提供盐酸曲美他嗪欧洲药典杂质标准品Trimetazidine for system suitabilityImp. A (EP) as Dihydrochloride: 1-(3,4,5- Trimethoxybenzyl)piperazine DihydrochlorideImp. B (EP): 1,4-Bis(2,3,4-trimethoxy-benzyl)piperazineImp. C (EP): 2,3,4-TrimethoxybenzaldehydeImp. D (EP): (2,3,4-Trimethoxyphenyl)methanolImp. E (EP) as Dihydrochloride: 1-(2,4,5- Trimethoxybenzyl)piperazine DihydrochlorideImp. F (EP) as Dihydrochloride: 1-(2,4,6-Tri- methoxybenzyl)piperazine DihydrochlorideImp. G (EP) as Hexahydrate: Piperazine HexahydrateImp. H (EP): Ethyl 4-(2,3,4-Trimethoxybenzyl)- piperazine-1-carboxylate1-Formyl-4-(2,3,4-trimethoxybenzyl)piperazine Hydrochloride
如题,俺第一次测盐酸左氧氟沙星,做有关物质时杂质A与左氧保留时间完全重叠,排除了乙酸铵、高氯酸钠等试剂滴原因,实在没辙咧,请教大虾帮忙。盐酸左氧氟沙星有关物质测定方法(来源:中国药典2010年版第一增补本): 有关物质 取本品,精密称定,加0.lmol/L盐酸溶液溶解并定量稀释制成每1ml中约含1.2mg的溶液,作为供试品溶液,精密量取适量,用0.1mol/L盐酸溶液定量稀释制成每1ml中含2.4ug的溶液,作为对照溶液。另精密称取杂质A对照品约18mg,置100ml量瓶中,加6mol/L氨溶液1ml与水适量使溶解,用水稀释至刻度,摇匀,精密量取2ml,置100ml量瓶中,用水稀释至刻度,摇匀,作为杂质A对照品溶液。照高效液相色谱法(附录V D)测定,用十八烷基硅烷键合硅胶为填充剂;以醋酸铵高氯酸钠溶液(取醋酸铵4.0g和高氯酸钠7.0g,加水1300ml使溶解,用磷酸调节pH值至2.2)-乙腈(85 :15)为流动相A,乙腈为流动相B;按下表进行线性梯度洗脱。柱温为40°C;流速为每分钟1ml。称取左氧氟沙星对照品、环丙沙星对照品和杂质E对照品各适量,加0.1mol/L盐酸溶液溶解并稀释制成每1ml中约含左氧氟沙星1.2mg、环丙沙星和杂质E各6ug的混合溶液,取10ul注人液相色谱仪,以294nm为检测波长,记录色谱图,左氧氟沙星峰的保留时间约为15分钟。左氧氟沙星峰与杂质E峰和左氧氟沙星峰与环丙沙星峰的分离度应分别大于2.0与2.5。量取对照溶液10ul注人液相色谱仪,以294mn为检测波长,调节检测灵敏度,使主成分色谱峰的峰高约为满量程的20%。精密量取供试品溶液、对照溶液和杂质A对照品溶液各10ul,分别注人液相色谱仪,以294nm和238nm为检测波长,记录色谱图。供试品溶液色谱图中如有杂质峰,杂质A(238nm检测)按外标法以峰面积计算,不得过0.3%。其他单个杂质(294nm检测)峰面积不得大于对照溶液主峰面积(0.2%),其他各杂质(294nm检测)峰面积的和不得大于对照溶液主峰面积的2.5倍(0.5%)。供试品溶液色谱图中任何小于对照溶液主峰面积0.1倍的峰可忽略不计。时间(分钟) 流动相A(%) 流动相B(%) 0 100 0 18 100 0 25 70 30 39 70 30 40 100 0 50 100 0
副产盐酸中含有大量的杂质成分,我估计了一下,肯能含有硫酸根,乙酸,氯乙酸,二氯乙酸等成分,硫酸根可以用氯化钡定量,但其他杂质怎么测定呢?有比色法或滴定法可以进行检验的吗?请各位大侠帮忙解决一下,谢谢!
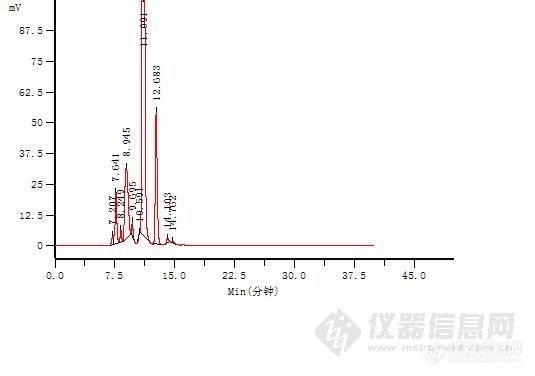
高效分子排阻色谱法测定注射用盐酸头孢替安高分子杂质头孢替安是杀菌性头孢菌素类广谱抗生素,头孢替安不但对革兰氏阳性菌有效,而且对革兰氏阴性菌。如流感嗜血杆菌,大肠杆菌、克雷白氏菌、奇异变形杆菌等的作用更强。对肠杆菌,枸橼酸杆菌、吲哚阳性变形杆菌等,也有抗菌作用头孢替安在肺中药物浓度较高,其它脏器和肌肉也有一定的浓度。临床应用于敏感菌所导致的感染,如肺炎、支气管炎、胆道感染、腹膜炎、尿路感染以及手术后或外伤引起的感染和败血症等。其基本结构同已上市的的头孢菌素类抗生素一样,头孢替安也会形成高分子聚合物,也会在临床使用中引发速发型过敏反应。对患者危害极大。已有的注射用盐酸头孢替安国家药品标准未将盐酸头孢替安高分子聚合物列为检定项目,国内的药学研究也未见头孢替安高分子聚合物的研究和报道。从临床用药安全性考虑,根据中国药典2010年版二部附录凝胶色谱原理。采用常用的葡聚糖凝胶G-10检测聚合物时由于头孢替安分子结构自身的原因,头孢替安不能完全缔合,因些我们采用高效分子排阻色谱法,以球状蛋白色谱用亲水硅胶为填充剂 TOSOH TSKgelG2000SW(7.5*300mm),测定注射用盐酸头孢替安高分子杂质1.仪器与试剂(1)仪器:岛津LC-10ATvp泵 岛津SPD-10AVP紫外可见光多波长检测器 浙大2010色谱数据工作站 色谱柱:TOSOH TSKgelG2000SW(7.5*300mm) (2)试剂: 乙腈 (色谱纯,天津市四友生物医学技术有限公司) 磷酸氢二钠(分析纯,北京化学试剂公司) 磷酸二氢钠(分析纯,北京化学试剂公司)双蒸水 (自制)2 色谱条件色谱柱:TOSOH TSKgelG2000SW(7.5*300mm)流动相:磷酸盐缓冲液(p H:6.8[/color
二氮杂菲测水中阴离子表面活性剂---定容某市环境监测中心 董捷 姜程程(图片无法显示,文章未完,请下载附件给予批评指正,谢谢)1. 范围本标准GB/T 5750.4-2006规定了用二氮杂菲萃取分光光度法测定生活饮用水及其水源水中的阴离子合成洗涤剂。本法适用于生活饮用水及其水源水中阴离子合成洗涤剂的测定。本法最低检测质量为2.5μg。若取水样100mL水样测定,则最低检测质量浓度为0.025mg/L(以十二烷基苯磺酸钠计)。生活饮用水及其水源水中常见的共存物质(mg/L)对本标准无干扰:Ca2+、NO3-(400)、SO42-(100)、Mg2+(70)、NO2-(17)、PO43-(10)、F-(7)、SCN-(5)、Mn2+、Cl2(1)、Cu2+(0.1)。阴离子表面活性剂质量浓度为0.1 mg/L时,会产生误差为-28.4%的严重干扰。2. 原理水中阴离子合成洗涤剂与Ferroin(Fe2+与二氮杂菲形成的配合物)形成离子缔合物,可被三氯甲烷萃取,于510nm波长下测定吸光度。3. 仪器3.1 分液漏斗,250mL。3.2 302B自动液液萃取仪。3.3 VIS-723G分光光度计。4. 试剂4.1 三氯甲烷4.2 二氮杂菲溶液(2g/L):称取0.2g二氮杂菲(C12H8N2•H2O,又名邻菲罗啉),溶于纯水中,加2滴盐酸(ρ20=1.19 g/mL),并用纯水稀释至100mL。4.3 乙酸铵缓冲溶液:称取250g乙酸铵(NH4C2H3O2),溶于150 mL纯水中,加入700 mL冰乙酸,混匀。4.4 盐酸羟胺-亚铁溶液:称取10g盐酸羟胺,加0.211g硫酸亚铁铵溶于纯水中,并稀释至100mL。4.5 十二烷基苯磺酸钠标准储备溶液:称取0.500g十二烷基苯磺酸钠(C12H25-C6H4SO3Na,简称DBS),溶于纯水中,定容至500 mL。4.6 十二烷基苯磺酸钠标准使用溶液:取十二烷基苯磺酸钠标准储备溶液(4.5)10.00mL于1000mL容量瓶中,用纯水定容。5. 分析步骤5.1 分别取0mL,0.5mL,1.00mL,2.00mL,3.00mL,5.00mLDBS标准使用溶液(4.6)于100mL容量瓶中,加纯水至100mL,混匀。5.2 按如下步骤进行实验操作。5.2.1 加入标准溶液于六个分液漏斗中分别加入配置好的100 mL DBS标准使用液,如图1。图1. 100 mLDBS标准使用液5.2.2 加入2 mL二氮杂菲溶液于六个分液漏斗中加入2 mL二氮杂菲溶液(4.2),萃取强度设为50Hz,运行时间60s,停止时间0s,萃取一次将标准使用液与二氮杂菲混匀,如下图2。 图2. 二氮杂菲与标准液混匀5.2.3加入10 mL缓冲液于上述分液漏斗中再加入10 mL缓冲液(4.3),萃取强度设为50Hz,运行时间60s,停止时间0s,萃取一次使其混匀,如下图3。 图3. 缓冲液混匀图5.2.4 加入1.0mL盐酸羟胺-亚铁溶液于六个分液漏斗中一次加入1.0mL盐酸羟胺-亚铁溶液(4.4),萃取强度设为50Hz,运行时间60s,停止时间0s,萃取一次使其混匀,如下图4。 图4.盐酸羟胺-亚铁混匀图5.2.5 加入10mL三氯甲烷 于上述分液漏斗中加入10mL三氯甲烷,萃取强度设为50Hz,运行时间60s,停止时间30s,萃取两次使其混匀,如下图5。 图5. 三氯甲烷萃取图5.2.6 静置分层静置数分钟,使得三氯甲烷与水相分层,如下图6。 图6. 静置分层5.2.7 放液,定容 于分液漏斗颈部塞入一小团脱脂棉,慢慢旋开活塞,使得三氯甲烷成滴滴入干燥的10mL比色管中,并将少量三氯甲烷加入分液漏斗中清洗脱脂棉,并合此三氯甲烷并于比色管中,最后用三氯甲烷定容至10mL,混匀,以供测定。如图7所示。图7. 放液图5.3 于510nm波长,用3cm比色皿,以三氯甲烷为参比,测定吸光度。5.4 绘制工作曲线。未完,详细资料请下载附件,谢谢!
混合溶液是盐酸羟胺、邻二氮杂菲和缓冲溶液,刚配出来就有淡淡的橘黄色,有人遇到过吗?虽然有做空白,但是有没有消除的办法呢?
我用盐酸萘乙二胺法做亚硝酸盐检测时,发现当我加入盐酸萘乙二胺后,出现红色显色反应,但是过30s左右,颜色随即消失,这好象不符和一般的显色时间的原理,请问这可能是什么原因,谢谢!
水质检测用的盐酸萘乙二胺在北京哪个地方能买到?
按照国标的盐酸萘乙二胺方法测定亚硝酸盐的含量,配制盐酸萘乙二胺的时候发觉很难溶解,溶剂上会出现一些类似于沉淀的物质,放置几天后,发现瓶子底部有一层白色的类似于粉末的物质,摇匀后,溶剂呈现出浑浊的状态,这是什么原因呢?
HPLC法测定盐酸左西替利嗪有关物质1.基本简介左西替利嗪是西替利嗪的 R-对映体,也是西替利嗪的活性成份。左西替利嗪与组胺H1受体的亲和力比西替利嗪高2倍,比 S-对映体(右西替利嗪)约高30倍。其化学名称为R-(-)2--1-哌嗪基]乙氧基]乙酸二盐酸盐。 2.仪器设备、试剂与对照品2.1仪器设备:waters e2695高效液相色谱仪赛托利斯 CPA225D分析天平色谱柱:Dikma Platisil Silica柱 4.6mm×250mm×5um2.2试剂:硫酸(AR)广州化学试剂厂乙腈(HPLC)赛默飞科技有限公司2.3 对照品:主成分自身对照法3.色谱条件流动相:乙腈-水-5.5%硫酸(935:62:3)波长:230nm 流速:1.0ml/min 温度:30℃洗脱方式:等度 进样体积:20ul4.样品制备4.1 供试品溶液制备精密称取本品约20mg,加流动相溶解并定量制成每1ml中约含0.2mg的溶液,即为供试品溶液。4.2对照溶液制备 精密量取供试品溶液1ml,置100ml容量瓶中,加流动相溶解并稀释至刻度,摇匀,即为对照溶液。4.3测定法4.3.1 精密量取20μl对照溶液注入液相色谱仪,记录色谱图。对照溶液中,主峰理论塔板数不得小于5000,拖尾因子不得大于2.0。4.3.2 精密量取对照溶液与供试品溶液各20ul,分别注入液相色谱仪,记录色谱图至主峰保留时间的3倍。供试品溶液色谱图中如有杂质峰,按自身对照法计算各杂质的含量。5. 结果讨论系统适用性结果报告:对照溶液图谱中,主峰理论塔板数13497,拖尾因子0.9。结果表明:Dikma Platisil Silica柱 能满足分析要求。PS:仅已此文献礼迪马科技25周年,祝愿迪马科技红红火火、蒸蒸日上![img]https://ng1.17img.cn/bbsfiles/images/2018/08/201808302052245314_8942_3170710_3.jpeg[/img][img]https://ng1.17img.cn/bbsfiles/images/2018/08/201808302052247005_3155_3170710_3.jpeg[/img][img]https://ng1.17img.cn/bbsfiles/images/2018/08/201808302052257616_5173_3170710_3.jpeg[/img][img]https://ng1.17img.cn/bbsfiles/images/2018/08/201808302052259556_225_3170710_3.jpeg[/img]
做了5遍,线性是一次比一次差,快崩溃了!所有玻璃器皿均用酒精洗完再用纯水洗数次晾干,而且加标样的时候也是小心翼翼,每次加完一种试剂也摇匀了,最后萃取的时候也注意了震荡次数和幅度一致,但是线性就是很差,经常是低浓度的颜色很深,高浓度的反而低,不成梯度,每次就最后3,4个点呈线性,实在郁闷!所用试剂呢都是现配,其中醋酸铵潮解比较厉害,部分已呈液体,不知道会不会对PH构成影响,(这里的缓冲液PH是否3.1~4.4,还请高人解释下),盐酸羟胺也潮解了,二氮杂菲稍微好点,之前用这些做铁的时候线性还可以,不知道为什么做阴离子就是不行!不知道是哪些因素影响了显色,有没有用此法做成功的人指导下,万分感谢!
盐酸萘乙二胺 怎么溶解
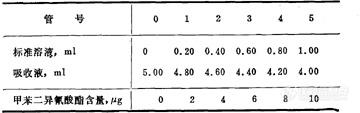
空气中甲苯二异氰酸酯的测定方法 盐酸萘乙二胺比色法 1 原理甲苯二异氰酸酯水解产生相应的芳香胺,芳香胺与亚硝酸钠重氮化后,再与盐酸萘乙二胺偶合生成紫红色,比色定量。2 仪器2.1 冲击式吸收管。2.2 抽气机。2.3 流量计,0~1L/min。2.4 具塞比色管,25ml。2.5 恒温水浴箱。2.6 分光光度计。3 试剂3.1 吸收液:量取25ml盐酸(20=1.19g/ml)与500ml水混匀,加入250ml二甲基甲酰胺,用水稀释成1L,临用前配制。3.2 亚硝酸钠-溴化钠溶液:称取3g亚硝酸钠和5g溴化钠,用80ml水溶解后,稀释成100ml。3.3 氨基磺酸铵溶液,100g/L。3.4 盐酸萘乙二胺溶液,10g/L。3.5 标准溶液:于25ml量瓶中加入5ml二甲基甲酰胺,准确称量,加入1~2滴甲苯二异氰酸酯,再准确称量,两次称量之差即为甲苯二异氰酸酯的质量,然后用二甲基甲酰胺稀释到刻度。计算1ml溶液中甲苯二异氰酸酯的含量,再用二甲基甲酰胺稀释成1ml=100微克甲苯二异氰酸酯的溶液。取5.0ml上述溶液于50ml量瓶中,加入7.50ml二甲基甲酰胺及1.25ml盐酸,混匀后用水稀释至刻度,配成1ml=10微克甲苯二异氰酸酯的标准溶液。4 采样将装有10ml吸收液的冲击式吸收管以1L/min的速度抽取50L空气。5 分析步骤5.1 对照试验:同采样,将吸收管装好吸收液带至现场,但不抽取空气,照样品分析。5.2 样品处理:采样后,用吸收管中的吸收液洗涤进气管内壁3次,由每个吸收管中各量取5.0ml样品溶液,分别放入比色管中,供测定用。5.3 标准曲线的绘制:按表2配制标准管。表2 甲苯二异氰酸酯标准管的配制[img]http://ng1.17img.cn/bbsfiles/images/2007/05/200705201410_52373_1625938_3.jpg[/img]向各标准管中各加入0.2ml亚硝酸钠-溴化钠溶液(3.2)混匀,放置1min,再加0.2ml氨基磺酸铵溶液(3.3),激烈振摇,待气泡消失后,放置2min,加入1ml盐酸萘乙二胺溶液(3.4),充分混匀后,在20℃水浴中,放置20min,于波长560nm下比色,以甲苯二异氰酸酯含量对吸光度作图,绘制标准曲线。5.4 测定:按5.3相同的操作条件,将处理后的样品进行测定,由标准曲线上查出甲苯二异氰酸酯含量。6 计算X=2C/V0式中:X——空气中甲苯二异氰酸酯的浓度,mg/m3;C——吸收管中所取样品溶液中甲苯二异氰酸酯的含量,?g;V0——标准状况下的样品体积,L。7 说明7.1 本法的检测限为0.9?g/5ml。测定范围为2~10微克/5ml。当甲苯二异氰酸酯的浓度为2、4、6、8、10微克/5ml时,变异系数分别为7.1%、5.8%、6.5%、5.6%、5.8%。7.2 采样后样品可保存3天。7.3 吸收液应临用前配制,因久置后其中盐酸逐渐与二甲基甲酰胺作用而失去酸性。
做盐酸川芎嗪氯化钠注射液时(标准号为WS1-(X-113)-2003Z)检不出有关物质流动相:1%醋酸钠溶液-甲醇波长302(最大紫外吸收296)我不知道为什么要选择302主峰出来很漂亮,理论板数、托尾因子都符合规定不知道是没有杂质还是标准有问题(原料药不检有关)包栝到市面上买回来的样品出检不出有关物质请教大家!给点意见!
电子级别的试剂如盐酸是如何做到杂质很低的,工艺上有什么联系?
用盐酸萘乙二胺能做出来吗
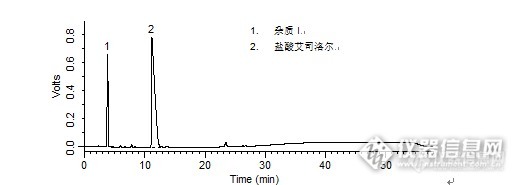
1. 杂质I2. 盐酸艾司洛尔 盐酸艾司洛尔样品制备 制备方法有关物质衍生溶液:取盐酸艾司洛尔对照品约10 mg,置10 mL量瓶中,加入1 mol/L盐酸溶液1 mL,放置30分钟,加1 mol/L的氢氧化钠溶液1 mL使中和,用流动相A 稀释至刻度,摇匀。分析条件 色谱柱Diamonsil C18(2) 250 x 4.6 mm,5 μm (Cat#:99603)流动相流动相A:乙腈:甲醇:磷酸盐缓冲液(取磷酸二氢钾3.0 g,加水至650 mL)=15:20:65流动相B:甲醇梯度流速1 mL/min柱温30 ℃检测器UV 222 nm进样量20 μL 色谱图有关物质衍生溶液http://ng1.17img.cn/bbsfiles/images/2016/04/201604211737_591070_708_3.png 峰号 保留时间 min 峰面积 μV*s 峰高 μV 理论塔板数 N USP拖尾因子 分离度 1 3.842 6280189 655879 2747.059 0.670 -- 2 11.157 29271705 784686 1512.532 5.026 10.154 本品种同时使用了SpursilC18色谱柱,在药典规定条件下进行检测,满足药典要求。
HPLC法测盐酸普萘洛尔含量,采用紫外检测器,用安捷伦-SB-C18柱,以乙腈:0.05M磷酸二氢钾30:70为流动相,但出来的色谱峰拖尾,且峰形对称性差。本人试过加酸加碱来改善拖尾,但效果不明显,特此求助,望有关人士能解我燃眉之急,非常感谢!!
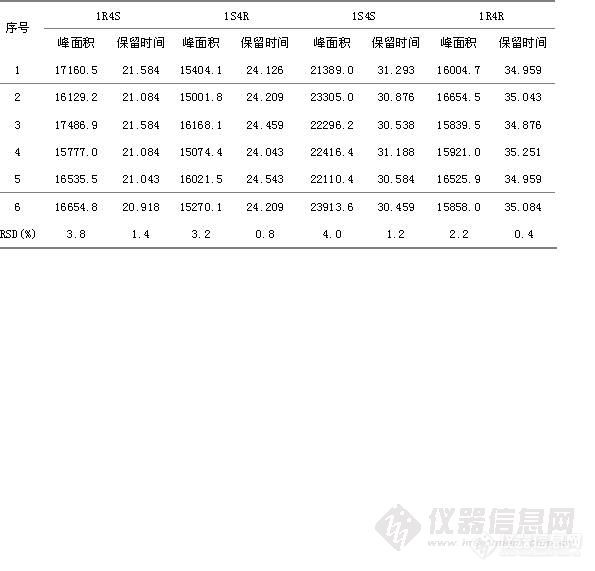
HPLC法测定盐酸舍曲林片的有关物质 摘要:目的:建立盐酸舍曲林片有关物质的测定方法,其外标法测定其四种光学异构体,自身对照法测定其它有关杂质。方法:采用依利特Hypersil ODS2色谱柱(4.6mm×150mm,5μm);流动相为磷酸二氢钠缓冲液 β-环糊精12.8g,HP-β-环糊精17.3g,加水至1000ml]-乙腈[font=Times New Roman]-[font=宋体]三乙胺([font=Times New Roman]800[font=宋体]:[font=Times New Roman]200[font=宋体]:[font=Times New Roman]10[font=宋体])[font=Times New Roman],[font=宋体]用磷酸调节[font=Times New Roman]p H [font=宋体]为[font=Times New Roman]2.5±0.5为流动相,流速为每分钟1ml/min,检测波长为214nm。结果:建立了β-环糊精、HP-β-环糊精手性流动相拆分盐酸舍曲林光学异构体,的方法,结论:该方法专属性好,无干扰,盐酸舍曲林异构体与主峰分离良好。可作为控制盐酸舍曲林片质量的方法。关键词:HPLC外标法;HPLC自身对照法;盐酸舍曲林片;光学异构体; β-环糊精; HP-β-环糊精。盐酸舍曲林是一种选择性抑制[font=Times New Roman]5-[font=宋体]羟色胺再摄取的抗抑郁药([font=Times New Roman]SSRI[font=宋体]),为新一代抗抑郁药的代表药物,目前已跃成为抗抑郁治疗的一线药和首选药。盐酸舍曲林由美国辉瑞公司开发,并于[font=]1990[font=宋体]年[font=]12[font=宋体]月最初在英国上市,目前为止已经在[font=]30[font=宋体]个国家陆续上市。盐酸舍曲林(商品名:郁尔复片剂)于[font=Times New Roman]1996[font=宋体]年[font=Times New Roman]12[font=宋体]月获得中国药品行政保护,并于当年在中国以片剂形式(不含原料药)上市,用于治疗精神抑郁症。现在国内各大医院、药店均有销售。盐酸舍曲林在中国无专利保护。[font=Times New Roman]2004[font=宋体]年[font=Times New Roman]6[font=宋体]月该行政保护终止。由于盐酸舍曲林存在4种光学异构体,在合成工艺过程中可能会引入中间杂质盐酸舍曲林顺式左旋异构体,盐酸舍曲林反式左旋异构体,反式右旋异构体,因其均为手性异构体,眼前其异构体检测方法为毛细管电泳法,且只对其异构体顺式左旋异构体进行检测,其它杂质检测采用反相HPLC,且其盐酸舍曲林主峰不能很好的与其相邻异构体分开,参照相关文献,并经方法学研究,建立了盐酸舍曲林有关物质的检测方法,采用HPLC外标法测定[
请各位大侠帮忙,我有个副产盐酸的样品,其中含有乙酸,氯乙酸,二氯乙酸等,我想通过其他方法将杂质检测,但不知道怎样才能去除盐酸,保证杂质酸不反应,拜托大家帮忙,先谢谢了!
1 范围 本标准规定了氧化铝中三氧化二铁含量的测定方法。 本标准适用于氧化铝中三氧化二铁含量的测定。测定范围:0.005%-0.100%, 2 方法原理 三价铁用盐酸经胺还原为二价铁,在乙酸一乙酸钠缓冲溶液中加人邻二氮杂菲使形成络合物,于分光光度计波长 510nm处测量其吸光度,借以测定三氧化二铁含量。 3 试剂 3.1 硼酸:优级纯。 3.2 无水碳酸钠:优级纯。 3.3 硝酸(3.00mol/L), 3.4 盐酸羟胺溶液(10g/L) 3.5邻二氮杂菲溶液(1g/L):称取1g邻二氮杂菲溶于1.5mL--2.5mI冰乙酸中(ρl.05g/mL),移入1000mL容量瓶中,用水稀释至1000mL,混匀。 3.6 缓冲溶液(pH=4.9):称取272g乙酸钠(CH3COONa3H20)溶于500mL水中,加人240mL 冰乙酸(pl.05g/mL),用水稀释至1000mL,混匀。 3.7 三氧化二铁标准贮存溶液:称取0.50g三氧化二铁(含量)99.99%,预先于600℃灼烧2h,并于干燥器(4.4)中冷却至室温)置于150mL烧杯中,沿杯壁加人20mL盐酸(ρ.19g/mL),盖上表皿微热使全部溶解,冷却至室温,将溶液移人1000mL容量瓶中,用水洗净烧杯,洗液并人容量瓶中,用水稀释至刻度,混匀。此溶液1mL含0.5mg三氧化二铁。 3.8 三氧化二铁标准溶液:移取25.00mL三氧化二铁标准贮存溶液(3.7)于500mL容量瓶中,加人30.0ml硝酸((3.3),用水稀释至刻度,混匀。此溶液1mL含0.025mg三氧化二铁,用时配制。 4 仪器、装置及器具 4.1铂坩埚:30mL,带盖。 4.2 分光光度计。 4.3 电热板:用调压器控制加热温度不高于250℃ 4.4 干燥器:用新活性氧化铝作干燥剂。 5 试样 5.1 试样应通过0.125mm孔径筛网。 5.2试样预先在300℃士100℃烘干2h,置于干燥器((4.4)中冷却至室温。 6 分析步骤 6.1 试料 称取约0.50g试样(5),精确至0.0001g 6.2 测定次数 独立地进行两次测定,取其平均值。 6.3 空白试验 随同试料 (6.1)做空白试验 6.4 测定 6.4.1将试料(6.1)置于铂坩埚(4.1)中,加人0.500g硼酸(3.1)和1.300g碳酸钠(3.2),用铂勺搅匀。盖卜坩埚盖,置于约700℃的高温炉中,升温至1000℃熔融20min,取出稍冷 空白试验直接在1000℃熔融2min-3min,取出稍冷。 6.4.2 向坩埚中加人沸水,加热至近沸,使熔块全部溶解,将溶液移人预先盛有22.3mL(随同试料空白则为12.6mL)硝酸(3.3)的150m工聚四氟乙烯烧杯中,坩埚用热水冲洗二次,用聚四氟乙烯棒搅拌使沉淀尽量溶解,坩埚用3ml一硝酸(3.3)和热水充分洗净,洗涤液并人烧杯中,盖上表皿,置电热板(4.3)上加热至沉淀全部溶解,取下,置冷水槽中冷却至室温。将溶液移人100ml容量瓶中,用水洗净烧杯,洗液并人容量瓶中,并用水稀释至刻度,混匀(此溶液也可用以测定二氧化硅量)。 6.4.3 分取50.00 mL试液于100mL容量瓶中[当三氧化二铁含量大于0.05%时,取25.00m工试液,补加1.5mI硝酸(3.3)加水至约50mL。并随同做对应的空白试验)。 6.4.4 向容量瓶中加人5.0mL盐酸经胺溶液(3.4),混匀。加人5.0mL邻二氮杂菲溶液(3.5)和25.0mL缓冲溶液((3.6),用水稀释至刻度,棍匀,放置10min。 6.4.5 将部分溶液移人3cm吸收池中,以水为参比,于分光光度计波长510nm处,测量其吸光度,将测得的吸光度减去随同试料空白试验溶液的吸光度后,从工作曲线上查出相应的三氧化二铁量。 6.5 工作曲线的绘制 于一组100mL容量瓶中,分别加人 0, 1.00, 2.00 ,3.00, 4.00, 5.00ml三氧化二铁标准溶液(3.9),加人3.0mI.硝酸((3.3),用水稀释至体积约50mL,混匀,以下按6.4.4进行。将部分系列标准溶液移人3cm吸收池中,以水为参比,于分光光度计波长510nm处,测量系列标准溶液的吸光度。以所测吸光度减去试剂空白溶液的吸光度,以三氧化二铁量为横坐标,吸光度为纵坐标,绘制工作曲线。 7 分析结果的计算 按下式计算三氧化二铁含量w(Fe2O3)(%): 式中:m1-----自工作曲线上查得的三氧化二铁量,单位为毫克(mg) m0-----试样的质量,单位为克(g) V1------分取试液的体积,单位为毫升(mL) V0------试液的总体积,单位为毫升(mL), 8.1 重复性 在重复性条件下获得的两次独立测试结果的测定值,在以下给出的平均值范围内,这两个测试结果的绝对差值不超过重复性限((r),超过重复性限(r)的情况不超过5%,重复性限((r)按以下数据采用线性内插法求得: w(Fe2O3) (%) 0.0087 0.0222 0.046 8 重复性限r (%) 0.0005 0.0003 0.001 3 8.2 允许差 实验室之间分析结果的差值应不大于表1所列允许差。 w(Fe2O3) 允 许 差 0.005--0.015 0.004 0.015--0.030 0.005 0.030--0.050 0.006 0.060--0.100 0.010 9 质量保证与控制 应用标准样品或控制样品,每月至少对本标准的有效性校核一次。当失效时应找出原因。纠正后重新进行校核。
求教一下做过乙酸乙酯萃取纯金然后测杂质含量的朋友,含有1mol/L盐酸的乙酸乙酯怎么配制?

盐酸洛美利嗪含量测定方法研究本品为二苯哌嗪类钙通道阻滞剂,具有选择性的脑血管舒张作用。毒理研究遗传毒性:微生物回复突变试验、染色体畸变试验和小鼠微核试验结果均为阴性。下面主要针对盐酸洛美利嗪的含量测定方法进行研究。 一、容量法洛美利嗪为有机碱,可与高氯酸发生酸碱中和反应。1.指示剂选择和滴定终点的确定精密称取盐酸洛美利嗪约0.2g,加入15ml冰醋酸,振摇使溶解,加入5ml醋酸酐及5ml醋酸汞试液,加入1滴结晶紫指示液,并用电位计指示电位的变化,描绘滴定曲线。试验证明,当电位发生突跃时,溶液呈黄绿色。以高氯酸滴定液(0.1mol/L)滴定,并将滴定的结果用空白校正。每1ml高氯酸滴定液(0.1mol/L)相当于27.073mg的盐酸洛美利嗪。2.指示剂滴定法与电位滴定法含量测定的结果比较精密称取10份样品,每份约0.2g,加入15ml冰醋酸,振摇使溶解,加入5ml醋酸酐及5ml醋酸汞试液,加入1滴结晶紫指示液,其中五份做电位法滴定,另外五份做指示剂法确定终点,分别计算含量,数据见表1,从数据可知,电位法和指示剂法结果基本一致。用指示剂指示终点的三批样品的结果见表1。http://ng1.17img.cn/bbsfiles/images/2012/12/201212251617_415393_2583865_3.jpg3.重复性试验及中间精密度试验三天内对同一批样品分别按80%、100%、120%三个水平各称取二份,指示剂滴定法测定其含量,结果见表2, 结果表明本法重复性及精密度较好。http://ng1.17img.cn/bbsfiles/images/2012/12/201212251618_415394_2583865_3.jpg二、高效液相色谱法(HPLC)1.色谱条件及系统适用性试验(1)色谱条件:色谱柱:以十八烷基硅烷键合硅胶为填充剂(Xtimate C18),250×4.6mm,5um。流动相:甲醇-0.03mol/L磷酸氢二钾缓冲液(用磷酸调节pH4.0)(85:15),使用前经0.45μm有机滤膜抽滤并脱气。检测波长:225nm流速:1.0ml/min进样体积:20μl(2)系统适用性试验:精密称取干燥恒重的对照品约25mg置50ml量瓶中,用流动相溶解并稀释至刻度,摇匀作为贮备液。精密量取贮备液5.0ml置50ml量瓶中,精密量取20ml注入液相色谱仪,记录色谱图,连续进样6次,计算精密度。结果见表3。http://ng1.17img.cn/bbsfiles/images/2012/12/201212251619_415395_2583865_3.jpg由试验结果可知,RSD小于1%,表明该色谱条件下精密度良好,系统适用性符合规定。2.线性关系精密称取干燥恒重的对照品约25mg置50ml量瓶中,用流动相溶解并稀释至刻度,摇匀作为贮备液。精密量取贮备液3.0、4.0、5.0、6.0、7.0和8.0ml置50ml量瓶中,用流动相稀释定容,摇匀作为溶液1、2、3、4、5和6,各精密量取20μl注入液相色谱仪。以标准溶液的浓度作为横坐标,色谱峰峰面积为纵坐标,绘制标准曲线。结果见表4。http://ng1.17img.cn/bbsfiles/images/2012/12/201212251620_415396_2583865_3.jpghttp://ng1.17img.cn/bbsfiles/images/2012/12/201212251612_415391_2583865_3.jpg3.含量测定方法及测定结果精密称取本品适量,用流动相制成每1ml中约含50mg盐酸洛美利嗪的溶液,作为供试品溶液。另称取经恒重的对照品,同法制成每1ml中约含50mg对照品溶液。按前述高效液相色谱条件,分别量取对照品溶液和供试品溶液各20ml注入色谱仪,记录色谱图,按外标法计算含量。三批样品的HPLC法含量测定结果见表7-16。三批样品的含量测定结果见表5. http://ng1.17img.cn/bbsfiles/images/2012/12/201212251620_415397_2583865_3.jpg三、结果讨论分别采用容量法和高效液相色谱法测定三批样品的含量,可以看出两种方法的准确度、精密度等均能满足盐酸洛美利嗪含量检测的要求。其中容量法相对简单,系统误差小,故采用容量法作为含量测定的方法。
请问论坛里的专家、朋友用ICP-OES如何检测浓盐酸(GR)、浓硫酸(GR)中的杂质元素,有人检测过吗?
请问论坛中的各位专家、朋友,你们用ICP-OES检测过浓盐酸(GR),浓硫酸(GR)中的杂质元素吗?如何检测的?
HJ 479-2009 环境空气 氮氧化物(一氧化氮和二氧化氮)的测定 盐酸萘乙二胺分光光度法[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=181815]HJ 479-2009 环境空气 氮氧化物(一氧化氮和二氧化氮)的测定 盐酸萘乙二胺分光光度法.pdf[/url]
方法要求以辛烷基硅胶为填充剂,0.2%磷酸氢二铵溶液(2mol/l磷酸调pH至7.0)-乙腈=50:50为流动相,盐酸氨溴索与杂质B分离度不小于4.0.用我司一代C8,5um,250*4.6mm色谱柱实验,结果主峰与杂质B的分离度刚刚满足4.0的要求,客户担心用一段时间后,分离度会下降,如果遇到类似的情况,大家是选择稍加调整方法提高分离度,还是继续筛选其他厂家,其他规格的色谱柱呢?
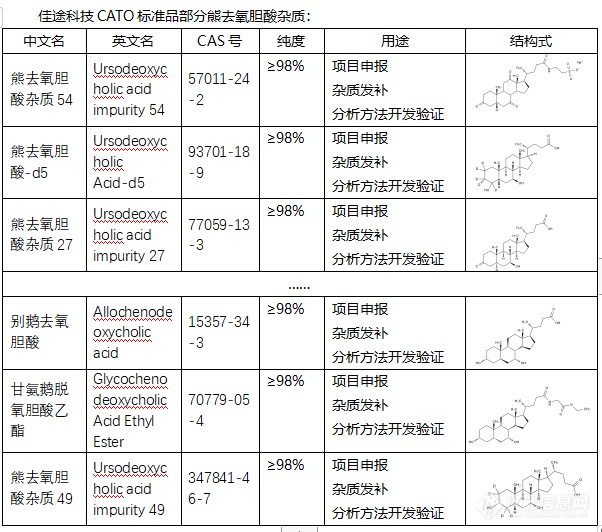
[font=宋体]◇关于熊去氧胆酸杂质[/font][font=宋体]熊去氧胆酸杂质是[/font][font=微软雅黑]是一种亲水的二羟胆汁酸[/font][font=Helvetica][color=#333333],[/color][/font][font=宋体][color=#333333]味道[/color][/font][font=Helvetica][color=#333333]无臭,[/color][/font][font=宋体][color=#333333]但是[/color][/font][font=Helvetica][color=#333333]味苦。[/color][/font][font=宋体][color=#333333]它在[/color][/font][font=Helvetica][color=#333333]医学上[/color][/font][font=宋体][color=#333333]效果是[/color][/font][font=Arial][color=#1f1f1f][font=宋体]促进胆汁分泌[/font][/color][/font][font=Helvetica][color=#333333]增加,[/color][/font][font=宋体][color=#333333]并且[/color][/font][font=Arial][color=#1f1f1f][font=宋体]抑制胆酸[/font][/color][/font][font=宋体][color=#1f1f1f]然后[/color][/font][font=Arial][color=#1f1f1f][font=宋体]诱导的[/font][/color][/font][font=宋体][color=#1f1f1f]其细[/color][/font][font=Arial][color=#1f1f1f][font=宋体]胞[/font][/color][/font][font=宋体][color=#1f1f1f]使它[/color][/font][font=Arial][color=#1f1f1f][font=宋体]凋亡[/font][/color][/font][font=宋体][color=#1f1f1f],[/color][/font][font=Helvetica][color=#333333]有利于胆结石中的胆固醇逐渐溶解。[/color][/font][font=宋体][color=#333333]它主要有以下三种作用:一[/color][/font][font=Arial][color=#333333][font=宋体]、[/font][/color][/font][font=微软雅黑][color=#666666]促进胆固醇排泄[/color][/font][font=微软雅黑][color=#666666];[/color][/font][font=微软雅黑][color=#666666]二[/color][/font][font=Arial][color=#333333][font=宋体]、[/font][/color][/font][font=微软雅黑][color=#666666]调节免疫功能[/color][/font][font=微软雅黑][color=#666666];[/color][/font][font=微软雅黑][color=#666666]三[/color][/font][font=Arial][color=#333333][font=宋体]、[/font][/color][/font][font=宋体][color=#333333]保护受损的胆管上皮细胞。但是,[/color][/font][font=微软雅黑][color=#666666]除此之外,还具有利胆的作用[/color][/font][font=微软雅黑][color=#666666]。[/color][/font][font=宋体][font=Calibri]CATO[/font][font=宋体]标准品提供的熊去氧胆酸杂质[/font][/font][font=微软雅黑][color=#333333]为核心产品[/color][/font][font=微软雅黑][color=#333333],[/color][/font][font=微软雅黑]对[/font][font=微软雅黑]肝功能[/font][font=微软雅黑]有明显的改善作用,在临床上[/font][font=微软雅黑]可用于治疗胆汁淤积性疾病[/font][font=微软雅黑],[/font][font=微软雅黑]在医药市场上有很大的前景。[/font][img=,602,532]https://ng1.17img.cn/bbsfiles/images/2024/02/202402042119305646_3395_6381607_3.png!w602x532.jpg[/img]
HPLC法测定盐酸氟桂利嗪胶囊的含量 摘要目的:建立高效液相色谱法测定盐酸氟桂利嗪胶囊的含量。方法:用C18柱,以乙腈-水(320mL中加入十二烷基硫酸钠2.0g和磷酸1.0mL)(68:32)为流动相,流速为1.0Ml/min,检测波长为254nm。结果:盐酸氟桂利嗪在10~90μg/mL浓度范围有良好线性关系,r=0.9999,方法回收率为99.6%(n=7),RSD为0.8%。结论:该方法是一种简便、灵敏、准确的分析法,能有效地控制盐酸氟桂利嗪胶囊的质量。关键词:高效液相色谱法;盐酸氟桂利嗪;含量HPLC Determination of Content of Flunarizine Hydrochloride CapsulesZHU Bi—jun,ZHAO Hal—rong,TIAN Hui,WAN Kai—huaAbstract Objective:To establish HPIJC method for the determination of Flunarizine Hydrochloride capsules.Method:Using C18 column,acetonitrile-water(add 2.0 g sodium laurylsulfate and 1mL phosphoric acid to 320mL water)(68:32)as the mobile phase.The flow rate was 1.0 mL/min;the detection wavelength was 254nm.Results:The calibration curve was linearin the range of 10~90μg /mL for geniposide(r=0.9999).The average recovery(n=7)was 99.59%(RSD=0.76%).Conclusion:111e method is simplicity,accurate and sensitive,and can control the quality of this preparation effectively.Key words:HPLC;Flunarizine Hydrochloride:content
因为牛奶样品消解后还要用消解液测蛋白含量,只能用盐酸进行消解。这样能消解完全吗?谁用盐酸消解过类似的样品吗?







 我要推广仪器
我要推广仪器
 下载APP
下载APP