目前我们实验室按欧洲药典标准检测克拉霉素含量、有关物质,所用的色谱柱是欧洲药典推荐的一款色谱柱:Kromasil 100-3.5-C18 100*4.6 ,目前该色谱柱使用寿命很短,一般不到100小时,请生产厂家在中国的代理商到实验 室指导也未找到问题所在,求助各位大侠问题可能出在哪里?或推荐相同规格能满足欧洲药典检测克拉霉素要求的色谱柱。
各位大侠好,我在做克拉霉素分散片时,紫外测定吸收度最后才0.25,这样的结果可信吗?请问紫外吸收度要在什么范围才最好啊?有没有规定必须在这个范围呢?你们有谁做国这个检品啊?急 谢谢了哈!!!
各位大侠好,我在做克拉霉素分散片时,紫外测定吸收度最后才0.25,这样的结果可信吗?请问紫外吸收度要在什么范围才最好啊?有没有规定必须在这个范围呢?你们有谁做国这个检品啊?急 谢谢了哈!!!
最近在做克拉霉素有关物质检查,却发现很多奇怪的现象,让我百思不解,写出来跟大家讨论讨论。仪器:岛津2010A/C(一新一旧),25cm的C18柱两根,210nm,05版药典二部方法,流动相为缓冲盐:乙腈(6:4),缓冲盐中加0.2%三乙胺,用磷酸调pH至5.5,柱温45度,分析时间30min。奇怪现象如下:1。柱温为室温(20度)时,主峰在6min左右出峰,柱温为45度时,主峰在8-9min出峰,有点反常。2。平衡好柱子后进样,系列进样,前几个样走得比较正常,比如到第3个样,第十几分钟后基线出现个大包(疑似进气泡的现象),连续3针基线都是波浪式的,但过了这几针却又好了。大家都说是检测池进气泡了,但是我每次做这个样品的时候都出现这个情况,而且流动相我每次都要超声10min,使用新的仪器(刚买不到1个月)也这样,而这台仪器做其它样品的时候一切正常,所以,让我很头疼,不知道有没有跟我碰到一样的情况。3。样品用流动相溶解,当进空白溶剂(即流动相)时,在2.4min出现一个小峰,峰高大约在1mAU,克拉霉素原料在2.4min也出现杂质峰,峰高要大一点,而克拉霉素片用的辅料也在2.4min出峰,所以做有关物质时就犯难了,这个峰怎么处理,也不能当溶剂或辅料峰扣掉,不扣的话,这个杂质峰就已经超标了。
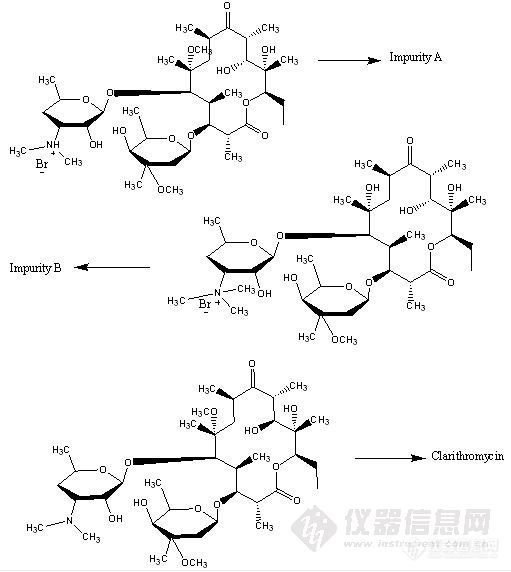
单位工艺生产出来的克拉霉素在主峰的相对保留时间0.84处出现了一个超过0.1%的杂质,现在需要对其进行结构确证。对这个未知杂质,我们了解的情况是,(一)这个杂质对EP/USP液相条件很是敏感的,主要体现在流动相的PH值上,EP/USP液相条件的缓冲液PH值是4.4,当我们把PH值调到4.0时,其他峰的保留时间基本没有变化,提前约0.2~0.3min,但是该杂质的保留时间提前约2min;当把PH调到5.0时,其他峰的保留时间也是基本不变,但是该杂质在主峰之后出峰了,大约延后了约4min。(二)将该杂质接出来做[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url],最明显的离子峰是748.4,但是这个峰是不是分子离子峰不是很确定,继续对这个离子做多级[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url],其主要碎片情况与克拉霉素一样。EP/USP液相条件:溶液A— 4.76 g磷酸二氢钾至1000ml水中,用磷酸) (l - 10) 或氢氧化钾(45% w/v) 调PH至4.4溶液B— 乙腈. Time (minutes) Solution A (%) Solution B (%) 0 75 2532 40 6034 40 6036 75 2542 75 25 流速是1.1ml/min然后我们根据748.4的离子峰,以及工艺,推测了几个可能的杂质,希望大家帮我看看,哪个可能,或者是不是还有其他的可能呢? [img]http://ng1.17img.cn/bbsfiles/images/2010/01/201001280805_199202_1638724_3.jpg[/img]
克拉的数值是确定一颗宝石价值多少的重要因素,所以说,宝石的克拉值越高,它的价值就越大。 克拉一词最早起源于古希腊文,它是地中海东岸的一种角豆树的种子(稻子豆)的名字。这种树豆荚结褐色果仁,长约十五厘米。 角豆树有一种奇特的现象,无论长在何处,它所结的果仁每一颗重量均一致,1粒果仁就是1克拉。于是,这种果实就被用来作为测量重量的砝码,用来称贵重和细微的物质。1颗宝石与多少粒种子的重量相等就有多少克拉。到了1907年,国际上商定其为宝石的计量单位。克拉作为宝石的计量单位,在现行的国际标准中作为法定的计量单位。它的换算公式为:1克拉=200毫克=0.2克。
【序号】:2【作者】:陈争明【题名】:水凝胶缓释克拉霉素纳米新材料在鼻窦炎抗炎中的作用和机制研究【期刊】:中国人民解放军海军军医大学【年、卷、期、起止页码】:2022【全文链接】:https://kns.cnki.net/kcms2/article/abstract?v=6xaVI2TORM2vC_L_QOfZqPXWOObXxPnwULVtR4uAzT8FTVB0gpO7NcYOjMpBVPOUi94Vux3LTdZBhtAr1ADf6Zn2zLX5SBXlfJ5cj64E-1tQMiO_9W4mYkPxwT3crUUuAuDxPXSVG9a-G00hdA_SgGph9x7lxnYH9H_q3404VOk=&uniplatform=NZKPT&language=CHS
在珠宝界,宝石的计量单位常常用 “克拉”,这是怎么来的呢? 克拉作为重量单位,起源于欧洲地中海边的一种角豆树的种子。角豆树有一个奇特的现象,不管它长在什么地方,它所结的果仁,每一颗重量均精确一致。在历史上这种果实就被用来作为测定重量的砝码,久而久之便成了一种重量单位,用它来称量贵重和细微的物体。 1907年,国际上商定克拉为宝石和黄金的计量单位。1914年,国际上把 “克拉”的标准重量定为200毫克。钻石以克拉计重在世界上是法定的。其它一些高档宝石如红宝石、蓝宝石、祖母绿、碧玺、海蓝宝、金绿宝石等,目前也都使用克拉作为计量单位。以实际重量乘上每克拉单价,就是某颗宝石的价格。
不知道建立质谱方法时,对标准品的纯度要求如何?看到一篇文献上这么写道:实验样品及试剂交沙霉素片( 200mg/片),麦迪霉素片(100mg/片),红霉素片(125mg/片),克拉霉素片(250mg/片),罗红霉素胶囊(50mg/粒)以前总觉得标准品都要购自Sigma这样的大公司,才有保证。不知道像上面这样从药店购买的药品可不可行?
GB 29685-2013 食品安全国家标准 动物性食品中林可霉素、克林霉素和大观霉素多残留的测定 气相色谱—质谱法
GB 29686-2013 食品安全国家标准 猪可食性组织中阿维拉霉素残留量的测定 液相色谱-串联质谱法
我们刚买了台液相色谱仪,准备用来检测产品中的棒曲霉素,但不知道那里(我现在在山西)卖棒曲霉素标准品,希望知道的朋友告诉一下。
想请教一下大家使用薄层定性鉴别用的对照品的时候是怎么称的?,如1mg/ml溶液或者更少的0.5mg/ml溶液的对照品是怎么称的?称多了不好,称少了好像天平的有一个最小称量值。
我们刚买了台液相色谱仪,准备用来检测产品中的棒曲霉素,但不知道那里(我现在在山西)卖棒曲霉素标准品,希望知道的朋友告诉一下。
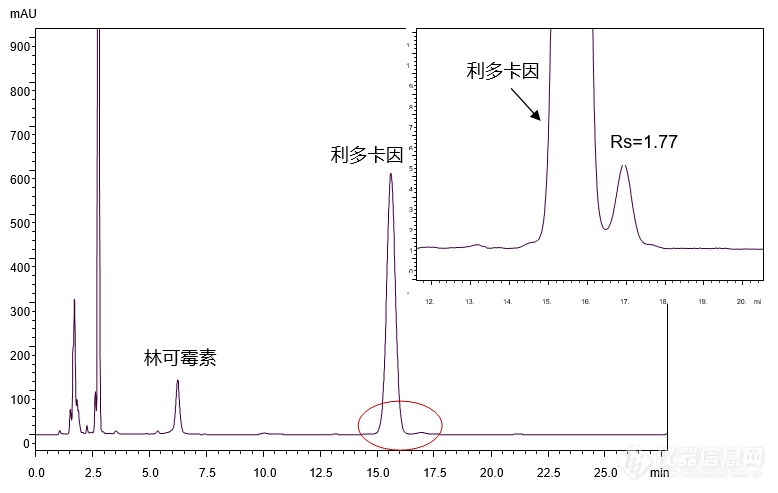
[align=center][b]【国家药品标准】林可霉素利多卡因凝胶的分析[/b][/align][align=center][b][/b][/align][align=right][b]——依据国家药品标准WS-10001-(HD-0140)-2002方法[/b][/align][b]林可霉素利多卡因凝胶[/b]为复方制剂,每克含林可霉素5毫克,利多卡因4毫克。适应症为用于轻度烧伤、创伤及蚊虫叮咬引起的各种皮肤感染。 [img=,193,127]http://ng1.17img.cn/bbsfiles/images/2018/07/201807260834522166_2994_2222981_3.gif!w193x127.jpg[/img] [img=,140,64]http://ng1.17img.cn/bbsfiles/images/2018/07/201807260834520028_3541_2222981_3.gif!w140x64.jpg[/img] 林可霉素 利多卡因 Lincomycin Lidocaine M.W.: 406.54 M.W.: 234.34客户提供林可霉素利多卡因凝胶样品,希望本实验室帮忙通过筛选色谱柱及调节分析条件,依据[color=#ff0000][b]国家药品标准WS-10001-(HD-0140)-2002[/b][/color]方法,实现林可霉素利多卡因凝胶样品的良好分析。首先,使用能在纯水条件下稳定使用的高极性色谱柱[color=#ff0000][b]CAPCELL PAK C[sub]18[/sub] AQ S5 4.6 mm i.d. × 150 mm[/b][/color],对林可霉素利多卡因凝胶样品进行分析,结果如图1所示,[color=#330099]利多卡因与其峰后杂质之间分离度为1.77[/color]。[align=center][img=,690,437]http://ng1.17img.cn/bbsfiles/images/2018/07/201807260858200006_8607_2222981_3.png!w690x437.jpg[/img][/align][align=center]图1 CAPCELL PAK C[sub]18 [/sub]AQ分析所得色谱图[/align]注:峰上标数字为分离度。[img=,528,205]http://ng1.17img.cn/bbsfiles/images/2018/07/201807260858202566_2695_2222981_3.png!w528x205.jpg[/img]为进一步提高利多卡因与其峰后杂质之间的分离度,在原条件基础上将柱温由30℃降低至25℃,并分别使用 CAPCELL PAK C[sub]18[/sub] AQ、CAPCELL PAK C[sub]18[/sub] MG及高含碳量ODS色谱柱SUPERIOREX ODS进行分析,结果如图2所示。[align=center][img=,690,490]http://ng1.17img.cn/bbsfiles/images/2018/07/201807260859201516_7229_2222981_3.png!w690x490.jpg[/img][/align][align=center]图2 25℃条件下不同色谱柱分析结果对比[/align]注:峰上标数字为分离度。[img=,637,223]http://ng1.17img.cn/bbsfiles/images/2018/07/201807260859204236_7198_2222981_3.png!w637x223.jpg[/img]如图2所示,在柱温25℃条件下使用三款色谱柱进行分析,其中,[color=#ff0000][b]CAPCELL PAK C[sub]18[/sub] AQ色谱柱分析结果最好,利多卡因与其峰后杂质分离得到最佳分离,分离度为4.23[/b][/color];[color=#330099][b]使用CAPCELL PAK C[sub]18[/sub] MG色谱柱进行分析时,利多卡因与其峰后杂质分离度为3.27[/b][/color];而使用SUPERIOREX ODS色谱柱分析时,利多卡因与其峰后杂质未得到有效分离。综上,在国家药品标准WS-10001-(HD-0140)-2002方法基础上,将色谱柱柱温由30℃降低至25℃,使用高极性色谱柱CAPCELL PAK C[sub]18[/sub] AQ及中等极性色谱柱CAPCELL PAK C[sub]18[/sub] MG进行分析,均可在25 min内完成林可霉素利多卡因凝胶样品的分析,并得到利多卡因与其峰后杂质之间的良好分离结果。[align=right][/align][align=right][/align][align=right] [/align][align=right]三耀精细化工品销售(中国)有限公司[/align][align=right]技术开发部[/align][align=right]地址:北京经济技术开发区宏达南路5号[/align][align=right]宏达利德工业园1栋418室[/align][align=right]邮编:100176[/align]
中文名称: 希波克拉底 外文名: Hippocrates of Chios 生卒年: 公元前460-公元前377 洲: 欧洲 国别: 古希腊 省: 小亚细亚科斯岛 希波克拉底(Hippcrates),古希腊著名医生,欧洲医学奠基人,被西方尊为“医学之父”。希波克拉底于公元前460年出生于小亚细亚科斯岛的一个医生世家,祖父、父亲都是医生,母亲是接生婆。在古希腊,医生的职业是父子相传的,所以希波克拉底从小就跟随父亲学医。父母去世后,他在希腊,小亚细亚,里海沿岸,北非等地一面游历,一面行医,从而增长了知识,接触了民间医学。那时,古希腊医学受到宗教迷信的禁锢。巫师们只会用念咒文,施魔法,进行祈祷的办法为人治病。公元前430年,雅典发生了可怕的瘟疫。对这种索命的疾病,人们避之唯恐不及。但希波克拉底却冒着生命危险前往雅典救治。他一面调查疫情,一面探寻病因及解救方法。不久,他发现全城只有每天和火打交道的铁匠没有染上瘟疫,他由此设想,或许火可以防疫,于是在全城各处燃起火堆来扑灭瘟疫。希波克拉底指出的癫痫病的病因被现代医学认为是正确的,他提出的这个病名,也一直沿用至今。希波克拉底对骨折病人提出的治疗方法,后来被证明是合乎科学道理的。为了纪念他,后人将用于牵引和其他矫形操作的臼床称为“希波克拉底臼床”。为了抵制“神赐疾病”的谬说,希波克拉底积极探索人的肌体特征和疾病的成因,提出了著名的“体液学说”,认为人体由血液、粘液、黄胆和黑胆四种体液组成,这四种体液的不同配合使人们有不同的体质。他把疾病看作是发展着的现象,认为医师所应医治的不仅是病而是病人;从而改变了当时医学中以巫术和宗教为根据的观念。主张在治疗上注意病人的个性特征、环境因素和生活方式对患病的影响。重视卫生饮食疗法,但也不忽视药物治疗,尤其注意对症治疗和预后。他对骨骼、关节、肌肉等都很有研究。他的医学观点对以后西方医学的发展有巨大影响。 现在看来,希波克拉底对人的[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质[/color][/url]的成因的解释并不正确,但他提出的[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质[/color][/url]类型的名称及划分,却一直沿用至今。那时,尸体解剖为宗教与习俗所禁止,希波克拉底勇敢地冲破禁令,秘密进行了人体解剖,获得了许多关于人体结构的知识。在他最著名的外科著作《头颅创伤》中,详细描绘了头颅损伤和裂缝等病例,提出了施行手术的方法。其中关于手术的记载非常精细,所用语言也非常确切,足以证明这是他亲身实践的经验总结。在他的题为《箴言》的论文集中,辑录了许多关于医学和人生方面的至理名言,如“人生矩促,技艺长存”;“机遇诚难得,试验有风险,决断更可贵”;“暴食伤身”:“无故困倦是疾病的前兆”;“简陋而可口的饮食比精美但不可口的饮食更有益”;“寄希望于自然”等,这些经验之谈脍炙人口,至今仍给人以启示。古代西方医生在开业时都要宣读一份有关医务道德的誓词:“我要遵守誓约,矢忠不渝。对传授我医术的老师,我要像父母一样敬重。对我的儿子、老师的儿子以及我的门徒,我要悉心传授医学知识。我要竭尽全力,采取我认为有利于病人的医疗措施,不能给病人带来痛苦与危害。我不把毒药给任何人,也决不授意别人使用它。我要清清白白地行医和生活。无论进入谁家,只是为了治病,不为所欲为,不接受贿赂,不勾引异性。对看到或听到不应外传的私生活,我决不泄露。”这个医道规范的制定者就是希波克拉底。20世纪中叶,世界医协大会又据此制定了国际医务人员道德规范。公元前377希波克拉底逝世,终年83岁。研究领域:医学相关作品:1、提出了著名的“体液学说”。认为复杂的人体是由血液、粘液、黄胆、黑胆这四种体液组成的,四种体液在人体内的比例不同,形成了人的不同[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质[/color][/url]:性情急躁、动作迅猛的胆汁质;性情活跃、动作灵敏的多血质;性情沉静、动作迟缓的粘[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url];性情跪弱、动作迟纯的抑郁质。人所以会得病,就是由于四种液体不平衡造成的。而液体失调又是外界因素影响的结果。所以他认为一个医生进入某个城市首先要注意这个城市的方向、土壤、气候、风向、水源、水、饮食习惯、生活方式等等这些与人的健康和疾病有密切关系的自然环境。2、制定了从医道德规范。3、《头颅创伤》4、论文集《箴言》
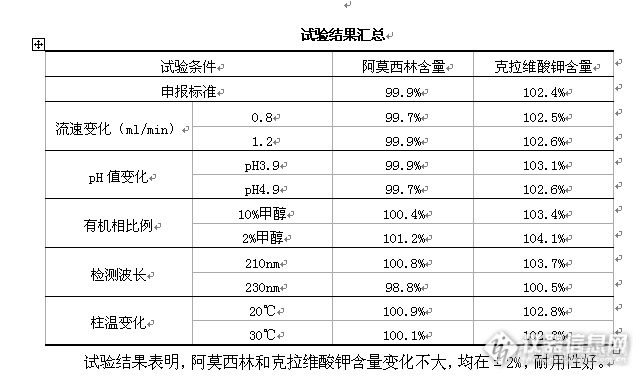
继”阿莫西林克拉维酸钾胶囊有关物质方法学”项目结束,整理的含量测定方法学。项目:含量测定(3.2.P.5.2.9)检查方法:照高效液相色谱法(中国药典2010年版二部附录Ⅴ D)测定试验条件:仪器:LC-2010CHT (SHIMADZU)万分之一电子天平(Sartorius ABS-124S型)工作站(LCsolution色谱工作站)色谱柱(填料:C18,规格:250mm×4.6mm,填料粒径:5μm)Xtimate C18 4.6*250 ,PN:Xt5B18425 ,SN:411101950UV检测器(检测波长:220nm)柱温:室温流动相:0.05mol/L磷酸二氢钠溶液(取磷酸二氢钠7.8g,加水900ml使溶解,用10%磷酸溶液或氢氧化钠试液调节pH值至4.4±0.1,加水稀释至1000ml)-甲醇(95:5)。流速:1.0ml/min。运行时间:约20分钟。系统适用性:取阿莫西林克拉维酸系统适用性试验对照品,加流动相溶解并稀释制成每1ml中含0.8mg的溶液,取20μl注入液相色谱仪,记录的色谱图应与标准图谱一致。具体试验操作:取装量差异项下的内容物适量,精密称取适量,加水适量,超声使溶解并定量稀释制成每1ml中含阿莫西林0.5mg的溶液,滤过,立即精密量取续滤液20μl注入液相色谱仪,记录色谱图;另分别精密称取阿莫西林对照品与克拉维酸对照品各适量,加水溶解并定量稀释制成每1ml中约含阿莫西林0.5mg和每1ml中含克拉维酸0.125mg的混合溶液,同法测定。按外标法以峰面积分别计算供试品中C16H19N3O5S和C8H9NO5的含量。计算公式:标示量百分含量(%)=××100%式中:Cs为对照品的浓度(mg/ml);At为供试液的主峰面积;Nt为供试液的稀释倍数;AS为对照品溶液的主峰面积;W为供试品取样量(mg)。“色”路蹒跚,藤下葡萄,某品种含量测定方法学之耐用性试验部分路蹒跚,藤下葡萄,某品种含量测定方法学之耐用性试验部分http://ng1.17img.cn/bbsfiles/images/2013/06/201306292159_448382_1621890_3.gif3.2.P.5.3.6.1波长选择本品含量测定检测波长参照中国药典2010年版二部收载的阿莫西林克拉维酸钾相关制剂质量标准含量测定项,即220nm。3.2.P.5.3.6.2流动相选择(色谱图见附件1122~1124)参照中国药典2010年版二部收载的阿莫西林克拉维酸钾相关制剂质量标准含量测定项,以0.05mol/L磷酸二氢钠溶液(取磷酸二氢钠7.8g,加水900ml使溶解,用10%磷酸溶液或氢氧化钠试液调节pH值至4.4±0.1,加水稀释至1000ml)-甲醇(95:5)为流动相。试验过程:系统适用性试验供试液:精密称取阿莫西林克拉维酸钾系统适用性对照品4.2mg至5ml量瓶中,加流动相适量超声使溶解并稀释至刻度,摇匀,滤过,取续滤液作为供试液;对照品溶液:精密称取阿莫西林对照品29.1mg和克拉维酸钾对照品7.3mg至50ml量瓶中,加水适量超声使溶解并稀释至刻度,摇匀,滤过,取续滤液作为供试液;精密量取上述供试液各20μl注入高效液相色谱仪,记录色谱图,典型色谱图见下图:http://ng1.17img.cn/bbsfiles/images/2013/06/201306292200_448383_1621890_3.gifhttp://ng1.17img.cn/bbsfiles/images/2013/06/201306292200_448384_1621890_3.gifhttp://ng1.17img.cn/bbsfiles/images/2013/06/201306292201_448385_1621890_3.gif3.2.P.5.3.6.3进样精密度试验(色谱图见附件1125~1130)

http://ng1.17img.cn/bbsfiles/images/2013/02/201302021634_424293_1621890_3.jpg3.2.P.5.3.2 有关物质色谱图见附件8~234。有关物质检查方法验证概要项目验证结果流动相选择流动相A为0.01mol/L磷酸二氢钾溶液(用2mol/L氢氧化钠溶液调节pH值至6.0),流动相B为0.01mol/L磷酸二氢钾溶液(用2mol/L氢氧化钠溶液调节pH值至6.0)-乙腈(20:80)。专属性各破坏条件下,产生的杂质与主成分能够有效分离,主成分峰未检出不纯物。线性和范围无相关研究内容。定量限、检测限阿莫西林以17.9分钟±1分钟基线计算信噪比,当S/N≒3时,检测限为1.48ng(浓度为0.074µg/ml),当S/N≒10时,定量限为4.92ng(浓度为0.246µg/ml);克拉维酸以6.3分钟±1分钟基线计算信噪比,当S/N≒3时,检测限为1.27ng(浓度为0.063µg/ml),当S/N≒10时,定量限为4.22ng(浓度为0.211µg/ml)。准确度无相关研究内容。精密度自身对照溶液连续进样6次,阿莫西林和克拉维酸峰面积和保留时间精密度RSD均小于0.5%溶液稳定性本品供试品溶液室温放置8小时稳定性较好,阿莫西林和克拉维酸钾主峰面积的RSD值和归一化含量RSD值均小于2.0%;当室温放置8小时后,杂质个数和杂质含量没有明显变化。耐用性磷酸盐pH值在5.5~6.0,检测波长230±2nm,流速、柱温耐用范围较宽。3.2.P.5.3.2.1 流动相选择(色谱图见附件8~10)参照中国药典2012年版二部收载的阿莫西林克拉维酸钾相关制剂有关物质项和新药转正标准第75册收载的阿莫西林克拉维酸钾胶囊质量标准WS1-(X-024)-2007Z有关物质项,流动相体系采用流动相A为0.01mol/L磷酸二氢钾溶液(用2mol/L氢氧化钠溶液调节pH值至6.0),流动相B为0.01mol/L磷酸二氢钾溶液(用2mol/L氢氧化钠溶液调节pH值至6.0)-乙腈(20:80)。根据初步拟订的流动相体系进行梯度洗脱选择试验,试验结果统计见下表图。梯度洗脱方式(一)时间(分钟)流动相A(%)流动相B(%)tR + 0982tR + 207030tR + 22982tR + 32982梯度洗脱方式(二)时间(分钟)[font=宋
请问那里可以买到氯霉素的标准品,尤其是在广东地区,价格怎样?供货周期多久?
如何才能买到多西环素(强力霉素)的两个代谢产物:4-epidoxycycline和6-epidoxycycline的标准品?
氯霉素和克伦特罗各地检测标准不同,但总有个大致统一的标准吧,但不知道大家有何高见?
求购棒曲霉素和5-羟甲基糠醛标准品!!!我的联系方式:niupf0406@163.com
现行有效标准:GB/T 8381.9-2005 饲料中氯霉素的测定 气相色谱法GB/T 9695.32-2009 肉与肉制品 氯霉素含量的测定GB/T 18932.19-2003 蜂蜜中氯霉素残留量的测定方法 液相色谱-串联质谱法GB/T 18932.20-2003 蜂蜜中氯霉素残留量的测定方法 气相色谱-质谱法GB/T 18932.21-2003 蜂蜜中氯霉素残留量的测定方法 酶联免疫法GB/T 20756-2006 可食动物肌肉、肝脏和水产品中氯霉素、甲砜霉素和氟苯尼考残留量的测定 液相色谱-串联质谱法GB/T 21108-2007 饲料中氯霉素的测定 高效液相色谱串联质谱法GB/T 21165-2007 肠衣中氯霉素残留量的测定 液相色谱-串联质谱法GB/T 22338-2008 动物源性食品中氯霉素类药物残留量测定GB/T 22959-2008 河豚鱼、鳗鱼和烤鳗中氯霉素、甲砜霉素和氟苯尼考残留量的测定 液相色谱-串联质谱法GB 29688-2013 食品安全国家标准 牛奶中氯霉素残留量的测定 液相色谱-串联质谱法(2014-1-1实施)SN/T 1864-2007 进出口动物源食品中氯霉素残留量的检测方法 液相色谱-串联质谱法SN/T 2058-2008 进出口蜂王浆中氯霉素残留量测定方法 酶联免疫法SN/T 2063-2008 进出口蜂王浆中氯霉素残留量的检测方法 液相色谱串联质谱法SN/T 2289-2009 进出口化妆品中氯霉素、甲砜霉素、氟甲砜霉素的测定 液相色谱-质谱/质SN/T 3235-2012 出口动物源食品中多类禁用药物残留量检测方法 液相色谱-质谱/质谱法SN/T 4537.2-2016 商品化试剂盒检测方法 氯霉素 方法二(2017-7-1实施)(暂无文本)SB/T 10386-2004 畜禽肉中氯霉素的测定SC/T 3018-2004 水产品中氯霉素残留量的测定 气相色谱法农业部781号公告-1-2006 动物源食品中氯霉素残留量的测定 气相色谱-质谱法农业部781号公告-2-2006 动物源食品中氯霉素残留量的测定高效液相色谱-串联质谱法农业部781号公告-10-2006 蜂蜜中氯霉素残留量的测定 气相色谱-质谱法(负化学源)农业部958号公告-13-2007 水产品中氯霉素、甲砜霉素、氟甲砜霉素残留量的测定 气相色谱法农业部958号公告-14-2007 水产品中氯霉素、甲砜霉素、氟甲砜霉素残留量的测定 气相色谱-质谱法农业部1025号公告-21-2008 动物源食品中氯霉素残留检测 气相色谱法农业部1025号公告-26-2008 动物源食品中氯霉素残留检测 酶联免疫吸附法农业部2483号公告-8-2016 饲料中氯霉素、甲砜霉素和氟苯尼考的测定 液相色谱-串联质谱法(2017-4-1实施)DB34/T 821-2008 动物组织中氯霉素的残留测定——酶联免疫吸附法DB34/T 1361-2011 饲料中氯霉素的测定—气相色谱质谱法已废止标准:SN 0199-1993 出口肉中甲砜霉素残留量检验方法(已废止)SN 0215-1993 出口禽肉中氯霉素残留量检验方法(已废止)SN 0341-1995 出口肉及肉制品中氯霉素残量检验方法(已废止)SN/T 1604-2005 进出口动物源性食品中氯霉素残留量的检验方法 酶联免疫法(已废止)SN/T 1966-2007 水产品中氯霉素残留检测方法 放射受体分析法(2015-12-31废止)DB23/T 856-2004 动物性食品中氯霉素残留量的测定方法(高效液相色谱法)(2014-10-14废止)(暂无文本)DB33/T 542-2005 水产品中氯霉素残留量的测定方法 气相色谱-二级质谱法(已废止)DB43/T 343-2007 动物组织中氯霉素残留量的测定(2015-5-1废止)(暂无文本)DB44/T 543-2008 水产种苗中氯霉素残留量的测定 气相色谱法(2016-10-31废止)(暂无文本)DB44/T 568-2008 水体中氯霉素残留量的测定—气相色谱法(2016-10-31废止)(暂无文本)DB44/T 571-2008 底泥中氯霉素残留量的测定—气相色谱法(2016-10-31废止)(暂无文本)DB51/T 408-2004 牛奶中氯霉素残留量检测方法--酶免疫法(2015-5-31废止)(暂无文本)DB51/T 409-2004 鸡蛋中氯霉素残留量检测方法--高效液相色谱法(2017-3-1废止)(暂无文本)DB51/T 469-2005 鸡蛋中氯霉素残留检测方法-酶联免疫吸附测定(ELISA)法(2015-5-31废止)DB51/T 568-2006 牛奶中氯霉素残留检测方法-气相色谱(GC)法(2017-3-1废止)(暂无文本)DB51/T 676-2007 牛奶中氯霉素残留检测方法-高效液相色谱-串联质谱(LC-MS-MS)法(2015-5-31废止)DB51/T 797-2008 鸡蛋中氯霉素残留量测定-高效液相色谱法-串联质谱 (LC-MS-MS) 法(2017-3-1废止)(暂无文本)
最近家里新买了花生,但花生红衣上有些褐色斑点 以为是发霉了,不知道有没有简易方法鉴别一下有没有发霉啊?就是测下有没有黄曲霉素!
买的展青霉素有证书的液体标准品也需要浓度校准吗?望老师不吝赐教
各位老师都是做食品安全的高手,想请教一下,除了眼观手摸意外,有没有知道检测水果打蜡的方法?或者如何鉴别用的是食用石蜡还是工业石蜡??国家目前有没有出具相应的标准?
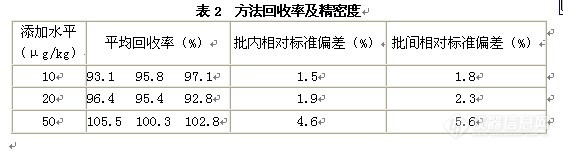
高效液相色谱串联质谱测定猪肉中新型兽药泰拉霉素摘要 建立了猪肉中新型兽药泰拉霉素的液相色谱串联质谱(HPLC-MS/MS)联用确证方法。样品用甲醇+0.1%磷酸(70:30 v/v)提取,离心后用PCX固相萃取小柱净化,Symmetry®C8色谱柱分离,串联质谱多反应监测(MRM)模式下分析,内标法定量。方法的线性范围为10~500μg/kg,检出限为5.0μg/kg,在10、20和50μg/kg 3个浓度水平进行添加实验,平均回收率为93.1%~105.5%,批内相对标准偏差为1.5%~4.6%,批间相对标准偏差为1.8 %~5.6%。 关键词 高效液相色谱串联质谱,猪肉,泰拉霉素,残留 泰拉霉素(Tulathromycin)是一种新近上市且为动物专用的大环内酯类半合成抗生素,分子式为C41H79N3O12(分子量806)。我国农业部在2008年第957号公告中首次允许泰拉霉素在动物生产中使用。泰拉霉素主要用于放线杆菌、支原体、巴氏杆菌、副嗜血杆菌引起的猪、牛的呼吸系统疾病。具有用量少、一次给药、低残留和动物专用等众多优点。在我国,大环内酯类药物现行使用较为广泛的是泰乐菌素和替米考星,虽然这2种药物在生产中都取得了良好的效果,但随着使用时间的延长,我国很多地区出现了不同程度的耐药性,导致用量不断增大,但治疗效果却在逐步降低 ,而泰拉霉素药效均强于泰乐菌素和替米考星等市场广泛使用的大环内酯类药物。因此,泰拉霉素在畜禽生产中使用前景非常广阔,其残留分析方法研究也显得尤为必要。 目前国内外大环内酯类药物残留检测方法已有ELISA筛选法、薄层色谱法、气质联用法、高效液相色谱和高效液相色谱串联质谱法等众多方法。但泰拉霉素的残留检测方面研究较少,国内尚未见有相关报道。本研究建立了以罗红霉素(C41H76N2O15, 837)为内标的泰拉霉素HPLC-MS/MS方法,方法的定量限为10μg/kg,可以满足国内外有关法规对其残留检测的要求,可为残留监控提供技术支持。1 材料与方法1.1仪器与材料 Waters 2695 Quattro MicroTM API高效液相色谱串联质谱仪(美国Waters公司);固相萃取仪(美国SUPELCO公司)。 泰拉霉素标准品(Sigma公司);罗红霉素 (Sigma公司);乙腈(色谱纯);甲醇(色谱纯);其余试剂均为分析纯试剂。PCX固相萃取柱(3 mL, 60 mg,AGELA公司)。1.2标准溶液的配制 泰拉霉素和内标储备液:分别准确称取10mg标准品于10mL容量瓶中,用0.05M K2HPO4/乙腈(75:25,v/v; pH6)定容,混匀后置冰箱冷藏储存,有效期3个月。 标准工作溶液的配制:吸取储备液1mL于100mL容量瓶中并用0.05M K2HPO4/乙腈(75:25,v/v; pH6)定容,再从中吸取1mL于50mL容量瓶中得0.2mg/L,用于添加(置冰箱冷藏储存,有效期2个月)。内标物罗红霉素的添加溶液配制方法同泰拉霉素。1.3样品制备 称取2.0g组织样品于50mL的聚四氟乙烯塑料管中,加入10mL甲醇+0.1%磷酸提取液(70:30 v/v)和50μL内标工作液,匀质1 min后5000rpm转速离心2min,收集上清液过已经分别用3mL甲醇和3mL提取液润洗过的60mg/3mL的PCX小柱,待全部过柱后,再用3mL水、3mL甲醇淋洗,最后用4mL4%的氨化甲醇洗脱,50℃水浴氮气吹干后用1mL流动相定容,进行HPLC-MS/MS分析。1.4仪器分析条件 液相色谱条件:Symmetry®C8 5μm 3.9mmx20mm,流动相:乙腈:0.1%甲酸水溶液=70:30;柱温:30℃,进样室温度15℃,进样量:10μL,流速为0.3mL/min。质谱条件:电离模式:ESI(+);检测方式:多级反应检测(MRM);毛细管电压:4.2 KV;锥孔电压:20 V;RF透镜电压:0.2 V;离子源温度:110 ℃;脱溶剂气温度:350 ℃;锥孔气流速:100 L/h;脱溶剂气流速:600 L/h;二级碰撞气:氩气。1.5添加回收率实验 添加回收率实验添加浓度为10、20和50µg/kg,实验步骤同1.3。2 结果与讨论2.1样品的提取和净化 泰拉霉素含3个氨基基团,为弱碱性化合物,易溶于酸性溶液和极性溶剂中,在pH 6~8的水溶液中较稳定,在pH 9条件下均不稳定。对泰拉霉素残留的提取和净化方法的设计主要依据其弱碱性、脂溶性和酸碱不稳定性。本实验比较了4种提取液:乙腈-0.1%偏磷酸(70:30 v/v)、甲醇-0.1%偏磷酸(70:30 v/v)、乙腈-0.1%磷酸(70:30 v/v)、甲醇-0.1%磷酸(70:30 v/v)对泰拉霉素的提取效果。结果表明,甲醇-0.1%磷酸(70:30)提取液对泰拉霉素的提取效果较好,回收率高于其他提取液,故本方法选择甲醇-0.1%磷酸(70:30)为样品提取液。 液-液分配(LLE)和固相萃取(SPE)是大环内酯类药物的2种主要净化手段,其中LLE操作较麻烦且回收率较低、重复性较差,所以本研究采用固相萃取技术净化,并比较了3种SPE净化小柱:SUPELCO C18小柱、Oasis MCX小柱和PCX小柱, 由于Oasis MCX小柱和PCX小柱兼有阳离子交换和疏水作用机制,基于泰拉霉素的弱碱性和脂溶性的理化特性,Oasis MCX小柱和PCX小柱的净化效果好于C18小柱。Oasis MCX小柱和PCX小柱提取效率没有明显差别,但PCX小柱成本低,所以,本方法净化选择PCX小柱。2.2色谱质谱条件的优化[fo
如题克拉克-鲁布斯缓冲溶液的配制方法
如题:求助克拉克-鲁布斯缓冲液配制(pH=1~5),谢谢
克拉克—鲁布斯(Clark—lubs)缓冲溶液的配制 2.1.1 pH7.0的克拉克—鲁布斯(Clark—lubs)缓冲液的配制 分别量取1.0mL氢氧化钠(0.1mol/L)溶液,12.5mL硼酸(0.4mol/L)溶液,12.5mL氯化钾(0.4mol/L)溶液,注入100mL容量瓶,稀释至刻度。2.1.2 pH9.0的克拉克—鲁布斯(Clark—lubs)缓冲液 分别量取20.8mL氢氧化钠(0.1mol/L)溶液,12.5mL硼酸(0.4mol/L)溶液,12.5mL氯化钾(0.4mol/L)溶液,注入100mL容量瓶,稀释至刻度。2.1.3 pH10.0的克拉克—鲁布斯(Clark—lubs)缓冲液 分别量取43.7mL氢氧化钠(0.1mol/L)溶液,12.5mL硼酸(0.4mol/L)溶液,12.5mL氯化钾(0.4mol/L)溶液,注入100mL容量瓶,稀释至刻度。







 我要推广仪器
我要推广仪器
 下载APP
下载APP