由于日本现增加对食品中呋喃硫威和禾草丹检测,那位大侠有方法共享一下,谢谢![em24]
各位大虾:经我们调查发现,以上三种农药在茶叶、花草中给滥用,现在面对日本肯定列表制度,需要检测茶叶中的苄嘧磺隆(Bensulfuron-methyl)、禾草丹(Thiobencarb)、阿维菌素(Abamectin)。哪位大虾有相应的检验方法,请提供。最好有[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]的方法,我们目前只有[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url](已经准备购入液相)。另外,禾草丹(Thiobencarb)、阿维菌素(Abamectin)的标准品在哪里可以买到?
有同时测吡氟禾草灵和精吡氟禾草灵的吗?我们当时测的有点混了,如果有用MS测的请提供他们分别的离子,谢谢!吡氟禾草灵和精吡氟禾草灵的英文名称分别是fluazifop和fluazifop-p-butyl.
厦门欣椿食品有限公司出口日本的葱中吡氟禾草灵(fluazifop-butyl)检出0.2ppm(基准值0.01ppm)超标,中国出口日本的葱检查频度提高到30%抽检
在用5009.134的标准做禾草敌标准中说禾草敌响应和含硫浓度的平方成正比,想请问的是,在做校正曲线的时候,应该选择的校正曲线类型是哪个呢?

10,抽取5个版友);中奖名单:dyd3183621(注册ID:dyd3183621)馨语(注册ID:huangdm)莫名其妙(注册ID:moyueqiu)20071940xu(注册ID:20071940xu)捌道巴拉巴巴巴(注册ID:v3082413)http://ng1.17img.cn/bbsfiles/images/2017/03/201703151507_01_1610895_3.jpghttp://ng1.17img.cn/bbsfiles/images/2017/03/201703151507_02_1610895_3.jpg【注意事项】同样的答案,每人只能发一次PS:该贴浏览权限为“回贴仅作者和自己可见”,回复的版友仅能看到版主的题目及自己的回答内容,无法看到其他版友的回复内容。下午3点之后解除,即可看到正确答案、获奖情况及所有版友的回复内容。=======================================================================精恶唑禾草灵水乳剂方法:HPLC基质:药品应用编号:103035化合物:恶唑禾草灵 解草唑固定相:Platisil Silica色谱柱/前处理小柱:Platisil Silica 5u 250 x 4.6 mm样品前处理:试样溶液:称取含恶唑禾草灵0.05 g的试样,置于100 mL容量瓶中,加入80 mL流动相,在超声波中振荡5 min使其溶解,取出冷却至室温后,用流动相稀释至刻度,摇匀,过滤。色谱条件:流动相:正己烷:异丙醇=90:10(V/V) 流速:0.8 mL/min 柱温:30 ℃ 检测器:UV 230 nm 进样量:10 μL文章出处:天津应用实验室关键字:恶唑禾草灵,解草唑谱图:http://www.dikma.com.cn/Public/Uploads/images/12(30).PNG图例:1-恶唑禾草灵 2-解草唑
哪位老师测过禾草灵和吡氟禾草灵,按照国标法GB23200.8和GB23200.121,都测不出来。有什么需要注意的吗?谢谢
吡氟禾草灵相关的化合物一共有如下几种:1. Fluazifop2. Fluazifop-P3. Fluazifop-butyl4. Fluazifop-P-butyl5. Fluazifop-methyl其中1和2,3和4是同分异构体,3、4是1、2的丁基酯想请教大家,知道以上五种物质对应的中文名嘛?谢谢!实际检测过程中是检测其中哪几种物质?我看日本厚生省的关于吡氟禾草灵的检测方法分析的目标化合物是“吡氟禾草灵酸、吡氟禾草灵、精吡氟禾草灵酸、精吡氟禾草灵”四种物质,这四种物质分别对应的英文名称是?
麻烦问一下,有没有做5009.142的标准,吡氟禾草灵标准品怎么衍生,标准里面样品衍生了,没有说标准品的衍生过程
gc-2010检测禾草敌和稻瘟灵该用FPD还是ECD,我用FPD进样口260℃ 柱温150℃保留2分钟以每分钟10℃升到250℃保留6分钟,检测器温度260℃,但是小点0.05 0.1 0.2ug/ml的点不出峰,5ug/ml出峰。换ECD进样口260℃ 柱温150℃保留2分钟以每分钟6℃升到270℃保留6分钟,检测器温度300℃,稻瘟灵出峰,禾草敌不出峰。想问下我哪地方有问题,有哪位老师做过这两个么,指点一下,谢谢!
同行们:好,最近有朋友做精吡氟禾草灵吗,为什么出峰时间和影响值都不稳定,其图如下:图1中:4.5-5min中和9min提取离子都是精吡氟禾草灵,图2在5--5.5min和8.5-9.5min都是精吡氟禾草灵图3:精吡氟禾草灵的峰有没有出峰有人做过这种物质吗,这种物质是由对映体,S和R,但精吡氟禾草灵应该都是R型,所以我觉得也不应该出现这种问题呀,不知道是不是水解的问题,因为这是一种酯类化合物,但我觉得是这个问题也不应该忽高忽低!下周再试一下!朋友们有什么高见,敬请发表意见!
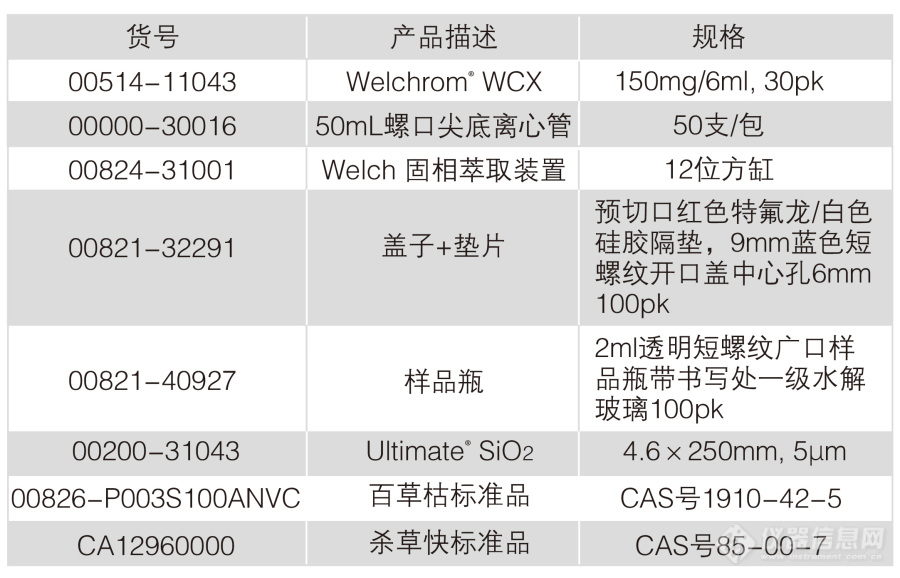
[align=center][b]剧毒除草剂“百草枯”和“杀草快”在水质中的检测(HJ914-2017)[/b][/align][b]百草枯。一个让人看了就心生恐惧的名字。[/b][align=center][img=,287,339]http://ng1.17img.cn/bbsfiles/images/2018/08/201808161550271107_510_932_3.png!w287x339.jpg[/img][/align]药如其名。百草枯是一种快速灭生性除草剂,具有触杀作用和一定内吸作用。能迅速被植物绿色组织吸收,使其枯死。[b]百草枯对人毒性极大,且无特效解毒药,口服中毒死亡率极高。[/b]目前已被[b]20[/b]多个国家禁止或者严格限制使用。我国在2016年7月1日停止水剂在国内销售和使用。[b]2016年,也就是两年前。[/b]那么,已经生产或使用的百草枯,是否还存在我们的水源中?[align=left]月旭科技推出“水质 百草枯和杀草快的检测”方案,可用于地表水、地下水和废水中的百草枯和杀草快的检测。[/align][b]适用范围[/b]适用于地表水,地下水和废水中百草枯和杀草快的测定。参考标准:《HJ914-2017水质 百草枯和杀草快的测定 固相萃取-高效液相色谱法》[b]溶液的配置[/b]标准储备液:百草枯(CAS:1910-42-5)市售100mg/L 1mL。杀草快:精确称取1mg,用甲醇溶解并定容到10mL,浓度为100mg/L。标准使用液(10mg/L):分别移取1mL 100mg/L百草枯和杀草快,用甲醇定容到10mL。硫酸溶液(1+1):量取50mL硫酸,缓慢加入50mL水中。含2%甲酸的乙腈溶液:量取2mL的甲酸,加入乙腈定容至100mL。甲酸铵溶液:称取12.6g甲酸铵溶于水中,加入20mL甲酸,用水定容至1000mL。氢氧化钠溶液:称取40g氢氧化钠,用水溶解并定容到100mL。[b]提取步骤[/b]水(或废水)样抽滤过0.45μm尼龙滤膜,于采样瓶中;移取200mL水(或废水)样于样品瓶中,用硫酸溶液或氢氧化钠溶液调节水样pH6-7,待净化。[b]SPE净化步骤[/b]SPE柱:月旭Welchrom WCX规格:150mg/6mL活化:5mL甲醇,5mL水,弃去上样:待净化液全部上样,弃去淋洗:4mL水(润洗样品瓶),弃去洗脱:15mL 2%甲酸乙腈溶液,收集于50mL离心管中,抽干氮吹至剩余1mL,用2%甲酸乙腈溶液定容至2mL,并过0.45μm滤膜,上HPLC检测。[b]色谱条件[/b]色谱柱:月旭Ultimate SiO2规格:4.6×250mm,5μm流动相:甲酸铵溶液:乙腈(60:40)流速:1.0mL/min柱温:40℃进样量:20μL检测波长:百草枯定量波长为257nm,辅助波长为290nm杀草快定量波长为309nm,辅助波长为290nm[b]色谱图或者加标回收率结果[/b][align=center][img=,690,242]http://ng1.17img.cn/bbsfiles/images/2018/08/201808161550467989_3820_932_3.jpg!w690x242.jpg[/img][/align][align=center]图1 杀草快对照0.5mg/L图谱[/align][align=center][img=,690,217]http://ng1.17img.cn/bbsfiles/images/2018/08/201808161550593099_7222_932_3.jpg!w690x217.jpg[/img][/align][align=center]图2 百草枯对照0.5mg/L图谱[/align][align=center][img=,690,217]http://ng1.17img.cn/bbsfiles/images/2018/08/201808161551125835_779_932_3.jpg!w690x217.jpg[/img][/align][align=center]图3 自来水加标5μg/L杀草快图谱[/align][align=center][img=,690,217]http://ng1.17img.cn/bbsfiles/images/2018/08/201808161551260796_5767_932_3.jpg!w690x217.jpg[/img][/align][align=center]图4 自来水加标5μg/L百草枯图谱[/align][align=center][img=,690,217]http://ng1.17img.cn/bbsfiles/images/2018/08/201808161551387188_9193_932_3.jpg!w690x217.jpg[/img][/align][align=center]图5 废水加标5μg/L杀草快图谱[/align][align=center][img=,690,217]http://ng1.17img.cn/bbsfiles/images/2018/08/201808161551554665_8402_932_3.jpg!w690x217.jpg[/img][/align][align=center]图6 废水加标5μg/L百草枯图谱[/align][align=center][/align][align=center][img=,520,306]http://ng1.17img.cn/bbsfiles/images/2018/08/201808161552140846_6979_932_3.jpg!w520x306.jpg[/img][/align][align=center]表1:加标回收率[/align][b]相关产品信息[/b][align=center][img=,690,442]http://ng1.17img.cn/bbsfiles/images/2018/08/201808161510524149_2688_932_3.jpg!w690x442.jpg[/img][/align]
[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质[/color][/url]测禾草灵不出峰为什么
大家好,有没有检测吡氟禾草灵的,在检测的时候有那些经验技巧!谢谢!
请问谁做过吡氟禾草灵?5009.142的方法简直是太变态了,哪位大侠指点一下?
向各位老师请教精恶唑禾草灵(骠马)的[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]测定方法,主要是如何提取?非常感谢![em31]
现有84.8%的乳氟禾草灵的药品,想要做薄层层析,但苦于没有找到合适的展开剂,请那位高手给指点一二,能提供三氟羧草醚的也可以,小弟不胜感激!
[em61] [em61] [em61] 我公司分析乳氟禾草灵原药,流动相比例为甲醇:乙腈:水:磷酸=30:40:30:0.2 ,样品用甲醇溶解,峰型还可以,我担心样品用甲醇溶解,分析结果会不会有误差,是否必须改为用乙腈溶?请高手们指点。。。。。。
[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=174581]GB 22618-2008 精噁唑禾草灵乳油[/url]
如何从土壤及植株中提取精恶唑禾草灵母体及衍生物?
如何从土壤及植株中提取精恶唑禾草灵母体及衍生物?
有没有人做过3-氨基-1,2,4-三氮唑(ATA、杀草强)液相检测的呀!!本人求助此物质的检测方法……………………谢谢分享
有网友问:最近我公司在检测烟草香精样品中的甲醇,所以想在您那边看看有没有类似的文章,以及遇到问题的解决方法。大家看看有什么好办法。
【生活中的仪器分析】样 品:蔬菜、水果、茶叶、茶粉等食品检测项目:草甘膦、草铵膦、氨甲基膦酸参考标准:SN/T 1923-2007检测仪器:a.WATERS液相色谱串联质谱仪:配有电喷雾(ESI)离子源(可用其他品牌作用等效的高效液相色谱质谱仪替代)b.Biotagevacmaster固相萃取仪c.IPRE Qclean PMG草甘膦专用固相萃取柱d.BiotageTurbovap LV 快速浓缩仪e.IKA MS3 涡旋混匀器g.TOMY-MX307离心机g.昆山超声波清洗器实验过程:1.提取及预处理称取2-5g(精确到0.001g)试样于50 mL聚丙烯离心管中,加入100μL内标液,加入20.0 mL水超声提取30min,于10000 r/min离心5min,取1.0 mL上清液于2mL子弹头离心管中,加入100μL酸度调节剂(注A),涡旋混匀,15000r/min离心5 min,待净化。注A:酸度调节剂配制方法:纯水+色谱纯甲醇+盐酸=160+40+13.4(V/V/V)2.固相萃取净化І将PMG-І柱(蓝柱)用2mL甲醇和2 mL 0.5%甲酸淋洗活化并自然滴干,将加入酸度调节剂处理的提取液(2)转移到小柱上,用5mL刻度试管收集流出液(1-2滴/秒),用1.0mL 0.5%甲酸洗柱并真空抽干,合并流出液,用移液枪吸取50%NaOH调pH7-9(用1-14pH试纸,根据样品不同约20-50μL),加水定容到3 mL刻度,混匀,待衍生。3.衍生步骤准确吸取600 μL净化液(3)于2mL子弹头离心管中,加入200 μL 5%硼砂溶液,边涡旋,边加入200 μL 25g/L FMOC-Cl乙腈溶液(注B),放置10min,加入50 μL甲酸,涡旋混匀,15000 r/min离心5min,吸取上清液准备过PMG-ІІ柱。注B:25 g/L FMOC-Cl乙腈溶液配制方法:称取0.25 gFMOC-Cl,溶解于10 mL色谱纯乙腈中。4.固相萃取净化ІІ 将PMG-ІІ柱(红柱)用2 mL甲醇和2 mL 0.5%甲酸淋洗并自然滴干,将上清液过PMG-ІІ柱,用3 mL水淋洗小柱,真空抽干5-10 min,再加入2 mL正己烷淋洗小柱,滴干后真空抽干5 min,最后用5 mL 5%氨水/甲醇洗脱小柱(1-2 mL/min)并用5 mL刻度试管收集流出液,45℃,氮气吹至近干,用20%乙腈定容1.0 mL,涡旋混匀,过0.2 μm PTFE膜后上机测试。5.测定5.1色谱条件a.色谱柱:Waters BEH-C18,1.7 μm,2.1 mm×100 mm;b.流动相:5mmol/L乙酸铵:乙腈梯度洗脱,梯度表见表1; 表1 流动相及梯度 时间(min)流速(mL/min)5mmol/L乙酸铵(%)乙腈(%)00.3901020.362384.40.362384.50.35956.50.35956.60.390109.00.39010c.检测器:串联四极杆质谱仪;d.柱温:35℃;e.进样量:10 μL。5.2质谱分析条件a)电离源:电喷雾正离子模式;b)毛细管电压:3.50KV;c)源温度:120℃;d)脱溶剂气温度:400℃;e)脱溶剂气流量:700L/h;f)碰撞室压力:2.7í10-3mbar;g)特征离子及参数见表2。 表 2 草甘膦和氨甲基膦酸的主要特征离子 化合物保留时间(min)母离子+(m/z)锥孔电压(V)子离子(m/z)碰撞能量(eV)草甘膦1.32392.215*88.02515214.01
顶空毛细管[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]法测定甘草酸单铵盐中乙醇残留量,用的是安捷伦7820A,现在有HP-5 DB-1301 DB-1三个柱子,请教一下应该用哪个柱子做乙醇残留啊
我尝试了一下,5ppm的沙蚕毒素草酸盐(溶于色谱纯甲醇)竟然不出峰.我的条件如下色谱柱:毛细管柱DB-1701、长30m、内径0.50μm、外径0.539mm温度:进样口220℃;检测器FPD300℃;柱温100-280程温(分二阶):一阶:初始温度100℃,初始时间2min,速率20℃/min,终止温度220℃,终止时间5min。二阶:初始温度220℃,初始时间0min,速率30℃/min,终止温度280℃,终止时间2min。气流:N2(100kpa)、空气(80kpa)、H2 (75kpa)我在网上查到的行业标准方法如下5.2 [url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]条件: 5.2.1 色谱柱:3mm(内径)×2m玻璃柱; 填装涂有1.5%OV-17Chromosorb W(80~100目)的担体。 5.2.2 温度 柱温:160℃; 汽化室温:200℃; FPD检测器温:170℃。 5.2.3 气体速度 载气(氮气)流速:70mL/min; 氢气流速:150mL/min; 空气流速:50mL/min。其中,沙蚕毒素草酸盐以甲醇溶解. 大家有什么建议吗?先谢啦
皮革中偶氮测定大家如何配置20%氢氧化钠甲醇溶液。
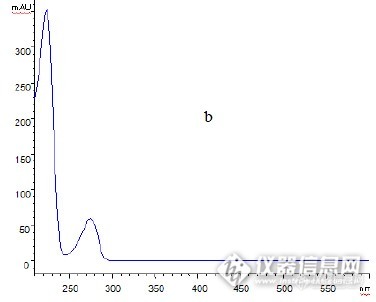
HPLC-DAD分析酸浆中木犀草素及木犀草素-7-β-D-葡萄糖甙成分酸浆(拉丁文名:Physali alkekengi L.)又名红菇娘、挂金灯、戈力、灯笼草、灯笼果、洛神珠、泡泡草、鬼灯等北方称为菇蔫儿、姑娘儿,以果实供食用。化学成分含酸浆苦素A(Physalin A)、酸浆苦素B、酸浆苦素C、木犀草素(Luteolin)及木犀草素-7-β-D-葡萄糖甙。果实含枸橼酸、草酸、维生素C、酸浆红色素(physalien)、酸浆醇(physanol)A,B。花萼含α胡萝卜素、酸浆黄质(physoxanthin)及叶黄素等,种子油的不皂化物中分得多种4α-甲基甾醇,主要为禾本甾醇(gramisterol)和钝叶醇(obtusifoliol)及4种新甾体。此外尚含多种4-脱甲基甾醇,如胆甾醇和24-乙基胆甾醇等。还含有多种三萜3β-一元醇,其中环木菠萝烷醇(cycloartanol)35%,环木菠萝烯醇(cycloartenol)27%、羊毛脂-8-烯-3β-醇(lanost-8-en-3β-ol)。木犀草素(luteolin)是一种天然黄酮类化合物,存在于多种植物中,具有抗炎、抗肿瘤、抗过敏等方面的作用。化学是如下:http://ng1.17img.cn/bbsfiles/images/2016/08/201608311303_607620_2217446_3.jpg目前,国内传统中药有效成分的提取方法普遍存在提取率低、杂质清除率不高、生产周期过长、能耗高、溶剂用量大等缺点。随着中药现代化进程的不断深入,许多现代高新技术不断地被应用到中药有效成分的提取和分离,使得中药有效成分的提取更高效和简便。超声-微波协同萃取技术直接将超声振动与开放式微波两种作用方式相结合,充分利用超声波振动的空化作用以及微波的高能作用,实现了低温常压条件环境下,对固体样品进行快速、高效、可靠的预处理,与常规提取方法相比,超声-微波协同萃取技术具有快速、节能、节省溶剂、污染小等优点。本实验应用超声-微波协同萃取法提取酸浆中的木犀草素及木犀草素-7-β-D-葡萄糖甙,采用高效液相-二极管阵列检测法(HPLC-DAD)测定提取物中木犀草素及木犀草素-7-β-D-葡萄糖甙的含量,药材中二者成分的含量分别为:1.200mg/g 和0.43mg/g,二个峰,木犀草素-7-β-D-葡萄糖甙峰位置分别为:221nm,270nm,木犀草素峰位置分别为:226nm,276nm,由于木犀草素-7-β-D-葡萄糖甙比木犀草素多了一个 β-D-吡喃葡萄糖基团,天麻素二个峰位置都发生了蓝移,样品中二个峰的光谱图与标准品二个峰的光谱图相同,可以进一步确定酸浆中含有木犀草素及木犀草素-7-β-D-葡萄糖甙。主要仪器与试剂主要仪器Agilent1100型四元梯度高效液相色谱仪(美国 Agilent 公司)Agilent TC-C18(ODS)色谱柱(5μm,4.6×250mm,美国 Agilent 公司)CW-2000 超声-微波协同萃取仪(新拓微波溶样测试技术有限公司)DJ-10A 型倾倒式粉碎机(上海隆拓仪器设备有限公司)RE-52AA 型旋转蒸发仪(河南巩义仪器厂)LXJ-IIB 型低速大容量多管离心机(上海安亭科学仪器厂)试剂木犀草素(中检所,含量98%;)木犀草素-7-β-D-葡萄糖甙(中检所,含量98%;)酸浆全草(采于黑龙江)除甲醇、乙腈为色谱纯(国药集团化学试剂有限公司),其余试剂除专门提到外,均为分析醇,实验用水为二次蒸馏水。实验方法供试品溶液的制备 精密称取酸浆粉末1.0g,置于超声-微波萃取仪玻璃容器中,加入50mL70%甲醇,开启超声微波,控制在恒温50℃下提取40min,萃取3次,合并提取液,浓缩至近干,残渣加入甲醇溶解,转移至10mL 量瓶中,加甲醇稀释至刻度,摇匀,过0.45μm 的微孔滤膜,取续滤液,即得。提取条件的考察溶剂的选择:精密称取酸浆粉末1.0g,置于超声-微波萃取仪玻璃容器中,分别用水、70%甲醇、70%乙醇溶液超声-微波协同萃取40min(n=3),萃取3次,合并提取液,浓缩至近干,残渣加入甲醇溶解,转移至10mL 量瓶中,加甲醇稀释至刻度,摇匀,过0.45μm的微孔滤膜,取续滤液,HPLC 测定萃取率。溶剂体积分数的选择:分别用体积分数为40%、50%、60%、70%、80%、90%和纯甲醇溶液超声-微波协同萃取30min(n=3),方法同上。溶剂用量的选择:分别用10mL、20mL、50mL、80mL、100mL70%甲醇提取,方法同上。提取时间的选择:分别用70%甲醇超声-微波协同萃取20min、30min、40min、50min、60min(n=3),方法同上。提取温度的选择:分别在40、45、50、55、60℃下用70%甲醇超声-微波协同萃取40min,方法同上。对照品溶液的制备 分别精密称取常温减压干燥12h 的木犀草素及木犀草素-7-β-D-葡萄糖甙对照品适量,加甲醇配制成木犀草素-7-β-D-葡萄糖甙为200μg/mL、木犀草素为100μg/mL 的混合对照品溶液,冷藏备用。色谱条件 色谱柱:Agilent TC-C18柱(5μm,4.6×250mm);流动相:A-0.1%乙酸水溶液;B-甲醇,线性梯度洗脱:0~30 min,3%~5% B;30~35 min,5%~20%B;35~40min,20%~20%B;检测波长:270nm;流速:1mL/min;柱温:30℃;进样量:20μL。结果与讨论提取条件的优化结果溶剂的优化结果:分别用水、70%甲醇、70%乙醇溶液超声-微波协同萃取30min(n=3),结果表明70%甲醇提取木犀草素-7-β-D-葡萄糖甙的量较高,而木犀草素的量差异不明显,因此选择70%甲醇提取。溶剂体积分数的优化结果:分别用体积分数为40%、50%、60%、70%、80%、90%和纯甲醇溶液超声-微波协同萃取30min(n=3),结果表明,在甲醇体积分数70%时,木犀草素-7-β-D-葡萄糖甙和木犀草素的提取率随着甲醇浓度的增加而增加;但当甲醇体积分数在70%以上时,木犀草素葡萄糖甙的提取率呈现下降趋势,木犀草素没有明显的变化。木犀草素葡萄糖甙属于一种苷,分子量小,极性较大,当甲醇体积分数过高时,溶液极性降低,使得极性较强的木犀草素葡萄糖甙不易溶出,而木犀草素极性相对木犀草素葡萄糖甙小,影响不明显,因此实验选择70%甲醇作为提取溶剂。溶剂用量的优化结果:分别用10mL、20mL、50mL、80mL、100mL70%甲醇提取,结果表明溶剂体积在50mL时木犀草素葡萄糖甙和木犀草素的提取率最高,之后随着溶剂用量的增加,木犀草素葡萄糖甙和木犀草素的提取率趋于稳定,因此溶剂用量选用50mL 进行提取 。提取时间的优化结果:分别用70%甲醇超声-微波协同萃取20min、30min、40min、50min、60min(n=3),结果表明超声-微波协同萃取时间从20~40min的过程中木犀草素葡萄糖甙和木犀草素的提取率逐渐增加;而提取时间超过40min之后,提取率反而逐渐下降。超声-微波协同萃取时间太长,植物中大量细胞细胞破碎,使得大量粘性物质等进入提取液,溶剂杂质增多、粘度增大,影响了有效成分的溶出,有效成分含量反而减少,因此选择提取时间为40min。提取温度的优化结果:分别在40、45、50、55、60℃下用70%甲醇超声-微波协同萃取40min,实验表明,提取温度在50~60℃的范围内,木犀草素葡萄糖甙和木犀草素的提取率没有明显差异,考虑到温度太高容易破坏活性成分,因此选择提取温度为50℃。流动相的考察在实验过程中,流动相首先考察了甲醇-水、乙腈-水等度洗脱对酸浆超声-微波协同萃取样品溶液进行分离,乙腈-水作为流动相时,出峰较快,不能较好地把木犀草素葡萄糖甙和木犀草素与其他杂质成分分离;甲醇-水作为流动相时,出现峰形拖尾现象,分离效果不理想。为改善上述现象,改用0.1%乙酸代替水并采用梯度洗脱,经过反复筛选之后,最终确定流动相组成为 A -0.1%乙酸水溶液, B -甲醇,洗脱程序为0~30 min , 3%~5% B;30~35 min ,5%~20% B ;35~40 min 20%~3% B,木犀草素葡萄糖甙和木犀草素和其他杂质成分能够很好的分离,得到较理想的色谱图。对照品溶液和酸浆萃取样品的HPLC-DAD 分析下图分别显示了在上述的色谱条件下,采用 DAD 进行检测得到的两种混合对照品及酸浆萃取样品的 HPLC 分离色谱图。图1色谱图中木犀草素葡萄糖甙和木犀草素的保留时间分别为18.74min, 26.87min,根据保留时间判断,图2中的 a、b 色谱峰分别初步鉴定为木犀草素葡萄糖甙和木犀草素。图3、4分别显示了混合对照品和酸浆萃取物中保留时间18.74min, 26.87min 的色谱峰进行 DAD 检测后得到的光谱图,木犀草素葡萄糖甙和木犀草素 UV 光谱图形状相似,出现 二个峰,木犀草素葡萄糖甙峰位置分别为:221nm,270nm,木犀草素峰位置分别为:226nm,276nm,由于木犀草素葡萄糖甙比木犀草素多了一个 β-D-吡喃葡萄糖基团,木犀草素葡萄糖甙二个峰位置都发生了蓝移,样品中二个峰的光谱图与
[align=center][/align][font=宋体]一、原理[/font][font=宋体]1.[/font][font=宋体]多糖的分级与纯化[/font][font=宋体]多糖纯化的实质是将粗多糖中的杂质(包括蛋白质、色素、低聚糖、无机盐等非糖物质)除去而获得分子量和极性均一的多糖组分。比较常用的方法有:醇沉法、超滤法和柱层析法等。其中柱层析法应用比较普遍,多用于样品的精细分级,具有操作简单,重现性好的优点,但样品上样量小。用于多糖分离的柱层析主要有两类:一类是通过分子量大小进行分级的凝胶柱层析,如Sephadex G-100(200、50、25、10)、SepharoseCL-6B等系列;另一类是离子交换层析,是根据多糖所带电荷的性质不同选择相应的离子交换柱对多糖进行分级。如待分离的多糖带有负电荷,可选择阴离子型的DEAE-纤维素柱或DEAE-Sepharose柱进行分级,以获得均一的多糖组分。[/font][font=宋体]通过热水煮提方法提取的多糖,通常是混合物,且分子量不均一,其单糖组成、分子结构和聚合度往往不同,可通过柱层析的方法,对其进行分级以达纯化的目的。本实验中,采用[/font]DEAE-[font=宋体]纤维素柱层析的方法对中草药中水溶性粗多糖进行分级纯化,分离纯化出各级分多糖,[/font][font=宋体]为后续研究多糖的结构特征和生物活性奠定基础。[/font][font=宋体]2.[/font][font=宋体]苯酚—硫酸法[/font][font=宋体]游离的寡糖、多糖中的己糖或糖醛酸在浓硫酸的作用下,脱水生成糠醛,再与苯酚作用起显色反应,在一定范围内其颜色深浅与糖的含量成正比,吸收值与糖含量呈线性关系。且己糖在[/font]490 nm[font=宋体]处(戊糖及糖醛酸在[/font]480 nm[font=宋体])有最大吸收,故用比色法测定多糖含量。该法简单,快速,灵敏,且颜色持久,同一台设备同一光源仅需制作一条标准曲线。[/font][font=宋体] [/font][font=宋体]二、步骤[/font]1.[font=宋体]中草药水溶粗多糖的提取流程[/font][font=宋体]中草药粉末[/font][font=Symbol][/font][font=宋体]蒸馏水浸泡过夜[/font][font=Symbol][/font][font=宋体]超声[/font]/[font=宋体]微波[/font][font=Symbol][/font][font=宋体]水浴浸提[/font][font=Symbol][/font][font=宋体]过滤去除药渣[/font][font=Symbol][/font][font=宋体]定容[/font][font=Symbol][/font][font=宋体]多糖含量测定[/font][font=宋体]加等体积[/font]95%[font=宋体]乙醇[/font][font=Symbol][/font]4[font=宋体]℃冰箱过夜,醇沉多糖[/font][font=Symbol][/font]5000r/min[font=宋体]离心去上清液[/font][font=Symbol][/font][font=宋体]粗多糖[/font][font=Symbol][/font][font=宋体]脱蛋白[/font][font=Symbol][/font][font=宋体]脱色素[/font][font=Symbol][/font][font=宋体]透析[/font][font=Symbol][/font][font=宋体]浓缩[/font][font=Symbol][/font][font=宋体]冷冻干燥得水溶多糖[/font][font=Symbol][/font]DEAE-[font=宋体]纤维素色谱柱[/font][font=Symbol][/font][font=宋体]收集多糖组分[/font][font=Symbol][/font][font=宋体]测定每[/font]50s[font=宋体]洗脱液的[/font]OD[font=宋体]值[/font][font=Symbol][/font][font=宋体]绘制横坐标为管数与纵坐标为[/font]OD[font=宋体]值的洗脱曲线[/font][font=宋体]根据洗脱曲线收集多糖溶液[/font][font=Symbol][/font]4[font=宋体]℃无水乙醇醇沉过夜[/font][font=Symbol][/font][font=宋体]离心[/font][font=Symbol][/font][font=宋体]多糖沉淀[/font][font=Symbol][/font][font=宋体]常规干燥[/font][font=Symbol][/font][font=宋体]纯度鉴定[/font][font=Symbol][/font][font=宋体]纯多糖[/font]2.[font=宋体]脱蛋白[/font][font=宋体]由于粗多糖中含有一定量游离的和结合的蛋白,为保证多糖的纯度,必须将蛋白质脱去。常用的脱蛋白方法有[/font]Sevag[font=宋体]法、三氯乙酸法和蛋白酶法(如链蛋白酶、胰蛋白酶、木瓜蛋白酶)等。[/font][font=宋体]([/font]1[font=宋体])[/font]Sevag[font=宋体]法[/font][font=宋体]根据蛋白质在有机溶剂(如氯仿)中易发生变性析出的特点[/font][font=宋体],把多糖配制成[/font]5%[font=宋体]的糖液,然后按照[/font]1:3[font=宋体]的体积比加入[/font]Sevag[font=宋体]试剂(氯仿∶正丁醇[/font]=4[font=宋体]∶[/font]1[font=宋体]),混匀后剧烈振荡,静置分层后吸去水层与溶剂层交界处的变性蛋白质,重复操作多次,直到除尽蛋白质为止。[/font][font=宋体]([/font]2[font=宋体])三氯乙酸法[/font][font=宋体]将粗多糖配成[/font]5%[font=宋体]的糖液,加入[/font]30%[font=宋体]三氯乙酸溶液,使三氯乙酸终浓度为[/font]15%[font=宋体],在[/font]4℃[font=宋体]冰箱中放置过夜,离心弃去沉淀,得无蛋白的多糖溶液,再用[/font]1 mol/L NaOH[font=宋体]溶液中和至[/font]PH[font=宋体]为[/font]7[font=宋体]。[/font][font=宋体]([/font]3[font=宋体])蛋白酶法[/font][font=宋体]将粗多糖配成[/font]5%[font=宋体]的糖液,用酸碱调[/font]pH[font=宋体]至中性,加链蛋白酶[/font]0.5 L[font=宋体],放恒温箱中[/font]37℃[font=宋体]保温[/font]24 h[font=宋体]后,加热升温至[/font]80℃[font=宋体]使酶失活,离心,得无蛋白的多糖溶液。[/font]3.[font=宋体]脱色[/font][font=宋体]([/font]1[font=宋体])[/font] [font=宋体]活性炭法[/font][font=宋体]将脱蛋白后的多糖配成[/font]5%[font=宋体]的糖液,用[/font]NaOH[font=宋体]溶液调[/font]pH[font=宋体]为[/font]4.5[font=宋体],加入活性炭粉末,于[/font]80℃[font=宋体]保温[/font]2 h[font=宋体]后过滤。反复进行[/font]3[font=宋体]次,直到溶液颜色不再降低为止。[/font][font=宋体]([/font]2[font=宋体])过氧化氢法[/font][font=宋体]将脱蛋白后的多糖溶液,用浓氨水调至[/font]pH[font=宋体]为[/font]8.0[font=宋体],逐滴滴加[/font]20%H[sub]2[/sub]O[sub]2[/sub][font=宋体]至溶液为浅黄色,[/font]50℃[font=宋体]水浴,保温[/font]2 h[font=宋体]后过滤。[/font]4.[font=宋体]透析脱盐[/font][font=宋体]将脱蛋白、脱色后多糖配成[/font]5%[font=宋体]溶液,置于透析袋中,先用自来水逆向流水透析[/font]72 h[font=宋体]后,再用蒸馏水透析[/font]24 h[font=宋体],每[/font]4 h[font=宋体]换水一次。[/font]5.[font=宋体]干燥[/font][font=宋体]将脱蛋白、脱色、脱盐后的中草药多糖溶液,水浴[/font]80℃[font=宋体]浓缩至[/font]10 mL[font=宋体],加三倍体积的无水乙醇过夜,离心取沉淀,经无水乙醇、乙醚洗涤后,常规干燥得中草药去蛋白去色素的多糖。[/font]6.[font=宋体]多糖含量测定[/font][font=宋体]采用苯酚[/font]-[font=宋体]硫酸法测定样品中的多糖含量。绘制标准曲线,根据葡萄糖标准曲线和样品吸光值计算其糖含量。[/font][font=宋体]标准曲线的制作:准确称取[/font]105℃[font=宋体]干燥恒重的标准葡萄糖[/font]50 mg[font=宋体]定容于[/font]500 mL[font=宋体]容量瓶中,加水至刻度,用移液管分别吸取[/font]0[font=宋体]、[/font]0.1[font=宋体]、[/font]0.2[font=宋体]、[/font]0.3[font=宋体]、[/font]0.4[font=宋体]、[/font]0.5[font=宋体]、[/font]0.6[font=宋体]、[/font]0.7[font=宋体]、[/font]0.8[font=宋体]、[/font]0.9[font=宋体]、[/font]1.0 mL[font=宋体]转入试管中,各以蒸馏水补至[/font]1.0 mL[font=宋体],每个浓度重复四个平行。然后分别向每支试管中加入[/font]4%[font=宋体]苯酚试剂[/font]1mL[font=宋体]及浓硫酸[/font]2 mL[font=宋体],摇匀,沸水浴中煮沸[/font]10 min[font=宋体],冷却至室温,放置[/font]10 min[font=宋体]后在[/font]λ= 480 nm[font=宋体]处测定吸光值[/font]A[font=宋体]。以吸光值[/font]A[font=宋体]为纵坐标,糖含量[/font]C[font=宋体]为横坐标,得糖的标准曲线。[/font][font=宋体]样品溶液制备:精密称取水溶性粗多糖[/font]5 mg[font=宋体],先加[/font]10 mL[font=宋体]蒸馏水溶解,然后定容至[/font]50 mL[font=宋体]。[/font][font=宋体]样品含量测定:用移液管吸取样品液[/font]1.0 mL[font=宋体],加入[/font]4%[font=宋体]苯酚溶液[/font]1 mL[font=宋体]及浓硫酸[/font]2 mL[font=宋体],按上述方法进行操作,测其吸光值,以标准曲线计算样品糖含量[/font][font=宋体]按下式计算中草药多糖的质量浓度与得率。[/font] [img=,300,41]https://ng1.17img.cn/bbsfiles/images/2021/11/202111021041176368_5305_3528941_3.gif!w300x41.jpg[/img]7.[font=宋体]分级[/font][font=宋体]([/font]1[font=宋体])[/font] DEAE-[font=宋体]纤维素的预处理和装柱[/font][font=宋体]将[/font]DEAE-[font=宋体]纤维素用蒸馏水充分浸泡溶胀后,倾倒除去水分,再用[/font]0.5 M NaOH[font=宋体]溶液浸泡[/font]1 h[font=宋体],用蒸馏水反复洗涤至[/font]PH[font=宋体]等于[/font]7[font=宋体]。再用[/font]0.5 M[font=宋体]的[/font]HCl[font=宋体]溶液浸泡[/font]1 h[font=宋体],用蒸馏水洗至中性,真空清除气泡,备用。[/font][font=宋体]装柱时,先将[/font]DEAE-[font=宋体]纤维素沿着玻璃棒缓慢倒入,以防产生气泡。柱子装好后,依次用[/font]5[font=宋体]倍柱体积的蒸馏水、[/font]3[font=宋体]倍柱体积的[/font]1.0 M NaCl[font=宋体]和[/font]5[font=宋体]倍柱体积的蒸馏水进行平衡,流速设定为[/font]20 cm/ h[font=宋体]。[/font][font=宋体]([/font]2[font=宋体])水溶性粗多糖的离子交换柱层析的线性梯度洗脱[/font][font=宋体]称取[/font]50 mg[font=宋体]水溶性粗多糖,溶于[/font]5 mL[font=宋体]蒸馏水中,在已平衡好的[/font]DEAE-[font=宋体]纤维素离子交换柱([/font]1.5 × 14 cm[font=宋体],[/font]Cl[sup]-[/sup][font=宋体]型)上进行上样,先用[/font]100 mL[font=宋体]蒸馏水做流动相进行洗脱,再用[/font]0~1.0 M NaCl[font=宋体]溶液[/font]300 mL[font=宋体]进行线形梯度洗脱,流速[/font]1.0 mL/min[font=宋体],每个试管收集[/font]3 mL[font=宋体]洗脱液,苯酚—硫酸法测定洗脱液中相对的糖含量分布。[/font]8.[font=宋体]纯度鉴定[/font][font=宋体]([/font]1[font=宋体])高效液相色谱法[/font][font=宋体]采用高效液相色谱仪系统,[/font] 10A[font=宋体]检测器,[/font]CLASS-Vp[font=宋体]工作站,[/font]TSK-gel G-3000PWXL[font=宋体]不锈钢色谱柱([/font]7.8 × 300 mm)[font=宋体],柱温为[/font]40℃[font=宋体]。[/font][font=宋体]取各级多糖样品溶于[/font]0.7 M Na2SO4[font=宋体]溶液中,其多糖终浓度为[/font]2 mg/mL[font=宋体],上样[/font]20 μL[font=宋体],流动相为[/font]0.7 M Na2SO4[font=宋体]溶液,流速为[/font]0.5 mL/min[font=宋体]。[/font][font=宋体]([/font]2[font=宋体])紫外光谱分析[/font][font=宋体]将水溶性粗多糖各级分多糖样品配成[/font]0.1 mg/mL[font=宋体]的溶液,用[/font]752PC[font=宋体]型紫外可见分光光度计于[/font]200-400 nm[font=宋体]处扫描检测。[/font][font=宋体]([/font]3[font=宋体])旋光仪分析[/font][font=宋体]不同的多糖具有不同的比旋度,它们在不同浓度的乙醇中具有不同溶解度,所以,如同一多糖水溶液经不同浓度的乙醇沉淀所得的沉淀物,具有相同比旋度,则证明该多糖为均一组分。[/font][font=宋体]将评估多糖样品溶于水,成为近似半饱和溶液,置于磁力搅拌器上,在搅拌下加乙醇,使溶液中乙醇浓度达到[/font]20%[font=宋体],搅拌片刻,使沉淀完全,离心得沉淀。上清液中再继续滴加乙醇,使溶液中乙醇浓度达[/font]40%[font=宋体],所产生的沉淀再经离心。前后两次沉淀,分别干燥后在相同条件下测定其水溶液的比旋度。[/font][font=宋体]将[/font]10 mL5%[font=宋体]饱和糖液用国产[/font]WXG[font=Symbol]-[/font]6[font=宋体]型自动旋光仪,钠灯光源,以蒸馏水调零点,[/font]1dm[font=宋体]管室温下测定。[/font]
龙胆草,原名:[url=https://baike.so.com/doc/1704429-1802107.html]龙胆[/url],别名:胆草、草龙胆、山龙胆,拉丁文名:Gentiana scabra Bunge. 龙胆科、龙胆属多年生草本,根茎平卧或直立,具多数粗壮、略肉质的须根。枝单生,直立,黄绿色或紫红色,中空,近圆形,具条棱,棱上具乳突,稀光滑。枝下部叶膜质,淡紫红色,鳞片形,先端分离,中部以下连合成筒状抱茎 产内蒙古、黑龙江、吉林、辽宁、贵州,陕西、四川、湖北、湖南、安徽、江苏、浙江、福建、广东、广西。生于山坡草地、路边、河滩、灌丛中、林缘及林下、草甸,海拔400-1700米。在苏联、朝鲜、日本也有分布。







 我要推广仪器
我要推广仪器
 下载APP
下载APP