
发现做三苯时候苯峰被干扰的问题,特发一帖,与各位分享。昨天在做三苯样品时候发现空白二硫化碳中含有少量苯,如下图一:http://ng1.17img.cn/bbsfiles/images/2015/01/201501091717_531833_2780210_3.png一开始以为是二硫化碳被污染,本人遂再开一瓶,进样后发现仍然有苯峰,如下图二:http://ng1.17img.cn/bbsfiles/images/2015/01/201501091718_531834_2780210_3.png为再次确定结果,正好手头GCMS空闲,上GCMS上走Scan以及SIM后发现没有苯的定量离子,所以我得出的结论为,二硫化碳未被污染,而是GC问题。立即动手,更换耗材,老化柱子,老化检测器。老化过程中发现基线有一些杂峰出来,让我更加坚信是GC端的问题。当我开开心心的再次走三苯样品的时候,悲剧出现了,苯峰没有被完全消除!如下图三:http://ng1.17img.cn/bbsfiles/images/2015/01/201501091718_531835_2780210_3.png今天接了一个要分将对/间二甲苯分开的单,所以拿出好久没用的DB-WAX的柱子,在另外一台GC上老化后直接上曲线走样,在做空白的时候惊讶的发现万恶的苯峰又来了,我判断问题仍然处在二硫化碳上。可是同一瓶二硫化碳走过MS,MS的灵敏度总不会低于FID吧?在思考的时候,样品走到1ppm浓度的三苯标液上,我发现苯峰分叉了!如下图四,图五:http://ng1.17img.cn/bbsfiles/images/2015/01/201501091718_531836_2780210_3.pnghttp://ng1.17img.cn/bbsfiles/images/2015/01/201501091718_531837_2780210_3.jpg证明空白中的那个被当做苯的杂质并不是苯,是一种跟苯有着相同保留时间的另一种物质!突然想起又一次其他实验室的过我们实验室来交流的时候有说过,正己烷有可能干扰到苯的测定。回想一下,早上更换装WAX柱子GC的洗液时正好看洗液是做有机氯农药时候用的正己烷!立即拿着刚测定有苯的空白进GCMS走了一个Scan,如下图六:http://ng1.17img.cn/bbsfiles/images/2015/01/201501091718_531838_2780210_3.png正己烷 57 43 的离子果然在!终于发现问题所在了!各位,要小心正己烷冒充苯啊!刚刚很多老师建议说极性柱子正己烷出峰应该在前面,这的确是我没想到的地方,找个空闲时间进一针正己烷试试,谢谢各位老师提醒!刚刚我用相同的条件进了一针正己烷,出峰时间在二硫化碳后面一点!看来还是我考虑得不周到!
采样人员带来碳管,一根碳管同时做苯,甲苯,二甲苯,乙酸乙酯,乙酸丁酯,丙酮,丁酮,正己烷。采样、检测依据是GBZ/T160里面的二硫化碳溶剂解吸法。采样流量都为0.1L/min 采样体积都为1.5L。非极性柱做估计 正己烷与乙酸乙酯、丁酮与乙酸乙酯分不开;极性柱做的话正己烷与二硫化碳分不开。GBZ/T160 里面的二硫化碳溶剂解吸法,采样流量体积都一样时,就能都采在一根碳管里吗?
用CEM的MARS-5微波消解仪做萃取试验的时候,能用正己烷+丙酮(1+1,体积比)做萃取溶剂吗?有讨论说微波系统中禁止使用酮、烃类物质,其中就包括正己烷和丙酮。可是标准SN/T1877.2-2007《塑料原料及其制品中多环芳烃的测定方法》中,前处理方法就是用的正己烷+丙酮(1+1,体积比)做溶剂的。会不会有危险?
想请问一下各位高手,在丙酮和正己烷重蒸过程中,重蒸装置上放温度计的地方需要个塞子,我用的是橡胶塞,考虑到橡胶中肽酸脂会污染正己烷,从而对有机氯农药的测定带来影响,我想用铝箔纸把橡胶塞包起来,避免影响,但是不知道铝箔与正己烷和丙酮蒸汽接触时会不会带来其他的影响,想请问一下高手们这种处理方法行吗?或者有更好的处理方法?谢谢!
甲胺、甲醇、三乙胺、正己烷、丙酮的分离用什么色谱柱(毛细)?
各位高人请教一下,做塑胶萃取时,使用 丙酮:正己烷 (1:1) 作为溶剂,微波萃取, 萃取后用正己烷定容时,有时会出现浑浊,这是什么原因呢? 谢谢!
[color=#444444][url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质联用[/color][/url],顶空进样,测正己烷溶液中的二氯乙烷和三氯乙烷,想同时测它们浓度,用什么做内标比较合适。[/color]
今天做一个原料药的正己烷残留测定,fisher色谱纯正己烷出了三个峰,百分比分别为18%、70%、12%;进分析纯的只有一个大峰(97%),两个溶剂最大的峰的保留时间是一致的。在网上看到很多都碰到这样的情况,色谱纯的正己烷会有三个峰出现,三个峰都是正己烷,那么这样的话定量该怎么么办呢?希望大家能讨论下。
我刚接触[url=https://insevent.instrument.com.cn/t/Mp]气相[/url],现在有个做正己烷溶剂残留的问题要解决,我用DB624的柱子,60度运行5分钟,在以20度/分升到180度,做的不是很理想,为了验证正己烷的保留时间,进了一针试剂正己烷,在2.437分时有一个相对较大的峰(我感觉应是平头峰),所以怀疑正己烷没出来。同时还要做乙酸乙酯的残留。请教各位,我的色谱条件有问题吗?有没有更好的条件!多谢了!
各们老师: 我用岛津GC-2014\中等极性色谱柱做丙酮与正己烷的混合溶液时,两个溶剂不能分开.请问用什么方法能够分开?谢谢指点!
环己烷、正己烷、二氯甲烷、丙酮对PAH的萃取能力排序是怎么样的呢?另外,怎么样直观的判断一种溶剂对PAH的萃取能力呢?向大家求教了[em09511]
我马上要做奶粉中维生素ADE了,想请大家帮个忙。为什么在检测中维生素D中用的是硅胶柱,而且流动相是环己烷和正己烷,我要是换成反相色谱柱,用甲醇做流动相可以吗?前处理还有简单的方法看见吗?谢谢各位了!非常着急!
啶虫脒在正己烷中的溶解度,到哪个网站去查,比较权威。啶虫脒在配标准溶液的时候,用丙酮定容好,还是用正己烷。
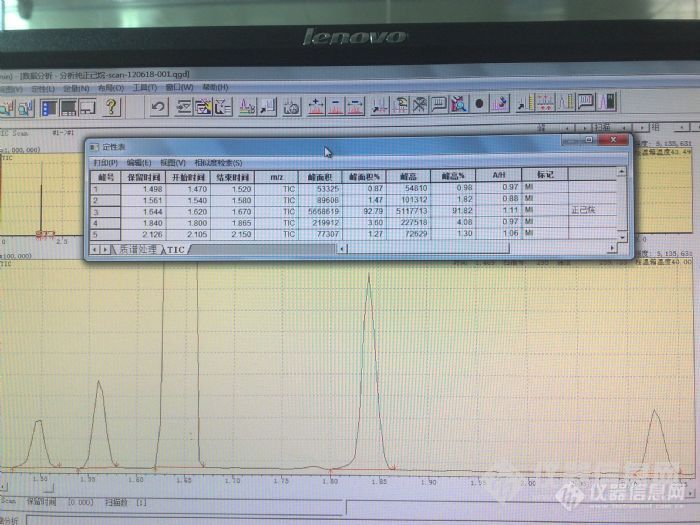
前段时间在安谱买了一瓶农残级正己烷,用于做溴类阻燃剂里面淋洗硅胶柱。下图左。右边的是在其它地方买的分析纯正己烷。http://ng1.17img.cn/bbsfiles/images/2012/06/201206201834_373654_1687966_3.jpg通常买回来的溶剂使用之前我都会先用GC-MS(气质联用仪——岛津QP-2010 Ultra)看看纯度如何。下面是农残级正己烷和分析纯正己烷的GC条件:http://ng1.17img.cn/bbsfiles/images/2012/06/201206201838_373657_1687966_3.jpg进样量是0.1μL。结果见下图。http://ng1.17img.cn/bbsfiles/images/2012/06/201206201840_373659_1687966_3.jpghttp://ng1.17img.cn/bbsfiles/images/2012/06/201206201841_373660_1687966_3.jpg从谱图和峰面积归一化法的结果(定性表)可以看出,安谱农残级正己烷的纯度比分析纯的要高出几个百分点,杂质峰也比分析纯的少了一个。安谱农残级正己烷瓶上标着纯度95%(GC)。从GC-MS的TIC结果来看还是比较吻合的。安谱农残级正己烷杂质峰都是正己烷的同分异构体(比如2-甲基-戊烷、3-甲基-戊烷等),不知正己烷的提纯工艺是怎样子的,感觉纯度还不是很高。
各位,GC做有机氯时,发现:标物为正己烷基体中9种有机氯(666、DDT、六氯苯),分别以正己烷和甲醇为基体稀释20倍,且分别向两种基体中加同量微量丙酮(丙酮为助溶正己烷/甲醇体系,为使两种体系具有可比性,所向两种稀释的标准中都加了同量微量丙酮),发现正己烷基体中9种出峰情况正常,但甲醇基体中γ-666,δ-666,op-ddt,pp'-ddt这四种物质出峰很低(2种DDT可明显判断是分解了,但γ-666和δ-666无法有效确证,因为666是有大π键是很稳定的),请各位大侠指教。(已排除衬管惰性降低的可能,这一点请各位不用考虑,而且多次实验表明:同样浓度,甲醇基体的有机氯标物中以上4种物质峰高明显比正己烷基体的有机氯标物要低很多)多谢!见附件中谱图,图中画圈的为9种目标组分,第三个较高的峰为六氯苯。欢迎可加QQ289638727沟通!
微波萃取土壤测“有机氯农药”要用到1:1丙酮+正己烷,但是仪器老师说ECD不能进丙酮,怎么办呢?
食品中粉锈宁残留量的测定方法 1.主题内容与适用范围 本标准规定了粮食、蔬菜和水果中粉锈宁残留量的分析方法。 本标准适用于使用过粉锈宁的粮食、蔬菜和瓜果的残留量分析。粉锈宁的最小检出限为2.8×10-10g。 2.原理 样品中粉锈宁残留物经提取、净化后用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法测定。 氮磷检测器对含氮化合物具有较好的灵敏度。粉锈宁及其主要代谢物羟锈宁与铷盐蒸汽相作用,产生CN-离子流,使检测器收集极的电信号发生变化,这种变化过程被记录下来。样品色谱图上粉锈宁和羟锈宁的峰高与标样色谱图相比,计算出粉锈宁残留量。 出峰顺序:粉锈宁,羟锈宁。 3.试剂 以下试剂除注明外,均为分析纯,试验用水均为蒸馏水。 3.1 丙酮。 3.2 二氯甲烷。 3.3 氯化钠。 3.4 无水硫酸钠,600℃烘4h备用。 3.5 活性炭(C.P.,粉状)。 3.6 三氯甲烷。 3.7 弗罗里硅土(Florisil)60-100目,进口分装。650℃烘5h,加3.5%水脱活,稳定4~6d。 3.8 农药标准溶液 精密称取粉锈宁和经锈宁标准品各50.00mg,加丙酮溶解,分别定容至100.0mL,作为贮备液,放在冰箱中保存。此溶液每毫升相当于粉锈宁或羟锈宁500μg。 临用时吸粉锈宁和羟锈宁贮备液等量混合,用丙酮逐级稀释为使用液,粉锈宁和羟锈宁浓度一般为1.00μg/mL。 4.仪器 4.1 小型粮食粉碎机。 4.2 高速组织捣碎机。 4.3 超声波发生器或往返式振荡器。 4.4 旋转蒸发器。 4.5 [url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url],具有氮磷检测器(铷珠)和微处理机。 5.分析步骤 5.1 提取 5.1.1 粮食:称30.0g粉碎后过60目的样品,置于500mL具塞三角瓶中,加入80mL丙酮和20mL蒸馏水,静置过夜后用振荡器提取1h,抽滤,残渣用30mL×2丙酮洗涤,合并提取液和洗涤液,在旋转蒸发器上浓缩(水浴40~45℃,下同),除去大部分丙酮,然后移至250mL分液漏斗中,依次加入50mL蒸馏水、10mL饱和氯化钠水溶液、40mL二氯甲烷,振摇2min,静置分层,下层有机相移至三角烧瓶中,水相用20mL二氯甲烷再提取一次,弃去水相,有机相经无水硫酸钠脱水(无水硫酸钠10g左右)后收集在250mL梨形瓶中,置旋转蒸发器上浓缩近干,取下梨形瓶,置水浴上使二氯甲烷挥发尽,用少量丙酮多次洗涤梨形瓶内壁,洗涤液收集至刻度试管中,用丙酮定容至5mL,供[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]测定。 5.1.2 瓜果和蔬菜:称取经组织捣碎机捣碎的样品匀浆50.0g于500mL具塞三角瓶中,加活性炭1g,丙酮80mL,摇匀,置超声波发生器上提取10min,抽滤,残渣加100mL丙酮浸泡过夜,再用超声波发生器提取30min,用20mL×2丙酮洗涤残渣和容器,合并提取液与洗涤液,置旋转蒸发器上浓缩至100mL左右,移至分液漏斗中,加30mL蒸馏水、12mL饱和氯化钠水溶液、40mL二氯甲烷,以下按 5.1.1“振摇2min”起操作。 5.2 净化 如测定时有干扰峰影响定量结果,样液可用柱层析法进一步净化。 玻璃层析柱内径1.2cm,长30cm,底部加1.5cm高的无水硫酸钠,称10g混合吸附剂(弗罗里硅土+活性炭,W∶W=9∶1),以1∶30丙酮-氯仿混合液湿法装柱,上层再加1.5cm高的无水硫酸钠,加入提取浓缩液,以150mL 1∶30丙酮-氯仿混合液淋洗(粮食用45mL 1∶19丙酮-氯仿混合液+85mL 1∶49丙酮-氯仿混合液淋洗)。收集淋出液于梨形瓶中,在旋转蒸发器上浓缩近干,水浴使三氯甲烷挥发尽,用丙酮定容,供[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]测定。 5.3 [url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]测定 5.3.1 色谱条件 5.3.1.1 色谱柱:内径3mm,长1.6m,玻璃柱,内装涂有4%OV-17和4%OV-210混合固定液的80~100目Chromosorb W AW-DMCS。 5.3.1.2 气流量:载气为氮气70mL/min;氢气3.6mL/min;空气160mL/min。 5.3.1.3 温度:柱温220℃;汽化室(进样口)290℃;检测室290℃。 5 灵敏度、准确度和精密度 粉锈宁和羟锈宁最小检出限分别为2.8×10-10g和5×10-10g。最低检出浓度:粮食样品粉锈宁为0.01mg/kg,羟锈宁为0.02mg/kg,瓜果蔬菜样品粉锈宁为0.006mg/Kg,羟锈宁为0.01mg/Kg。 粉锈宁-羟锈宁方法回收率:添加浓度0.04~5.0mg/kg范围时为84.0%~103%;检测变异系数(CV)为4.2%~9.4%。 6.说明:样品一般不经柱层析净化,但如需经柱净化,对不同批号的弗罗里硅土,应预先做淋洗曲线,以保证农药得到充分的回收
1.主题内容与适用范围 本标准规定了粮食、蔬菜和水果中粉锈宁残留量的分析方法。 本标准适用于使用过粉锈宁的粮食、蔬菜和瓜果的残留量分析。粉锈宁的最小检出限为2.8×10-10g。 2.原理 样品中粉锈宁残留物经提取、净化后用气相色谱法测定。 氮磷检测器对含氮化合物具有较好的灵敏度。粉锈宁及其主要代谢物羟锈宁与铷盐蒸汽相作用,产生CN-离子流,使检测器收集极的电信号发生变化,这种变化过程被记录下来。样品色谱图上粉锈宁和羟锈宁的峰高与标样色谱图相比,计算出粉锈宁残留量。 出峰顺序:粉锈宁,羟锈宁。 3.试剂 以下试剂除注明外,均为分析纯,试验用水均为蒸馏水。 3.1 丙酮。 3.2 二氯甲烷。 3.3 氯化钠。 3.4 无水硫酸钠,600℃烘4h备用。 3.5 活性炭(C.P.,粉状)。 3.6 三氯甲烷。 3.7 弗罗里硅土(Florisil)60-100目,进口分装。650℃烘5h,加3.5%水脱活,稳定4~6d。 3.8 农药标准溶液 精密称取粉锈宁和经锈宁标准品各50.00mg,加丙酮溶解,分别定容至100.0mL,作为贮备液,放在冰箱中保存。此溶液每毫升相当于粉锈宁或羟锈宁500μg。 临用时吸粉锈宁和羟锈宁贮备液等量混合,用丙酮逐级稀释为使用液,粉锈宁和羟锈宁浓度一般为1.00μg/mL。 4.仪器 4.1 小型粮食粉碎机。 4.2 高速组织捣碎机。 4.3 超声波发生器或往返式振荡器。 4.4 旋转蒸发器。 4.5 气相色谱仪,具有氮磷检测器(铷珠)和微处理机。 5.分析步骤 5.1 提取 5.1.1 粮食:称30.0g粉碎后过60目的样品,置于500mL具塞三角瓶中,加入80mL丙酮和20mL蒸馏水,静置过夜后用振荡器提取 1h,抽滤,残渣用30mL×2丙酮洗涤,合并提取液和洗涤液,在旋转蒸发器上浓缩(水浴40~45℃,下同),除去大部分丙酮,然后移至250mL分液漏斗中,依次加入50mL蒸馏水、10mL饱和氯化钠水溶液、40mL二氯甲烷,振摇2min,静置分层,下层有机相移至三角烧瓶中,水相用20mL二氯甲烷再提取一次,弃去水相,有机相经无水硫酸钠脱水(无水硫酸钠10g左右)后收集在250mL梨形瓶中,置旋转蒸发器上浓缩近干,取下梨形瓶,置水浴上使二氯甲烷挥发尽,用少量丙酮多次洗涤梨形瓶内壁,洗涤液收集至刻度试管中,用丙酮定容至5mL,供气相色谱测定。 5.1.2 瓜果和蔬菜:称取经组织捣碎机捣碎的样品匀浆50.0g于500mL具塞三角瓶中,加活性炭1g,丙酮80mL,摇匀,置超声波发生器上提取10min,抽滤,残渣加100mL丙酮浸泡过夜,再用超声波发生器提取30min,用20mL×2丙酮洗涤残渣和容器,合并提取液与洗涤液,置旋转蒸发器上浓缩至100mL左右,移至分液漏斗中,加30mL蒸馏水、12mL饱和氯化钠水溶液、40mL二氯甲烷,以下按 5.1.1“振摇2min”起操作。 5.2 净化 如测定时有干扰峰影响定量结果,样液可用柱层析法进一步净化。 玻璃层析柱内径1.2cm,长30cm,底部加1.5cm高的无水硫酸钠,称10g混合吸附剂(弗罗里硅土+活性炭,W∶W=9∶1),以1∶30丙酮-氯仿混合液湿法装柱,上层再加1.5cm高的无水硫酸钠,加入提取浓缩液,以150mL 1∶30丙酮-氯仿混合液淋洗(粮食用45mL 1∶19丙酮-氯仿混合液+85mL 1∶49丙酮-氯仿混合液淋洗)。收集淋出液于梨形瓶中,在旋转蒸发器上浓缩近干,水浴使三氯甲烷挥发尽,用丙酮定容,供气相色谱测定。 5.3 气相色谱测定 5.3.1 色谱条件 5.3.1.1 色谱柱:内径3mm,长1.6m,玻璃柱,内装涂有4%OV-17和4%OV-210混合固定液的80~100目Chromosorb W AW-DMCS。 5.3.1.2 气流量:载气为氮气70mL/min;氢气3.6mL/min;空气160mL/min。 5.3.1.3 温度:柱温220℃;汽化室(进样口)290℃;检测室290℃。 5 灵敏度、准确度和精密度 粉锈宁和羟锈宁最小检出限分别为2.8×10-10g和5×10-10g。最低检出浓度:粮食样品粉锈宁为0.01mg/kg,羟锈宁为0.02mg/kg,瓜果蔬菜样品粉锈宁为0.006mg/Kg,羟锈宁为0.01mg/Kg。 粉锈宁-羟锈宁方法回收率:添加浓度0.04~5.0mg/kg范围时为84.0%~103%;检测变异系数(CV)为4.2%~9.4%。 6.说明:样品一般不经柱层析净化,但如需经柱净化,对不同批号的弗罗里硅土,应预先做淋洗曲线,以保证农药得到充分的回收
步骤:先用分散剂(水、乙醇、饱和氯化钾)将牙膏样品涡旋打散,再加入正己烷涡旋萃取。正常情况都是分层明显的。可这次遇到个奇葩样品,静置很久也不分层,加入氯化钠也无效果。于是把样液转移到离心管离心处理,结果下层是水状,上层变成凝胶状了。怎么办呢?[img]https://ng1.17img.cn/bbsfiles/images/2022/11/202211091249004122_1633_2561261_3.png[/img][img]https://ng1.17img.cn/bbsfiles/images/2022/11/202211091249003415_8086_2561261_3.png[/img]
发现23200.10的净化步骤中的正己烷-丙酮溶剂没说比例啊!怎么办?[img]https://ng1.17img.cn/bbsfiles/images/2021/11/202111031102443449_5710_3931817_3.png[/img][img]https://ng1.17img.cn/bbsfiles/images/2021/11/202111031102443127_3501_3931817_3.png[/img]
各位老师好,请问:用高效液相色谱做溴氰菊酯时,流动相环己烷可以用正己烷代替吗?
请问正己烷和丙酮的共沸点是多少啊?怎么计算?谢谢
测的是牙膏中的香精。样品前处理很简单,向牙膏里加分散液和玻璃珠,涡旋打散均质,再加入正己烷,涡旋萃取。分散液主要成分是水和乙醇。白色牙膏处理后都是正己烷层在上。可是有一个透明蓝色牙膏处理后分层超级慢,等了好久才分出一点点,还是在下层,上层是不透明的牙膏分散液。等了快一小时了,下层有一小部分跑上层了,中间是牙膏分散液,下边是另一部分正己烷层。感觉好像牙膏分散液层阻挡了正己烷上去。有人遇到类似情况的吗?怎么解决?
我用索式萃取对塑料薄膜萃取过后(正己烷做萃取液),请问怎么才能彻底清除塑料薄膜上面的正己烷呢?现在用的方法是丙酮超声15分钟,结果好像不行。请指教,谢谢。
请问一下,丙酮和正己烷有开封保质期吗?如果有是多少了?谢谢!
昨天看见一位老师在做761方法时,用的正己烷是加了氯化钠的。他的体会是加盐可以有效去除正己烷中的微量水分。我的疑问:正己烷中可能有水么?正己烷的水可以用这个方法去除么?
我现在有生育酚四种同分异构体的储备液,是溶解在乙醇里面的。但是我的样品时溶解在正己烷里面的,所以想用正己烷做溶剂来制作标曲。因此有几个问题想请教一下。1、生育酚的乙醇储备液(1000 ug/mL,只有1 mL)可以直接用正己烷稀释到使用浓度(100 ug/mL)吗?2、如果不能直接稀释,请问可以使用旋蒸去除乙醇后再使用正己烷溶解吗?因为没有氮气,怕旋蒸过程会造成生育酚损失,可以添加BHT来保护生育酚吗?3、使用紫外检测器,生育酚标曲的浓度范围一般配置为多少呢?1-100 ug/mL吗?
问题:话说,如果我用气质做有机磷,我的单标或者混標标液用什么溶剂稀释啊?丙酮还是正己烷好呢?求解求解,我记得之前不知道在哪里看来的,说气质打丙酮不好方法1-我用丙酮乙酸乙酯方法2-用正己烷,而有机氯用丙酮方法3-有机磷用气相fpd,我们有机磷用丙酮方法4-上气质,最好用基质,其次用定容溶剂大家觉得呢?
[color=#444444]谁做过正己烷中苯残留的测定,有没有大概的实验方法啊?我从网上找了一个没太看懂,高手们指导一下吧!谢谢![/color][color=#444444]操作条件 [/color][color=#444444]柱填充剂 在177~250μm的[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]分析用载体上加入含相当10%载体的聚乙二醇6,000的氯仿溶液,再把氯仿蒸发、干燥。 [/color][color=#444444]柱管 内径3~4mm 长2~3m的不锈钢管或玻璃管。[/color][color=#444444]柱温度 50~70℃的一定温度。 [/color][color=#444444]检测器 氢氮离子化检测器。 [/color][color=#444444]载气用氮气 调正分离管温度及载气流速使苯能在约5min流出[/color][color=#444444]取本品5mL,基保加入内部标准物质溶液50mL混合,做检液,另外取对照物质溶液50mL,内部标准溶液50mL混合做标准溶液。此二液在如下条件进行[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]分析,检液的峰高(H)和内部标准物质的峰高(HS)的比H/H,不超过标准溶液峰高(H’)和内部标准物质的峰高(HS’)的比H’/HS’。 [/color][color=#444444]内部标准物质溶液是取甲基异丁基酮0.5mL,加正己烷至100mL,对照物质溶液是取0.25mL\加正己烷至100mL。[/color]
三聚氰胺检测中,脂肪含量高用 三氯乙酸溶液饱和的正己烷 怎么配置和添加操作







 我要推广仪器
我要推广仪器
 下载APP
下载APP