请问老师:如何分析盐水溶液中的苯胺和多胺(2,2-二氨基二苯基甲烷,2,4-二氨基二苯基甲烷,4,4-二氨基二苯基甲烷,N-甲基二氨基二苯基甲烷)含量??含量在几个到几十个PPM
如何分析多胺(2,2-二氨基二苯基甲烷,2,4-二氨基二苯基甲烷,4,4-二氨基二苯基甲烷,N-甲基二氨基二苯基甲烷)中水分的含量(应该是几十个PPM)??如果用KF滴定法怎么消除其碱性太强的现象??如果用水杨酸酸中和的话是不是生成水???我们现用的分析方法是加水杨酸然后滴空白再加样品测定水含量.这种方法有问题吗???
请问老师,如果用光谱的方法进行盐水溶液中的苯胺和多胺(2,2-二氨基二苯基甲烷,2,4-二氨基二苯基甲烷,4,4-二氨基二苯基甲烷,N-甲基二氨基二苯基甲烷)含量的测定,它们的吸收峰是多少???
我按gb17378.5《海洋监测规范:沉积物》中测有机碳的方法,配制苯基代邻氨基苯甲酸指示剂,将0.5克苯基代邻氨基苯甲酸溶于2g/L的碳酸钠溶液中。但是苯基代邻氨基苯甲酸只溶解了一点点,加热也不行。做过相关实验的同行,请指点一二。
0.2g N-苯基代邻氨基苯甲酸0.2g碳酸钠的100ml水中,加热溶解是先溶解0.2g碳酸钠,还是0.2g N-苯基代邻氨基苯甲酸,有顺序吗?再问一下,热水温度控制在多少?
苯基代邻氨基苯甲酸是什么颜色的,今天买来的苯基代邻氨基苯甲酸有的怪怪的
最近要做这两个物质的检测, 不知道有没有老师在做的给点意见和相关资料,谢谢苯乙酮 98-86-2 2-苯基-2-丙醇 617-94-7
N-苯代邻氨基苯甲酸与N-苯基邻氨基苯甲酸这二种试剂是不是一样的。那位教师告诉我一下。谢谢!
N-苯基邻氨基苯甲酸中的“N”读成“恩”还是“蛋”
N-苯基邻氨基苯甲酸配指示剂无法溶解,不知道怎么回事,用热水也试了
有人知道荧光增白剂2氨基2苯乙烯含量如何测定吗
俺想用氨基苯乙酮做显色剂,不知道有人用过吗?还存在邻,间,对的问题,不知道大家用的哪个?
慕尼黑上海分析生化展,迪马科技最新推出了Inspire 苯基系列色谱柱,一直忙未给大家及时普及,今天来说说第一款Inspire PFP ( 五氟代苯基) 是Inspire 液相色谱柱家族新成员,针对分离极性化合物过程中的保留时间和分离度问题而特别设计。Inspire PFP 凭借其优异的选择性,可为极性化合物、复杂天然产物、位置异构体和其它相关化合物在C18 和C8 色谱柱上的分离提供一个替代和补充。Inspire PFP 具有U 型色谱分离特性,适用于正相、反相和亲水作用色谱三种分离模式,并具有多种作用机理,因而能够同时分离检测不同极性化合物的混合物,为目前难以解决的复杂极性和亲水性样品的分离分析提供了强有力的工具,可轻松解决其它色谱柱面临的分离难题,为用户实现强极性分析物的优异选择性提供一种更加便捷的途径。同时也为色谱工作者使用简单流动相,避免使用极端pH 条件和准备复杂流动相提供了可能性。Inspire PFP 色谱柱特点• 五氟代苯基硅烷键合在高纯硅胶基质上• 具有U 型色谱分离特性,适用于正相、反相和亲水作用色谱三种分离模式• 对极性化合物具有独特的保留能力• 良好的峰形、超高的柱效、分离度和使用寿命• 适用于芳环类化合物或长共轭体系化合物的分离• 优异的批次重现性增强位置异构体分离能力官能团位置的微小差异可以极大的影响分子性能,在许多情况下,传统的C18色谱柱根本无法扑捉到这种细微的差异。然而,Inspire PFP的多功能选择性却可以区分由于分析物内部微小位置变化而导致的分析物的空间位阻变化还是分析物的偶极矩偏移。色谱柱如图所示 规格 150 × 4.6 mm, 5 μm 流动相0.1% 甲酸乙腈溶液:0.1% 甲酸水溶液 = 40:60 流速1.5 mL/min 柱温室温 检测器 UV 254 nm 样品1. 3,4-二甲氧基苯酚 2. 2,6-二甲氧基苯酚3. 3,5-二甲氧基苯酚4. 2,6-二氟苯酚5. 2,4-二氟苯酚6. 2,3-二氟苯酚7. 3,4-二氟苯酚8.3,5-二甲基苯酚9.2,6-二甲基苯酚10.4-氯-3-甲基苯酚11.4-氯-2-甲基苯酚12.3,4-二氯苯酚13.3,5-二氯苯酚http://www.dikma.com.cn/u/image/2016/09/06/1473147613188048.jpg苯氧酸类化合物分子上卤素的加入可以从根本上增强化合物的极性,而极性的变化通常伴随着反相色谱柱在保留时间和分离能力上困难的增加。此时使用InspireTM PFP 是解决保留问题的最有效的方法。InspireTM PFP利用偶极-偶极和氢键作用更好地保留,区分和分离极性卤化化合物。色谱柱 如图所示规格 150 × 4.6 mm, 5 μm流动相乙腈:0.1% 甲酸水溶液 = 50:50流速1.0 mL/min柱温室温检测器UV 220 nm样品1. 苯氧乙酸2. 邻氯苯氧乙酸3. 对氯苯氧乙酸4. 2,4-二氯苯氧乙酸5. 2,4,5-三氯苯氧乙酸6. 2,4,5-三氯苯氧丙酸http://www.dikma.com.cn/u/image/2016/09/06/1473147817102957.jpg类固醇通过整合偶极-偶极、π-π和氢键机理,InspireTM PFP实现标准反相条件下极性化合物的最佳分离。色谱柱 如图所示 规格 150 × 4.6 mm, 5 μm 流动相甲醇:水 = 60:40 流速1.5 mL/min 柱温室温 检测器UV 254 nm 样品1.泼尼松龙3.地塞米松5.氢化可的松21-乙酸酯7.可的松-21-乙酸酯2.泼尼松4.皮质酮6.11-α羟孕酮8.11-酮孕甾酮http://www.dikma.com.cn/u/image/2016/09/06/1473148006619700.jpg甲基苯乙酮异构体目标分析物上的基团位置变化可以影响化合物的偶极矩,这种变化可以很容易被高电负性的氟原子和其它保留机理察觉,因此InpireTM PFP可以有效地用于分离甲基苯乙酮的位置异构体。色谱柱 如图所示规格 150 × 4.6 mm, 5 μm流动相甲醇:水 = 50:50流速1.0 mL/min柱温室温检测器UV 254 nm样品1. 邻 -甲基苯乙酮2. 对 -甲基苯乙酮3. 间 -甲基苯乙酮http://www.dikma.com.cn/u/image/2016/09/06/1473148212903667.jpg核苷酸和核苷色谱柱 如图所示规格 150 × 4.6 mm, 5 μm流动相0.1% 甲酸水溶液流速1.0 mL/min柱温室温检测器UV 220 nm样品1. 胞嘧啶2. 5'-CMP3. 5'-UMP4. 5'-GMP5. 尿苷6. 胸腺嘧啶 http://www.dikma.com.cn/u/image/2016/09/06/1473148405914511.jpg抗胃酸药色谱柱 如图所示规格 150 × 4.6 mm, 5 μm流动相乙腈:20 mM 磷酸氢二钾(pH 7.0) = 20:80流速1.0 mL/min柱温室温检测器UV 220 nm样品1.法莫替丁2.西咪替丁3.尼扎替丁4.雷尼替丁 http://www.dikma.com.cn/u/image/2016/09/06/1473148597658988.jpg氧化应激标记物色谱柱 如图所示规格 150 × 4.6 mm, 5 μm[/
SVHC中的4,4-二氨基二苯基甲烷和可分解致癌芳香胺中的4,4-二氨基二苯甲烷是同一个吗??有老师说不同,如果不是,cas号分别是什么??能给解释下不?
大神们,帮忙指点一二,配置苯基代邻氨基苯甲酸不溶解啊,如果加热,温度多少度合适,加热能彻底溶解吗?需要过滤吗?
请教大家一下,一种用于固相萃取的试剂:二苯基硼酸二氨基乙酯(DPB或DPBEA)大家用过没有。我在文献上看到sigma公司有卖,可是我没有买到。有谁知道还有其他哪个公司可以买到,或者有其它的替代试剂没有。谢谢大家!!
最近扩项,按照HJ/T 39-1999《 固定污染源排气中氯苯类的测定方法》——无水乙醇洗脱—气相色谱法,对于氯苯类的采样,需要一种很奇特的富集剂。标准原文如下:5.13 富集剂:二乙烯苯与乙基苯乙烯共聚物类多孔高分子小球型载体,比表面积约400㎡/g,颗粒度0.45-0.9mm。事先在脂肪提取器中用无水乙醇(5.1)处理8个小时。晾干后于80℃烘8h,备用采样用的富集柱:于40mm×5mm(内径)的硬质玻璃柱中,填装0.5g富集剂(5.13)并于两段塞少量玻璃棉,或视样品浓度,适当增加柱长度。
本人小小化验菜鸟一枚,最近要检测乙基三苯基溴化磷纯度,尝试过FID[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]检测,但是不出峰,跪求各位大侠支招,给一个检测方法,在此先谢过大家啦
想问trans-stilbene oxide是叫反-氧化-1,2-二苯乙烯还是叫反-2,3-二苯基环氧乙烷,什么地方可以买到
由于蓝月亮 荧光增白剂事情发生,害得我要去查找国外对于荧光增白剂使用的法律法规和限量。命苦,网上有难找。那位朋友有 联苯乙烯二苯基二磺酸二钠——CBS-X, 在欧洲,日本,美国等发达国家的使用情况。如这些国家的法规方面是否有禁用的相关文件。不胜感激。
GB223.4-2008钢铁中锰的滴定法测定中,指示剂N苯基邻氨基苯甲酸是消耗六价铬吗?是的话硫酸亚铁铵的浓度计算中我觉的应该加上校正值,而不是减去,可国标中是减去的,为什么呀?最后计算锰时也是减去校正值的,我觉得应该是加上才对呀?请哪位老师解释一下。
本人从事职业卫生行业,现遇GBZ/T 160.67-2004工作场所空气有毒物质测定 异氰酸酯类化合物中 二苯基甲烷二异氰酸酯(MDI)测定的问题。 具体内容是MDI经吸收液采集会水解生成 4,4-二氨基二苯基甲烷(MDA),标准中叙述在碱性条件下使用甲苯萃取,利用七氟丁酸酐的衍生化,经色谱柱的分离ECD检测器检测。 具体问题系,一、MDA 由MSDS知本身微溶于甲苯和水,标准上说利用甲苯配制标准溶液,但仅仅取50mgMDA溶于50mL甲苯极其难溶解,使用水来溶解也是难于溶解。故通过本人经验和相应的实验依据,认定国标系错误。 请做过同仁给予标准溶液配制指点。二、上述国标本身赘述样品处理和标准曲线的处理本人怀疑本身就有问题,从而造成在下在GC上使用了各种的分离条件(柱子、分析条件等)找不到梯度的目标峰。 请同仁给予样品处理和标准系列处理方面正确方法的指导,请再给我一个GC的分析条件。本人使用30m*0.25*0.25规格的柱子。三、请处理过相同难题的同仁,深切恳请给予色谱图参考,网上相关文献极为匮乏。请色谱同仁给予指导,和处理要点。
在测定不锈钢中铬时,加入指示剂N-苯基邻氨基苯甲酸,进行指示剂校正时,指示剂是消耗六价铬吗?那为什么GB223.4-2008滴定法测锰中,计算硫酸亚铁铵浓度时和最后计算锰浓度时都是减去校正值,我觉得应该加上才对啊,谁能解释解释?
用高效液相色谱分析:邻氯苯基环戊酮; 2-吡啶甲醛;方酸; 2,5-二氧基-4-碘苯乙胺; 苯瞵酸; 戊亚酮胺的含量的检测条件,恳请各位专家帮帮忙啊!
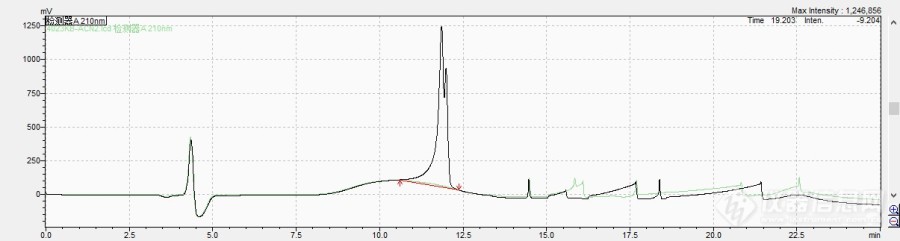
CAS:283159-95-5我用C18的柱子,流动相A、B分别是0.05%TFA的纯水、0.05%TFA的乙腈,流动相B的梯度是初始10%,5min升到60%并保持10min,15min后降到10%保持5min,谱图主峰分叉。[img=,690,184]https://ng1.17img.cn/bbsfiles/images/2024/01/202401271531592473_6839_4185279_3.jpg!w690x184.jpg[/img]请问(S)-3-氨基-3-(4-氯苯基)丙酸甲酯是手性化合物吗,需要什么类型色谱柱和流动相条件分析?
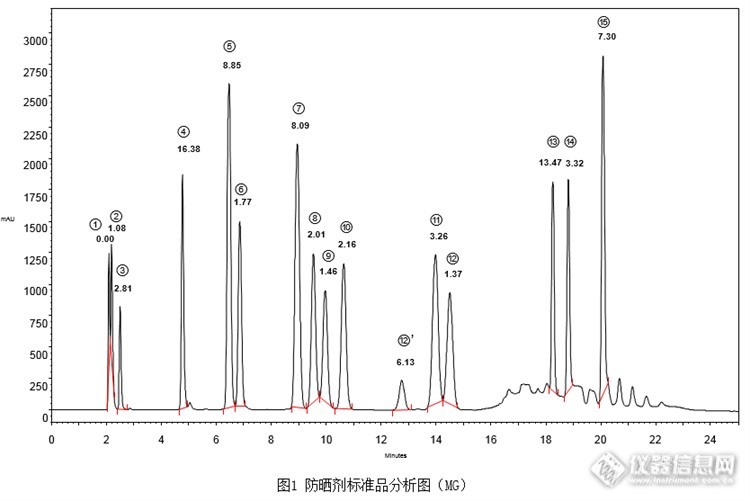
[align=center][b]2015版《化妆品安全技术规范》防晒剂检验方法-苯基苯并咪唑磺酸等15种组分[/b][/align][align=center][b]第一法(高效液相色谱-二极管阵列检测器法)[/b][/align]本次实验按照2015版《化妆品安全技术规范》中防晒剂检验方法的第一法(高效液相色谱-二极管阵列检测器法),对苯基苯并咪唑磺酸等15种防晒剂进行同时分析。15种防晒剂标准品按照《化妆品安全技术规范》配制成混合标准溶液,分别使用CAPCELL PAK C18 MG S5 4.6 mm i.d. × 250 mm,CAPCELL PAK C18 MGII S5 4.6 mm i.d. × 250 mm,CAPCELL PAK ADME S5 4.6 mm i.d. ×250 mm,CAPCELL PAK C18 AQ S5 4.6 mm i.d. × 250 mm以及SUPERIOREX ODS S5 4.6 mm i.d. × 250 mm五款色谱柱对混合标准溶液进行分析。其中,MG和MGII色谱柱得到相对较好结果,但两款色谱柱原流动相条件下,个别峰未实现基线分离。结果如图1、图2。[img=,690,460]http://ng1.17img.cn/bbsfiles/images/2017/08/201708170930_01_2222981_3.png[/img][img=,690,432]http://ng1.17img.cn/bbsfiles/images/2017/08/201708170930_02_2222981_3.png[/img]1:苯基苯并咪唑磺酸; 2:二苯酮-4和二苯酮-5; 3:对氨基苯甲酸; 4:二苯酮-3; 5:对甲氧基肉桂酸异戊酯6:4-甲基苄亚基樟脑; 7:PABA乙基己酯; 8:丁基甲氧基二苯甲酰基甲烷; 9:奥克立林;10:甲氧基肉桂酸乙基己酯; 12’:峰12的同分异构体; 11:水杨酸乙基己酯; 12:胡莫柳酯;13:乙基己基三嗪酮; 14:亚甲基双-苯并三唑基四甲基丁基酚; 15:双-乙基己氧苯酚甲氧苯基三嗪(按出峰顺序)[img=,690,304]http://ng1.17img.cn/bbsfiles/images/2017/08/201708170930_03_2222981_3.png[/img]为得到更好的分离效果,使用1支更新的MGII色谱柱,在原流动相条件基础上,对梯度进行调整,结果如图3所示。各峰分离度得到明显改善,但峰11和峰12分离度为1.43,仍未达到基线分离。[img=,690,425]http://ng1.17img.cn/bbsfiles/images/2017/08/201708170933_01_2222981_3.png[/img]1:苯基苯并咪唑磺酸; 2:二苯酮-4和二苯酮-5; 3:对氨基苯甲酸; 4:二苯酮-3; 5:对甲氧基肉桂酸异戊酯6:4-甲基苄亚基樟脑; 7:PABA乙基己酯; 8:丁基甲氧基二苯甲酰基甲烷; 9:奥克立林;10:甲氧基肉桂酸乙基己酯; 12’:峰12的同分异构体; 11:水杨酸乙基己酯; 12:胡莫柳酯;13:乙基己基三嗪酮; 14:亚甲基双-苯并三唑基四甲基丁基酚; 15:双-乙基己氧苯酚甲氧苯基三嗪(按出峰顺序)[img=,690,292]http://ng1.17img.cn/bbsfiles/images/2017/08/201708170933_02_2222981_3.png[/img]继续调整梯度条件,分析结果如4所示。在此条件下,各峰实现基线分离,得到良好分析结果。[img=,690,421]http://ng1.17img.cn/bbsfiles/images/2017/08/201708170935_01_2222981_3.png[/img]1:苯基苯并咪唑磺酸; 2:二苯酮-4和二苯酮-5; 3:对氨基苯甲酸; 4:二苯酮-3; 5:对甲氧基肉桂酸异戊酯6:4-甲基苄亚基樟脑; 7:PABA乙基己酯; 8:丁基甲氧基二苯甲酰基甲烷; 9:奥克立林;10:甲氧基肉桂酸乙基己酯; 12’:峰12的同分异构体; 11:水杨酸乙基己酯; 12:胡莫柳酯;13:乙基己基三嗪酮; 14:亚甲基双-苯并三唑基四甲基丁基酚; 15:双-乙基己氧苯酚甲氧苯基三嗪(按出峰顺序)[img=,690,307]http://ng1.17img.cn/bbsfiles/images/2017/08/201708170937_01_2222981_3.png[/img]接下来将色谱柱更换为MG色谱柱,在调整后的梯度条件下进行分析,结果如图5所示,同样可得到良好的分析结果。[img=,690,419]http://ng1.17img.cn/bbsfiles/images/2017/08/201708170938_01_2222981_3.png[/img]1:苯基苯并咪唑磺酸; 2:二苯酮-4和二苯酮-5; 3:对氨基苯甲酸; 4:二苯酮-3; 5:对甲氧基肉桂酸异戊酯6:4-甲基苄亚基樟脑; 7:PABA乙基己酯; 8:丁基甲氧基二苯甲酰基甲烷; 9:奥克立林;10:甲氧基肉桂酸乙基己酯; 12’:峰12的同分异构体; 11:水杨酸乙基己酯; 12:胡莫柳酯;13:乙基己基三嗪酮; 14:亚甲基双-苯并三唑基四甲基丁基酚; 15:双-乙基己氧苯酚甲氧苯基三嗪(按出峰顺序)[img=,690,291]http://ng1.17img.cn/bbsfiles/images/2017/08/201708170940_01_2222981_3.png[/img]
精馏得到三苯基膦,其中含有少量三苯基氧膦,想对其定量,使用乙腈-水做流动相,柱温25℃,测样时进3-5针,三苯基氧膦峰面积越来越大,猜测在测样过程中可能有三苯基膦氧化生成了三苯基膦,请问如何防止测样时三苯基膦氧化生成三苯基氧膦呢?
[color=#444444]用液相检测邻氨基苯乙酮时 前处理为什么要在100摄氏度加热5分钟,然后冷却到室温后再检测呢?[/color]
[color=#444444][color=#444444]各位大牛,最近用液相色谱检测三苯基磷和三苯基氧膦,流动相是水和甲醇1:4,流量1.5ml/min,C18柱子,含有三苯基磷的样品在12min左右出了一个峰,含有三苯基氧膦的样品在2min左右出了一个峰,改成梯度测试,三苯基磷在32min左右出了一个峰,三苯基氧磷在2min左右出了一个峰,我不能确定2min左右的峰是不是三苯基氧膦,有没有做过的,给个判断,或者给个液相条件,不甚感激![/color][/color]
求一苯基锡二苯基锡三苯基锡同时检测的GC-MS检测方法







 我要推广仪器
我要推广仪器
 下载APP
下载APP