2030451_3.jpg
现在吵的很热的就是莽草酸了是吧!想找一条提取分离途径过把研究瘾哈哈成全谢谢
莽草酸的紫外分析,测定其温度、光照、ph值稳定性研究。遇到如下问题,请高手解决:1、我在网上查的资料,莽草酸最大吸收波长在213-219nm之间,但我测的最大吸收波长在213nm以下,基本上在203nm左右,我是用水做溶剂的,请问这跟仪器有关吗?我用的是日立3010的机子!2、在做ph值的稳定性,请教高手,莽草酸显酸性,ph值约为5,做稳定性要做到几最合适,我设计的时候是从1-14的ph值。请问高手是否合理?3、我做的浓度梯度吸光值随浓度增加而增大,我用500ug/ml的浓度,最大吸光值有3点几,20ug/ml时在1点几到2点几之间,请问吸光值和浓度有关系吗?4、稳定性的研究不一定是吸光值要变小,增大也能反应出稳定性不好吗?我查的资料都是说稳定性不好,那么吸光值就小。以上问题请高手赐教,我是新手。谢谢
求莽草酸的紫外检测方法,我只知道在213nm处有最大吸收,但不知道该如何处理样品,如果直接测得话,不管多小的浓度,都是吸收太大!请专家指点一下~[em06]********************************************************************八角茴香可做治禽流感药物 中国茴香农收益大增http://news.china.com/zh_cn/domestic/945/20051123/12879126.html达菲是一种药物,能够减轻禽流感症状, 八角成了达菲主要成分莽草酸的来源.
反相离子对色谱法测定马尾松松针中莽草酸的含量 马廉举, 刘 新(重庆医科大学药学院, 重庆400016)摘要 目的: 建立反相离子对色谱法测定马尾松松针中莽草酸的含量方法。方法: 采用D iamonsil C18色谱柱( 250 mm @ 41 6 mm, 5 Lm ), 流动相为5 mmo l/L磷酸溶液( 先用2 mo l/L氢氧化钠调至pH 612, 再加入四丁基溴化铵, 使其浓度为1 mm o l/L)-甲醇( 90B10), 检测波长为217 nm, 流速为11 0 m l/m in, 柱温为25e 。结果: 莽草酸在5~ 300 Lg /m l范围内与峰面积呈良好的线性关系( r= 01 9999), 样品的平均回收率为97151%, RSD为0199%。结论: 此方法准确、简便, 适用于马尾松松针中莽草酸的定量分析。关键词 莽草酸; 马尾松松针; HPLC; 离子对
[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=102813]反相高效液相法测定八角茴香果实中的莽草酸[/url]

2010年版药典(一部)中,对益母草中盐酸水苏碱的测定有如下描述(以丙基酰胺键合硅胶为填充剂):http://ng1.17img.cn/bbsfiles/images/2011/01/201101080907_272670_801_3.jpg那么为什么要用丙基酰胺柱来测盐酸水苏碱呢?丙基酰胺硅胶基质的柱子是什么柱子呢? 首先我们要了解盐酸水苏碱的特性,盐酸水苏碱的极性极大,普通的反相色谱对它的保留分离能力较弱,通常在死时间里流出而无法得到分离,而亲水作用色谱HILIC能为极强性的化合物提供良好的保留,在此类化合物上应用广泛。 目前已有多种商品化的HILIC色谱柱,多为硅胶基质,键合不同极性基团,如丙基酰胺基,酰胺基,聚琥珀亚酰胺等极性基团,氨基键合硅胶柱由于使用寿命较短,键合相容易流失,造成保留 丙基酰胺键合硅胶克服了传统正相色谱柱在水相条件下不稳定的缺点,其常使用流动相是和反相色谱相同的水相缓冲液( 40%)及有机溶剂,但是其梯度条件通常是初始为高比例有机相,逐步加大水相含量;极性丙基酰胺键合硅胶的HILIC色谱柱在反相条件下,可以有效的保留极性化合物,是一种崭新的极性化合物HPLC分离解决方式.博纳艾杰尔推出的Venusil HILIC (丙基酰胺键合硅胶),就是一样一款非常适合于益母草中盐酸水苏碱测定的柱子,测定方法及谱图如下:色谱柱:Venusil HILIC (丙基酰胺键合硅胶),4.6×250mm,5µm,100Å(订货号:VH952505-0)流动相:乙腈-0.2%冰醋酸(80:20)流速:0.5mL/min柱温:25℃进样体积:20μL检测器:ELSD蒸发光散射检测器http://ng1.17img.cn/bbsfiles/images/2010/11/201011291710_262707_801_3.jpg益母草供试品含量测定色谱图(主峰保留时间:22.697min)
高效液相色普法测定甘草酸苷含量,甘草酸苷对照品峰面积与之前相比较偏高,导致含量偏高,请问有可能是哪些原因?

[align=center][img=,287,192]http://ng1.17img.cn/bbsfiles/images/2018/01/201801171022069711_9983_676_3.png!w287x192.jpg[/img][/align][align=left]八角俗称大料,颜色有红棕和黄棕色的,轻轻闻一下,有芳香味和甜味。八角常用于炖肉中,可除去肉的臭气,起添香作用,所以它又被称为大茴香。八角外形上并不仅限为8个角,还有10个角,或10多个角的,都叫八角,哪种八角好?[b]优质八角[/b]好的八角都是小胖子,质硬而脆,瓣角整齐完整,尖角平直,个大饱满,颜色为棕红色,果皮较厚,表面明亮有光泽,内表面两侧颜色较浅,背面粗糙有皱缩纹。通常为8个角,也有7个或9个角的。荚边有较大的裂缝,裂缝内藏着圆鼓鼓的籽粒。好的八角会发出甘草的香味,且香味较浓烈,味辛、甜。[b]劣质八角[/b]不好的八角角型细长,朵瘦,无籽,呈暗棕色或黑褐色,表面可能还会有虫斑。通常为10多个角,荚角角尖弯弯曲曲,角尖上翘,像个鸟嘴。劣质的八角气味较差。[/align][align=left][/align][align=center][img=,346,192]http://ng1.17img.cn/bbsfiles/images/2018/01/201801171022569325_3422_676_3.png!w346x192.jpg[/img][/align][align=left][b][color=#cc0000]八角和莽草的区分[/color][/b][/align][align=left]从外形上看,大料比莽草大; 从外形上看,莽草比大料小。[/align][align=left]大料有7到9个角,最常见的为8个角; 莽草有10到12个角,最常见的为10个角。[/align][align=left]大料角上的尖端是直的; 莽草尖端是钩状的。[/align][align=left]八果果柄较长,且果实外露; 莽草果柄较短,且果实比较封闭。[/align][align=left]八角味甜; 莽草味酸。[/align][align=left]这下不会出错了吧,要认真挑选哟,莽草是有毒的。[/align][align=left][/align]
摘要:建立胡黄连中香草酸和桂皮酸的含量测定方法。方法用双波长扫描法测定胡黄连中香草酸和桂皮酸的含量。结果香草酸。桂皮酸斑点峰面积3Il内稳定,香草酸回收率为103.86%,RSD=1.33%,桂皮酸回收率为103.16%,RSD=1.28%。结论该方法稳定,可行。具有实用性。 关键词:胡黄连 薄层扫描法 香草酸 桂皮酸 胡黄连具有保肝利胆、抗炎、抗真菌等药理作用。胡黄连含胡黄连素、胡黄连苷(I II III)、D-甘露醇、香草酸、肉桂酸、胡黄连醇成分。香草酸和桂皮酸是其中的两种抗菌成分。我们对胡黄连中香草酸、桂皮酸含量建立了薄层扫描法,以达到控制胡黄连的质量,从而为临床疗效提供保证。 1 仪器与试剂 药材:胡黄连,太原市药材公司;仪器:日本岛津CS--9301PC薄层扫描仪;手提式荧光灯(上海固村电光仪器厂);对照品:香草酸对照品(中国药品生物制品检定所);桂皮酸对照品溶液(省药检所提供e=0.604mg/50ml);硅胶GF254(青岛海洋化工厂)所用试剂均为分析纯。 2 实验条件 2.l 薄层层析条件:分别以石油醚-氯仿-丙酮-冰醋酸(10:4.4:10.1);正己烷-乙醚-冰醋酸(5:5:0.1);正己烷-氯仿-乙醚-冰醋酸(5:3:2:0.1)以及氯仿:甲醇(2:1)展开,多次比较发现正己烷。氯仿-乙醚-冰醋酸(5:3:2:0.4)分离效果好。 2.2 测定波长及主要扫描参数,分别对香草酸,桂皮酸对照品斑点在200nm-370nm扫描,在290nm处有最大吸收,350nm处无吸收,固定350nm为参比波长,290nm为测定波长。
最近做小儿七星茶颗粒甘草酸的测定时遇到了很奇怪的问题:对照品有峰,样品却只出杂质峰,开始怀疑是样品没有含量,可是拿以前做过的有含量的样品再做,却没峰了;后来吸一半对照品一半样品进样就出峰了,可是加对照品到样品中一起按标准处理后就又没有峰出来,怎么想都不明白问题出在哪里。所用的试剂换了好几次,也换人配了,结果还是一样没有,会不会是超声引起的呢,因为我们的超声机的功率只有80瓦,不过以前也做得出啊,大家帮帮忙,下面是标准【含量测定】 照高效液相色谱法(中国药典2005年版一部附录VI D)测定。 色谱条件与系统适用性试验 以十八烷基硅烷键合硅胶为填充剂;以甲醇-0.2mol/L醋酸铵溶液—冰醋酸(65:35:1)为流动相;检测波长为250nm。理论板数按甘草酸峰计算应不低于2000。 对照品溶液的制备 取甘草酸铵对照品适量,精密称定,加流动相制成每1ml含16μg的溶液,即得(折合甘草酸为15.672μg)。 供试品溶液的制备 取装量差异项下的本品内容物,混匀,研细,取约7g,精密称定,置50ml量瓶中,加流动相约45ml,超声处理(功率300W,频率40kHz)30分钟,放冷,加流动相至刻度,摇匀,滤过,取续滤液,即得。 测定法 精密吸取对照品溶液与供试品溶液各20μl,注入液相色谱仪,测定,即得。

俗称大料的八角顾名思义应该有8个瓣,是家庭必备的佐料,但是如果买到的大料多于8个角,那有可能是含有剧毒的莽草。 莽草与八角同属茴香科,但莽草属于一种剧毒药。执业中药师、金象大药房商品部赵部长介绍,八角有几种伪品,市场上有红茴香、多蕊红茴香、莽草、野八角、短柱八角。其中莽草、野八角和短柱八角都有10个以上的瓣。http://ng1.17img.cn/bbsfiles/images/2010/12/201012011358_263352_1641058_3.jpg 八角有多种伪品 莽草与八角同属茴香科,但莽草属于一种剧毒药。执业中药师、金象大药房商品部赵部长介绍,八角有几种伪品,市场上有红茴香、多蕊红茴香、莽草、野八角、短柱八角。其中莽草、野八角和短柱八角都有10个以上的瓣。 在这些伪品中,只有莽草有毒,10至13个瓣的应该就是莽草,果实瘦长,尖端还带钩,背面粗糙。“莽草闻起来气味、口味较弱,久尝后有麻舌的感觉。它确实有剧毒,人食用之后会出现昏迷、休克、口吐白沫等症状,最严重的后果甚至将直接致死。” 如何区别假八角,正宗的八角为红棕色,挑选时应找果实饱满的,除了通过8个角辨认,八角的种子要选黄褐色有光泽且气味浓郁的。“红茴香与八角最难区分,相对来说,红茴香果品较薄,外表比正常茴香粗糙,有皱纹,气味稍弱些,味酸。” 从外形上看,真正的八角呈褐棕色,角瓣整齐且往往都是半开不开的,露出里面的种子来。假八角往往色泽浅黄,角瓣不整齐,角的尖端带钩。还有一个方法是从味道上鉴别,正宗的八角味道辛辣,香味足,但不会麻嘴;假八角滋味淡,有一股类似柚叶、樟脑、松针的气味,有麻舌感。你到市场上,可以自己观察。
草酸亚铁的确定,是分析草酸根还是分析铁含量,那种分析方法更有效,如何分析?
市场真假大料混着卖http://www.people.com.cn/mediafile/pic/20101206/91/9766785055700268591.jpg八角茴香(大料)http://www.people.com.cn/mediafile/pic/20101206/81/10894704441264001393.jpg莽草冒充八角茴香(大料)的莽草,莽草有剧毒;如果在市场上买到和莽草一样的假大料,怎办?http://www.people.com.cn/mediafile/pic/20101206/32/751059241538477108.jpg短柱八角
原子荧光测铅有可能空白荧光强度达到580(负高雅270V灯电流60mA),正常情况是空白荧光强度280。北京的专家说一般铁氰化钾含铅。回来试验,发现不是铁氰化钾的质量问题,也不是盐酸含铅(我买过一瓶高纯的进口盐酸,和我蒸馏提纯前后的盐酸含铅空白一样好。)。我改用酒石酸代替草酸实验(可以采用酒石酸、柠檬酸、邻苯二甲酸、乙酸等代替草酸实验,效果和草酸一样,参阅《中国金属学会第13届分析测试学术年会论文集》2006年P37),证实了是草酸的问题。估计草酸含铅约10mg/kg,符合试剂标准20mg/kg要求。我的铁氰化钾、盐酸、草酸都是进口的,所以不要盲目迷信进口的试剂一定好。我以前用的铁氰化钾、盐酸、草酸都是国产的质量也很好。昨天用了一瓶广州化学试剂厂的铁氰化钾,和美国进口的铁氰化钾比较,铅的空白是一样的好。所以不是盐酸含铅过高的问题。我发帖是提醒朋友们注意到草酸这种络合剂很可能含铅,这是没有人提到过的情况。
请问用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]如何把乙酸、丙酸及草酸分析出?用酯化吗?

问题:四君子颗粒中甘草苷、甘草酸铵的检测对照品分析中甘草苷与甘草酸铵的分离度是?答案:62.445【活动奖励】因zgx3025(注册ID:v2844608)的答案不正确,所以取消本次获得的钻石币幸运奖(2钻石币):抽奖软件,当天随机抽取3个回答正确的版友ID号(最后一个ID号,截止至下午3:00),每人奖励2个钻石币mengzhaocheng(注册ID:mengzhaocheng)莫名其妙(注册ID:moyueqiu)http://ng1.17img.cn/bbsfiles/images/2016/03/201603031621_585902_1610895_3.pnghttp://ng1.17img.cn/bbsfiles/images/2016/03/201603031621_585903_1610895_3.png积分奖励:所有回答正确的版友奖励10个积分(幸运奖获得者除外)。【注意事项】同样的答案,每人只能发一次PS:该贴浏览权限为“回贴仅作者和自己可见”,回复的版友仅能看到版主的题目及自己的回答内容,无法看到其他版友的回复内容。下午3点之后解除,即可看到正确答案、获奖情况及所有版友的回复内容。=======================================================================四君子颗粒中甘草苷、甘草酸铵的检测样品制备制备方法1. 对照品:取甘草苷对照品、甘草酸铵对照品适量,精密称定,加甲醇制成每1 mL分别含甘草苷20 μg、甘草酸铵0.2 mg溶液,即得(甘草酸重量=甘草酸铵重量/1.0207)。2. 供试品:取本品装量差异项下的内容物3 g,精密称定,置具塞锥形瓶中,精密加入甲醇25 mL,密塞,称定重量,超声处理(功率250 W,频率40 KHz)30分钟,放冷,再称定重量,用甲醇补足减失的重量,摇匀,滤过,精密量取续滤液15 mL,蒸干,残渣加甲醇使溶解,移至5 mL量瓶中,加甲醇稀释至刻度,摇匀,滤过,取续滤液,即得。分析条件色谱柱Platisil ODS 250 x 4.6 mm,5 μm (Cat#:99503)流动相A:乙腈 B:0.05%磷酸溶液 梯度流速1.0 mL/min柱温30 ℃检测器UV 237 nm 进样量10 μL 色谱图对照品http://ng1.17img.cn/bbsfiles/images/2016/03/201603031020_585805_1610895_3.jpg 峰号 保留时间 min 峰面积 μV*s 峰高 μV 理论塔板数* N USP拖尾因子 分离度 1 15.739 771814 49202 22131.352 0.998 -- 2 36.170 766340 93054 391608.534 1.043 62.445 *药典要求理论板数按甘草苷峰计算应不低于5000供试品http://ng1.17img.cn/bbsfiles/images/2016/03/201603031021_585807_1610895_3.jpg 峰号 保留时间 min 峰面积 μV*s 峰高 μV 理论塔板数* N USP拖尾因子 分离度 1 15.784 475765 27766 18773.718 0.973 -- 2 36.033 152478 18510 403100.536 0.997 58.879 *药典要求理论板数按甘草苷峰计算应不低于5000本品种同时使用了Diamonsil C18、DiamonsilC18(2)两款色谱柱,在药典规定条件下进行甘草苷、甘草酸铵的检测,均满足药典要求。
有那位化学分析高手指教,草酸亚铁中硫酸根的测定方法,急
请问草酸亚铁的国家标准或行业标准那里有?
请问磷酸二氢铵、草酸亚铁的国家标准在那里可以找到?
草酸测水分选用卡尔费休水分仪的时候草酸会干扰并且附着在电极上方,在测定过程吸水导致测定结果偏大;选用快速水分仪105℃测定时间5min草酸会升华。请教下各位这个草酸的水分测定是不是可以用快去水分仪降低温度去测定,又或者是使用减压干燥80℃消失的重量就是水分这种方式去测定?
哪位高手知道,测试有机酸如,苹果酸,乙酸,草酸,丙酮酸,甲酸等用什么柱子啊和流动相啊?
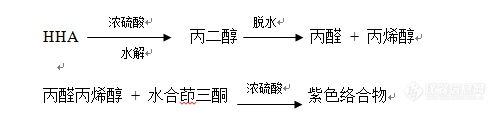
[align=center][b]羟丙基透明质酸质量标准的建立[/b][/align][align=center]杨桂兰,臧恒昌[b][/b][/align][b]摘要:[/b]透明质酸(HA)具有保湿、润滑、营养、修复和预防损伤等生理功能,在维持组织完整性方面和促进感染、损伤、胚胎发育过程中组织形成和重塑方面发挥重要作用。在化妆品、食品及医药领域的应用越来越广泛。但HA容易被体内透明质酸酶降解,体内留存时间短。研究者们期望通过对其进行修饰,得到抗酶解的HA衍生物,延长体内保留时间。修饰HA的衍生物近年来主要致力于将其修饰为两亲性衍生物,对抗酶解活性也有研究;这种亲油亲水性使其不仅能够降低降解速率,而且能够降低表面张力。其次,两亲性HA可以解决美容填充时HA分子量过大,黏度过高,注射困难的问题,修饰后的两亲性HA具有黏度降低(相同分子量相同浓度)的优点。HA两亲性衍生物也可作为生物可降解性的药物载体。 本文参考羟丙基淀粉取代度测定方法,建立了采用分光光度法测定羟丙基透明质酸(HHA)取代度的方法。同时摸索了HHA的抗酶解活性检测法、干燥失重、pH、蛋白含量及微生物等关键指标的测定方法。[b]关键词:[/b]透明质酸;羟丙基透明质酸[align=left] 本研究为确保自制羟丙基透明质酸的质量,特制定一系列产品的质量检验标准。[b]1分子量测定1.1材料[/b] NaCl(AR),NaN[sub]3[/sub] (CP) ,BSA(Roch);高效液相色谱仪,(美国Agilent);多角度激光光散射仪,DAWNEOS,美国Wyatt。[b]1.2方法[/b] 测定条件:流动相:0.2mol/L NaCl (包含0.02% NaN[sub]3[/sub]);流速:0.6ml/min,样品浓度:0.05 mg/ml;柱温:35 ℃,进样体积:500 μl。按照仪器操作规程进行操作。[b]2取代度测定2.1原理[/b][/align][align=center][b][img=,497,113]http://ng1.17img.cn/bbsfiles/images/2018/07/201807251603456954_6718_3389662_3.png!w497x113.jpg[/img][/b][/align][align=left][b]2.2材料[/b] HHA;水合茚三酮、1,2-丙二醇、浓硫酸、亚硫酸氢钠、可见分光光度计、具塞比色管(25 ml),容量瓶(100 ml、1000 ml)[b]2.3方法 [/b] 丙二醇标准溶液的配制:准确称量1.0g丙二醇溶液于1000 ml容量瓶中,加纯化水稀释至刻度,然后分别取2、4、6、8、10 ml于100 ml容量瓶中,定容至刻度,得到丙二醇含量分别为20、40、60、80、100 mg/ml的溶液。[b]2.3.1丙二醇标准曲线的制备[/b] 分别吸取上述丙二醇溶液0.5 ml于25 ml具塞比色试管中,置于冰浴中,逐滴加入4 ml浓硫酸(不宜加入过快,并不时震荡)混合均匀后置100 ℃的水中加热3 min(秒表控制),取出后立即放入冰浴中,冷却至15℃,沿管壁加入水合茚三酮试剂0.3 ml,边加边摇匀;在25 ℃的水浴中放置80 min,再用浓硫酸稀释至12.5ml(约7.7 ml浓硫酸)。缓慢倾倒混匀后(不要用混合器震荡),静置5 min,用1 cm比色皿于590 nm波长处测定溶液的吸光度,绘制吸光度—浓度曲线,拟合丙二醇标准曲线方程。 空白:以相同条件下不加丙二醇溶液作空白。[b]2.3.2试样的测定[/b] 分别称取0.05 g~0.1 gHHA及制备该批HHA所用HA粉末于100ml的量瓶中,量取25 ml的0.5 mol/L的硫酸,缓缓加入量瓶中。置于100℃水浴中加热,缓缓摇动,至试样完全溶解,冷却,用纯水定容,量取0.5ml此溶液置25ml比色管中,其余如上述丙二醇的配制方法。羟丙基含量和取代度算法分别如公式1、2所示。[/align][align=center][img=,411,64]http://ng1.17img.cn/bbsfiles/images/2018/07/201807251608249134_5590_3389662_3.png!w411x64.jpg[/img][/align][align=center]注: C:试样中丙二醇含量,由吸光度计算得出; m:取样量;0.7763:转换系数;[/align][align=center][img=,387,58]http://ng1.17img.cn/bbsfiles/images/2018/07/201807251610349286_9676_3389662_3.png!w387x58.jpg[/img][/align][align=center]注:6.9190:HA分子量/环氧丙烷分子量[/align][align=left][b]3抗HAase降解特性3.1材料[/b] 注射用透明质酸酶(HAase)(上海第一生化药业有限公司、1500单位/瓶);缓冲液(磷酸二氢钠:0.0057 g、磷酸氢二钠:0.0230 g、氯化钠:9.0 g、纯化水:1.0 kg);平氏黏度计,Φ1.0 mm、Φ2.0 mm;恒温水槽,上海仪表仪器厂; DK-8D数显恒温水浴锅,金坛市医疗器械厂。[b]3.2方法[/b] 称取HHA和对照HA各 0.5 g份于150 ml肖特瓶中,加入50 ml缓冲液,震荡至完全溶解。用氢氧化钠溶液或HCl溶液调节pH值6.0~7.2,取溶解液10.0 g,纯水稀释5倍;作为起始样品测黏度。取1500单位的酶用缓冲液稀释10倍,分别吸取40单位加入上述HA和HHA溶液中,摇匀,放入37℃的水浴中降解,24 h取样:称取10.0gHA溶液于50 ml容量瓶中,加入纯化水稀释至刻度线,加热煮沸2min,冷却至室温,测其在25℃下的运动黏度,算法如公式3所示。[/align][align=center] [img=,449,41]http://ng1.17img.cn/bbsfiles/images/2018/07/201807251629104708_3045_3389662_3.png!w449x41.jpg[/img][/align] 24小时黏度下降率Δη低于75%。[align=left][b]4透光率的测定4.1材料[/b] 紫外-可见分光光度计、电子天平(精度0.01g)[b]4.2方法[/b][/align][align=left] 取本品0.50g至盛有100 ml水的锥形瓶中,在冰箱中放置过夜,溶解后,纯水作为空白,参考紫外-可见分光光度计操作规程,550 nm波长处测定溶液的透光率。[/align][align=left][b]5pH的测定5.1材料[/b][/align][align=left] 电子天平(精度0.01g)、pH计、磁力搅拌器、磁子、100 ml锥形瓶、100 ml量筒、新沸放冷的纯化水。[/align][align=left][b]5.2方法[/b][/align][align=left][b]5.2.1 溶解[/b][/align][align=left] 称取供试品0.10 g,置锥形瓶中。加新沸放冷的水100 ml和磁子,将锥形瓶用封口膜封口,将锥形瓶置磁力搅拌器上搅拌约4小时,完全溶解,目测为均一透明溶液。[b]5.2.2 测定[/b][/align][align=left] 按照所用pH计的操作规程,先对pH计进行校准,之后将电极和温度探头深入被测溶液中,缓慢搅拌,读取pH值。[b]6运动黏度的测定6.1材料[/b][/align][align=left] 电子天平(精度0.1 mg);平氏黏度计(毛细管内径为1.0 mm ± 0.05 mm);恒温水浴:控温精度±0.01 ℃;秒表:分度0.01秒;振荡器。[b]6.2方法[/b] 称量样品0.1 g(折干),置100 ml容量瓶中,加水振荡至溶解后作为供试液。取毛细管内径为1.0mm ± 0.05 mm的平氏黏度计,加入5 ml供试液,置水浴中,25 ℃下放置15分钟后,秒表测定供试液流过黏度计两条线之间的时间,取两次测定的平均值按下式计算,即为供试品的运动黏度,计算方法如公式4所示。[/align][align=left] 运动黏度ν(mm[sup]2[/sup]/s)=[i]Kt [/i]公式(4)[/align][align=center]式中 [i]K[/i]为用已知黏度的标准液测得的黏度计常数,mm[sup]2[/sup]/s[sup]2[/sup];[/align][align=center][i]t[/i]为测得的平均流出时间,s;[/align][b]7干燥失重7.1材料[/b] 卤素水份测定仪,HHA样品;[b]7.2方法[/b][align=left] 取本品约1.0g,置HG53 型卤素水分测定仪托盘内。110 ℃测定15分钟,记录测定结果。[b]8细菌、霉菌及酵母菌测定8.1供试液制备 [/b][/align][align=left] 取34ml无菌磷酸盐缓冲液1瓶,将1500U HAase加入其中,用吸量管各吸取1ml分别加入至4个平皿中,作为阴性对照。再取样品1.5 g,加入到做完阴性对照的含有HAase的30 ml磷酸盐缓冲液中,42℃下振荡溶解,制得 1﹕20的供试品溶液。[b]8.2 细菌总数测定[/b](1)阴性对照试验将温度低于45℃溶化的营养培养基分别注入上述2个含有1 ml的磷酸盐缓冲液的平皿中,每个平皿约15~20 ml左右,凝固,倒置培养。均不得有菌生长。(2)样品测定用吸量管准确吸取上述1∶20的供试液2ml加入至8 ml的磷酸盐缓冲溶液中,混匀,作为1∶100的稀释级。向平皿中分别加入1∶20、1∶100的供试液各1 ml,向每个平皿注入温度低于45℃的事先溶化的营养琼脂约15~20 ml,待凝固后倒置放入培养箱中。每个稀释级均制备2个平板。[b]8.3 霉菌及酵母菌数测定[/b](1)阴性对照试验 分别注入向2个含有1ml的上述磷酸盐缓冲液的平皿中将温度低于45℃溶化的玫瑰红钠琼脂培养基,每个平皿约15~20 ml左右,凝固,倒置培养,均不得有菌生长。(2)样品测定 用吸量管准确吸取上述1∶20供试液2ml加入至8 ml的磷酸盐缓冲溶液中,混匀,作为1∶100的稀释级。各吸取1∶20、1∶100的稀释级的供试液1 ml加入至平皿中,注入温度不超过45 ℃的溶化玫瑰红钠琼脂培养基,每个平皿约15~20 ml,待凝固后,倒置培养。每个稀释级均制备2个平板。[b]8.4 结果[/b] 将营养琼脂培养基和玫瑰红钠琼脂培养基平板分别倒置于30~35℃、23~28℃生化培养箱中,营养琼脂平板培养3天,用于细菌计数;玫瑰红钠琼脂培养平板培养5天,用于霉菌、酵母菌计数,按照稀释比例,计算出每克样品中的微生物数。[b]9 结论[/b] 采用制定的质量标准对产品检验,结果表明,HHA能保持HA的润滑性和流动性,也具有明显的抗HAase降解的特性;克服了HA衍生物抗酶解但缺少润滑性的缺点,预期用途是开发成骨关节注射液或皮下注射填充剂用于美容,期望能够延长体内保留时间起到长效治疗的作用,减少患者注射次数,减轻患者痛苦。[/align][align=center]参考文献[/align] 赵凯, 刘丽艳, 刘婧婷. 分光光度法测定羟丙基淀粉取代度. 食品科学,2011, 32(22) : 201-203.[align=center][b][/b][/align][align=center][b][/b][/align]
草酸学名乙二酸,化学式HOOC-COOH。当我们从课本上了解了羧酸的化学性质,是不是也能推断出草酸的一般性质了?在我们身边,草酸一般用作除锈剂或者可以除去白衣衫上的墨水污迹,而它其实也是一种可以能致人死命的危险的化学物质。可是大家知道平时爱吃的巧克力中也含有草酸吗?不要慌张,这种危险情况极少出现。我们每天都通过许多不同渠道摄入草酸,草酸在很多食品中都有少量存在,而在少数食品中含量很高。可可就属于含量最高的食品之一,,每100克可可中含有500毫克草酸;绿色蔬菜中的草酸含量一般很高,每100克菠菜含600毫克,大黄含500毫克,甜菜、花生、茶中也有较多的草酸。平均一个人一天大约摄入150毫克草酸,而草酸的致死剂量是1500毫克左右。我们在普通的一天中会摄入这么多草酸吗?那么摄入草酸对我们人体有什么影响? 大黄在美国曾被称为“食用大黄”,在过去,人们常把它和糖放在一起炖了吃。大黄最出名的特性是治疗便秘,因为它能刺激肠道排出自然毒素——草酸。一碗炖烂的大黄里含有的草酸已经接近于使人中毒的剂量。第一次世界大战期间,由于有人把大黄叶当作蔬菜吃,以至于草酸中毒身亡。而吃巧克力则无须担心,无论你对巧克力多么喜爱,但巧克力中的草酸含量太低,就是你吃的无法下咽的时候,体内的草酸含量达不到让你腹泻的程度。 在大黄流行的时候,烹制大黄食品方法层出不穷,曾经使用铝锅来炖大黄,发现意想不到的好处:它能把铝锅“炖”的很干净。之所以有这样的效果,是因为草酸能把铝锅氧化膜和表面金属溶解掉。当然,这种方法还会使食者摄取铝元素造成潜在的危害。

在一次失效分析的过程中,发现别的实验室在分析我们自己的日常样品中,有比较明显的草酸的结果显示,但是自己内部的测试结果没有很好地显示出来。因为之前没有需要报告草酸这个项目,所以也没有买过草酸的标准溶液。为了研究内部是否可以测试草酸,需要另外购买草酸的标准液(Fig-1)。[align=center] [img=,435,422]https://ng1.17img.cn/bbsfiles/images/2023/09/202309011031011657_3849_2942222_3.jpg!w435x422.jpg[/img][/align] 等草酸标液到了之后,开始配置不同浓度的标液,从低到高开始尝试进样,但是都没有在谱图上发现明显的出峰。到了150ppm仍然没有检测到很明显的峰,这是比较不正常的。在观察谱图时,发现在22分钟左右有一个翘起来的部分,有点像峰(Fig-2)。但是因为之前的程序设定了运行时间都是22分钟,是不是因为运行时间太短,导致草酸的峰还没有出来呢。[align=center] [img=,690,357]https://ng1.17img.cn/bbsfiles/images/2023/09/202309011031471109_959_2942222_3.jpg!w690x357.jpg[/img][/align] 经过更改程序文件,把出峰时间改到26分钟,在22-23分钟左右看到一个非常明显的草酸出峰(Fig-3),果然是之前的运行时间太短了,导致草酸不能够显示出来。[align=center][img=,690,213]https://ng1.17img.cn/bbsfiles/images/2023/09/202309011032034570_2099_2942222_3.jpg!w690x213.jpg[/img][/align] 为了进一步,进行了草酸的标准曲线验证。在新程序条件运行下,草酸在不同浓度都可以显示出来,可以得到一个非常好标准曲线,R2的值达到0.99以上(Fig-4)。[align=center][img=,690,468]https://ng1.17img.cn/bbsfiles/images/2023/09/202309011032193998_3826_2942222_3.jpg!w690x468.jpg[/img][/align][align=center]另外,有继续进行了草酸的回收率实验。把草酸标准溶液加入到正常的7个阴离子溶液中,草酸能够明显检测出来(Fig-5),回收率大概是107%。[/align][align=center][img=,690,328]https://ng1.17img.cn/bbsfiles/images/2023/09/202309011032369340_1786_2942222_3.jpg!w690x328.jpg[/img][/align] [font=等线]在新程序下运行,发现在日常的产品中也可以明显检测出草酸(Fig-6)。这证明了的确是之前的运行时间太短,导致草酸检测不出来.[/font][align=center][img=,690,502]https://ng1.17img.cn/bbsfiles/images/2023/09/202309011032564960_9557_2942222_3.jpg!w690x502.jpg[/img][/align] 经过这次的草酸寻峰计,还得出了一个教训。在以后研究新的物质时,要提前好好查下相关的文献,因为不同的柱子和淋洗方法都会影响到物质的出峰。如果没有资料可查,需要尝试在尽可能的运行时间去检测,以免有峰遗漏。
甘草提取液,同样的条件,在Agilent ZOBAX Eclipse plus C18 和迪马铂金C18上跑出来的图相比,前者就硬是差一个最大的峰----甘草酸,用对照品进样也发现在前者上不出峰,后用100%乙腈冲很久才出来一大堆杂质峰,应该是吸附在柱子上了。请问这2种都是C18的柱子,为什么会有如此大的差别,前者为什么会产生强吸附?谢谢
我这里有几份分析资料,想和大家分享。内容含有分析条件及色谱图, 有需要的就下载吧!1. 分析食品中的丙酸2. 涂料中一般溶剂(二氯甲烷,乙苯_二甲苯)的分析3. 分析大蒜中的二烯丙基二硫(蒜素)
钼兰比色法测定磷 磷钨酸比色法测定尿酸 变色酸比色法测定草酸 五溴丙酮2硫脲比色法测定枸橼酸
我们是用草酸酸化提取出土霉素。怎样测定药品中草酸残余量呢?我用的是高效液相色谱。用的是反向离子柱。







 我要推广仪器
我要推广仪器
 下载APP
下载APP