急求标准对二氯苯、五氯化磷、乙酰氯、对甲苯磺酰胺、乙二醇甲醚、马来酸国标、化工标准、地方标准都行谢谢!![em0808]
丙烯酰胺(1000ppm左右),氧化双三丁基锡TBTO(800ppm左右)怎么不出峰?用的是DB-XLB非极性的柱子15*0.25*0.25开始丙稀酰胺,氧化双三丁基锡试过几十ppm,没出峰,浓度加大到800-1000还是没出峰。做这些物质有哪些标准啊?升温程序:40度-保持1.5min; 120度/Min-155-保持1min; 20度/min-300度-保持8min; 进样口和传输线分别是270,280度。MS程序:0-18MIN采集信号(其中1.8-2.3关闭灯丝避开溶剂)峰难看一点没关系,但为什么会不出来呢,是分解掉了?还是化合物不稳定?查不到什么资料另外2-甲氧基乙醇和2-乙氧基乙醇虽然出峰了,但峰超级难看,是用DB-XLB这种非极性的柱子做不好吗?哪个做过的,分享下经验和注意事项?http://simg.instrument.com.cn/bbs/images/brow/em09511.gif修改:不好意思,刚才SIM时没把二甲苯麝香的离子加进去,现在出来了,但20ppm峰不高,估计要买固体标物配高浓度的来做
如题:对氨基苯磺酰胺与对氨基苯磺酸在亚硝酸盐的测定中有区别吗?为什么水中亚硝酸盐的测定GB/T5750。5-2006中用到的是对氨基苯磺酰胺;而食品GB/T5009.34-2008中用到的却要求是对氨基苯磺酸.我们检测水的时候也是用对氨基苯磺酸,你们说对检测结果会有影响吗
磺胺药物对氨基苯磺酰胺的合成目的原理Ar-NHCOCH3 + 2HOSO2Cl → p-ClO2S-Ar-NHCOCH3+ HClp-ClO2S-Ar-NHCOCH3 + NH3 → p-CH3CONH-Ar-SO2NH2 + HClp-CH3CONH-Ar-SO2NH2 + H2O → p-H2N-Ar-SO2NH2 + CH2CO2H仪器药品乙酰苯胺(自制) 5g(0.037mol);氯磺酸(d=1.77) 22.5g(12.5ml,0.19mol);浓氨水(28%,d=0.9) 35ml 浓盐酸,碳酸钠。过程步骤(1)对乙酰氨基苯碘酰氯在100ml干燥的锥形瓶中,加入5g干燥的乙酰苯胺,在石棉网上用小火加热熔化。瓶壁上若有少量水气凝结,应用干净的滤纸吸去。冷却使熔化物凝结成块。将锥形瓶置于冰浴中冷却后,迅速倒入12.5ml氯磺酸,立即塞上带有氯化氢导气管的塞子。反应很快发生,若反应过于激烈,可用冰水浴冷却。待反应缓和后,旋摇锥形瓶使固体全溶,然后再在温水浴中加热10~15min使反应完全。将反应瓶在冷水中充分冷却后,于通风中在充分搅拌下,将反应液慢慢倒入盛75g碎冰的烧杯,用少量冷水洗涤反应瓶,洗涤液倒入烧杯中。搅拌数分钟,并尽量将大块固体粉碎,使成颗粒小而均匀的白色固体。抽滤收集,用少量冷水洗涤,压干,立即进行下一步反应。(2)对乙酰氨基苯磺酰胺将上述粗产物移入烧杯中,在不断搅拌中慢慢加入17.5ml浓氨水(在通风橱内),立即发生放热反应并产生白色糊状物。加完后,继续搅拌15min,使反应完全。然后加入19ml水,在石棉网上用小火加热10~15min,并不断搅拌,以除去多余的氨,得到的混合物可直接用于下一步合成。(3)对氨基苯磺酰胺(磺胺)将上述反应物放入圆底烧瓶中,加入3.5ml浓盐酸,在石棉网上用小火加热回流0.5h。冷却后,应得一几乎澄清的溶液,若有固体析出,应继续加热,使反应完全。如溶液呈黄色,并有极少量固体存在时,需加入少量活性炭煮沸10min,过滤。将滤液转入大烧杯中,在搅拌下小心加入粉状碳酸钠至恰呈碱性(约4g)。在冰水浴中冷却,抽滤收集固体,用少量冰水洗涤,压干。粗产物用水重结晶(每克产物约须12ml水),产量3~4g。熔点161~162℃。纯品对氨基苯磺酰胺为白色针状结晶,熔点163~164℃。注意事项1.氯磺酸对皮肤和衣服有强烈的腐蚀性,暴露在空气中会冒出大量氯化氢气体,遇水会发生猛烈的放热反应,甚至爆炸,故取用时需加小心。反应中所用仪器及药品皆需十分干燥,含有氯磺酸的废液不可倒入水槽,而应倒入废液缸中。工业氯磺酸常呈棕黑色,使用前宜用磨口仪器蒸馏纯化,收集148~150℃的馏分。2.酰磺酸于乙酰苯胺的反应非常剧烈,将乙酰苯胺凝结成快状,可使反应缓和进行,当反应过于激烈时,应适当冷却。3.在氯磺化过程中,将有大量氯化氢气体放出。为避免污染室内空气,装置应严密,导气管的末端要与接受器内的水面接近,但不能插入水中,否则可能倒吸而引严重事故!4.加入速度必须缓慢,必须充分搅拌,以免局部过热而使对乙酰胺基苯磺酰胺水解。这是实验成功的关键。5.尽量洗去固体所夹杂和吸附的盐酸,否则产物在酸性介质中放置过久,会很快水解,因此在洗涤后,应尽量压干,且在1~2h内将它转变为磺胺类化合物。6.粗制的对氨基苯磺酰氯久置容易分解,甚至干燥后也不可避免。若要得到纯品,可将粗产物溶于温热的氯仿中,然后迅速转移到事先温热的分液漏斗中,分出氯仿层,在冰水浴中冷却后即可析出晶体。纯品对氨基苯磺酰氯的熔点为149℃。7.为了节省时间,这一步的粗产物可不必分出。若要得到产品,可在冰水浴中冷却,抽滤,用冰水洗涤,干燥即可。粗品用水重结晶,纯品熔点为219~220℃。8.对乙酰胺基苯磺酰胺在稀酸中水解成磺胺,后者又与过量的盐酸形成水溶性的盐酸盐,所以水解完成后,反应液冷却时应无晶体析出。由于水解前溶液中氨的含量不同,加3.5ml盐酸有时不够,因此,在回流至固体全部消失前,应测一下溶液的酸碱性,若酸性不够,应补加盐酸回流一段时间。9.用碳酸钠中和滤液中的盐酸时,有二氧化碳产生,故应控制加热速度并不断搅拌使其逸出。磺胺是一两性化合物,在过量的碱溶液中也易变成盐类而溶解。故中和操作必须仔细进行,以免降低产量。分析思考 1.为什么在氯磺化反应完成以后处理反应混合物时,必须移到通风橱中,且在充分搅拌下缓缓倒入碎冰中?若在未倒完前冰就化完了,是否应补加冰块?为什么?2.为什么苯胺要乙酰化后在氯磺化?直接氯磺化行吗?3 .如何理解对氨基苯磺酰氨是两性物质?试用反应式表示磺胺与稀酸和稀碱的作用。
我按照10版药典的方法用气相检测糖精钠中的甲苯磺酰胺,两种对照品邻甲苯磺酰胺和对甲苯磺酰胺都没有出峰,所有的图只有溶剂峰—二氯甲烷出峰了。色谱柱用的是药典规定的OV-17.有没有人做过这个东西??求解~~
有谁做过糖精钠中邻甲苯磺酰胺和对甲苯磺酰胺?按2015版药典设置柱温180度。用fid检测器做,检测器设置为280,进样口250。只有溶剂峰,没见目标峰,有谁做过,请教一下色谱条件。
谁做过[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]或[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质[/color][/url]检测邻对苯基磺酰胺的,望指教。
中 华 人 民 共 和 国 国 家 标 准GB/T 5009.30—1996食品中叔丁基羟基茴香醚(BHA)与 代替GB 5009.30—852,6—二叔丁基对甲酚(BHT)的测定方法第二篇 薄层色谱法 8 原理 用甲醇提取油脂或食品中脂肪的抗氧化剂,用薄层色谱定性,根据其在薄层板上显色后的最低检出量,与标准品最低检出量比较而概略定量,对高脂肪食品中的BHT、BHA、PG能定性检出。 9 试剂 以下试剂均为分析纯。 9.1 甲醇。9.2 石油醚(30~60℃)。9.3 异辛烷。9.4 丙酮。9.5 冰乙酸。9.6 正己烷。9.7 二氧六环。9.8 硅胶G:(薄层用)。9.9 聚酰胺粉200目。9.10 可溶性淀粉。9.11 BHT、BHA、PG混合标准溶液的配制:分别准确称取BHT、BHA、PG各 10.0 mg,分别用丙酮溶解转入三个 10 mL 容量瓶中,用丙酮稀释至刻度。每毫升含 1.0 mg BHT、BHA、PG,吸取BHT(1.0 mg/mL)1.0 mL、BHA(1.0 mg/mL)、PG(1.0 mg/mL)各 0.3 mL 置同一 5 mL 容量瓶中,用丙酮稀释至刻度。此溶液每毫升含 0.20 mg BHT、0.060 mg BHA、0.060 mg PG。9.12 显色剂:2,6—二氯醌—氯亚胺的乙醇溶液(2 g/L)。 10 仪器 10.1 减压蒸馏装置。10.2 浓缩瓶具刻度的尾管。10.3 层析槽 a. 24cm×6cm×4cm, b. 20cm×13cm×8cm。10.4 玻璃板:5cm×20cm、10cm×20cm。10.5 微量注射器:10μL。 11 分析步骤 11.1 提取11.1.1 植物油(花生油、豆油、菜籽油、芝麻油):称取 5.00 g 油置 10 mL 具塞离心管中,加入 5 mL甲醇,密塞振摇 5 min,放置 2 min,离心(3000~3500r/min)5min,吸取上层清液置 25 mL 容量瓶中,如此重复提取共五次,合并每次甲醇提取液,用甲醇稀释至刻度。吸取 5 mL 甲醇提取液置一浓缩瓶中,于40℃水浴上减压浓缩至 0.5 mL,留作薄层色谱用。11.1.2 猪油:称取 5.00 g 猪油置 50 mL 具磨口的锥形瓶中,加入 25 mL 甲醇,装上冷凝管于75℃水浴上放置 5 min,待猪油完全溶化后将锥形瓶连同冷凝管一起自水浴中取出,振摇 30 s,再放入水浴 30 s;如此振摇三次后放入75℃水浴,使甲醇层与油层分清后,将锥形瓶连同冷凝管一起置冰水浴中冷却,猪油凝固,甲醇提取液通过滤纸滤入 50 mL 容量瓶中,再自冷凝管顶端加入 25 mL 甲醇,重复振摇提取一次,合并二次甲醇提取液,将该容量瓶置暗处放置,待升至室温后。用甲醇稀释至刻度。吸取 10 mL 甲醇提取液置一浓缩瓶中,于40℃水浴上减压浓缩至 0.5 mL,留作薄层色谱用。11.1.3 食品(油炸花生米、.酥糖、巧克力、饼干):按6.2测定脂肪的含量,并称取约 2.00 g 的脂肪视提取出的油脂是植物油还是动物性脂肪而决定提取方法。可按11.1.1或11.1.2操作。11.2 测定11.2.1 薄层板的制备11.2.1.1 硅胶G薄层板:称取 4 g 硅胶G置玻璃乳钵中,加 10 mL 水。研磨至粘稠状,铺成 5cm×20cm 的薄层板三块,置空气中干燥后于80℃烘1h,存放于干燥器中。11.2.1.2 聚酰胺板:称取 2.4 g 聚酰胺粉 0.6 g 可溶性淀粉置于玻璃乳钵中,加约 15 mL水,研磨至浆状铺成 10cm×20cm 的薄层板三块,置空气中干燥后于80℃烘 1 h,置干燥器中保存。11.2.2 点样11.2.2.1 用 10 μL 微量注射器在 5cm×20cm 的硅胶G薄层板上距下端 2.5 cm 处点三点:标准溶液 5 μL、样品提取液 6~30 μL、标准溶液 5μL。11.2.2.2 另取一块硅胶G薄层板点三点:标准溶液 5μL、样品提取液 1.5~3.6 μL、加标准溶液 5 μL。11.2.2.3 用 10 μL微量注射器在 10cm×20cm 的聚酰胺薄层板上距下端 2.5 cm 处点:标准溶液 5μL,样品提取液 10μL,加标准溶液 5 μL,边点样边用吹风机吹干,点上一滴吹干后再继续滴加。11.2.3 展开11.2.3.1 溶剂系统 硅胶G薄层板:正己烷—二氧六环—醋酸(42+6+3),异辛烷—丙酮—醋酸(70+5+12) 聚酰胺板: a.甲醇—丙酮—水(30+10+10) b.甲醇—丙酮—水(30+10+12.5) c.甲醇—丙酮—水(30+10+15) 对甲醇—丙酮—水系统,芝麻油只能用(a)、菜籽油用(b),食品用(c)。 展开系统中水的比例对花生油、豆油、猪油中PG的分离无影响。 将点好样的薄层板置预先经溶剂饱和的展开槽内展开 16 cm。11.2.3.2 展开11.2.3.2.1 硅胶G板自层析槽中取出薄层板置通风橱中挥干至PG标准点显示灰黑色斑点。即可认为溶剂己基本挥干,喷显色剂,置110℃烘箱中加热 10 min,比较色斑颜色及深浅,趁热将板置氨蒸气槽中放置 30s,观察各色斑颜色变化。11.2.3.2.2 聚酰胺板 自层析槽中取出薄层板置通风橱中吹干,喷显色剂,再通风挥干,直至PG斑点清晰。11.2.4 评定11.2.4.1 定性 根据样品中显示出的BHT、BHA、PG点与标准BHT、BHA、PG点比较Rf值和显色后斑点的颜色反应定性。如果样液点显示检出某种抗氧化剂,则样品中抗氧化剂的斑点必须与加入内标的抗氧化剂斑点重叠。 当点大量样液时由于杂质多,使样品中抗氧化剂点的Rf值略低于标准点。这时必须在样品点上滴加标准溶液作内标,比较Rf值。 表1 BHT、BHA、PG在薄层板上的最低检出量Rf值及斑点颜色 ---------------------------------┬----------------------------------------┬-----------------------------------------------┐ 薄 层 板 │ 硅 胶 G 板 │ 聚 酰 胺 板 │ 结果 ├----------------------------------------------------┼------------------------------------------------┤ │ Rf值 最低检出量 色斑颜色 │ Rf值 最低检出量 色斑颜色 │抗氧化剂 │ μg │ μg │---------------------------------┼--------------------------------------┼------------------------------------------------┤BHT │ 0.73 1 桔红→紫红 │ — — — │BHA │ 0.37 0.3 紫红→蓝紫 │ 0.52 0.3 灰棕 │PG │ 0.04 0.3 灰→黄棕 │ 0.66 0.3 蓝 │--------------------┴--------------------------------------------------┴------------------------------------------------┘ 注:PG在硅胶G板上定性及半定量不可靠,有干扰且Rf值太小,须进一步用聚酰胺板展开。
最近弄些邻对甲苯磺酰胺的检测,想知道他们的危害,就是找不到相关资料,希望各位大侠帮帮忙。
GB 26403-2011 食品安全国家标准 食品添加剂 特丁基对苯二酚
试问:硫酸铈能否氧化对叔丁基苯酚?可以直接电位滴定测对叔丁基苯酚的纯度吗?实验室电位滴定仪:梅特勒T50。若手动滴定,指示剂采用二苯胺磺酸钠还是邻二氮杂菲-亚铁?望指点
乳及乳制品中舒巴坦敏感β-内酰胺酶类药物检验方法指定检验方法4.乳及乳制品中舒巴坦敏感β-内酰胺酶类药物检验方法杯碟法1、范围本标准规定了乳及乳制品中舒巴坦敏感β-内酰胺酶类药物的检验方法。本标准适用于乳及乳制品中舒巴坦敏感β-内酰胺酶类物质的检验。本方法的检出限为4U/mL。2、原理该方法采用对青霉素类药物绝对敏感的标准菌株,利用舒巴坦特异性抑制β-内酰胺酶的活性,并加入青霉素作为对照,通过比对加入β-内酰胺酶抑制剂与未加入抑制剂的样品所产生的抑制圈的大小来间接测定样品是否含有β-内酰胺酶类药物。3、设备和材料除微生物实验室常规灭菌及培养设备外,其他设备和材料如下:3.1 抑菌圈测量仪或测量尺。3.2恒温培养箱:36℃±1℃。3.3 高压灭菌器。3.4 无菌培养皿:内径90 mm,底部平整光滑的玻璃皿,具陶瓦盖。3.5 无菌牛津杯:外径(8.0士0.1) mm,内径(6.0士0.1) mm,高度(10.0士0.1) mm。3.6 麦氏比浊仪或标准比浊管。3.7 pH计。3.8 无菌吸管:1mL(0.01mL刻度值),10mL(0.1mL刻度值)。3.9 加样器:5μL~20μL,20μL -200μL及配套吸头。4、培养基和试剂 除另有规定外,所用试剂均为分析纯,水为GB/T6682中规定的三级水。4.1 试验菌种:藤黄微球菌(Micrococcus luteus) CMCC(B) 28001,传代次数不得超过14次。4.2 磷酸盐缓冲溶液:按附录A中A.1规定。4.3生理盐水(8.5 g/L):按附录A中A.2规定。4.4 青霉素标准溶液:按附录A中A.3规定。4.5 β-内酰胺酶标准溶液:按附录A中A.4规定。4.6 舒巴坦标准溶液按附录A中A.5规定。。4.7 营养琼脂培养基:按附录A中A.6规定。4.8 抗生素检测用培养基Ⅱ:按附录A中A.7规定。5、操作步骤5.1 菌悬液的制备将藤黄微球菌接种于营养琼脂斜面上,经36士1℃培养18h-24 h,用生理盐水洗下菌苔即为菌悬液,测定菌悬液浓度,终浓度应大于1×1010 CFU/mL,4 ℃保存,贮存期限2周。5.2 样品的制备将待检样品充分混匀,取1 mL待检样品于1.5 mL离心管中共4管,分别标为:A、B、C、D,每个样品做三个平行,共12 管,同时每次检验应取纯水1 mL加入到1.5 mL离心管中作为对照。如样品为乳粉,则将乳粉按1:10的比例稀释。如样品为酸性乳制品,应调节pH值至6-7。5.3 检验用平板的制备取90mm灭菌玻璃培养皿,底层加10 mL灭菌的抗生素检测用培养基Ⅱ,凝固后上层加入5 mL含有浓度为1×108 CFU/mL藤黄微球菌的抗生素检测用培养基Ⅱ,凝固后备用。5.4 样品的测定按照下列顺序分别将青霉素标准溶液、β-内酰胺酶标准溶液、舒巴坦标准溶液加入到样品及纯水中:A 青霉素5 μL。B 舒巴坦25 μL、青霉素5 μL。C β-内酰胺酶25 μL、青霉素G5 μL。D β-内酰胺酶25 μL、舒巴坦25 μL、青霉素5 μL。混匀后,将上述A~D 试样各200 μL 加入放置于检验用平板上的4个无菌牛津杯中,36士1℃培养培养18~22 h ,测量抑菌圈直径。每个样品,取三次平行试验平均值。5.5 结果报告纯水样品结果应为:(A)、(B)、(D)均应产生抑菌圈;(A)的抑菌圈与(B)的抑菌圈相比,差异在3 mm以内(含3 mm),且重复性良好;(C)的抑菌圈小于(D)的抑菌圈,差异在3 mm以上(含3 mm),且重复性良好。如为此结果,则系统成立,可对样品结果进行如下判定:7.1 如果样品结果中(B)和、(D)均产生抑菌圈,且(C)与(D)抑菌圈差异在3 mm以上(含3 mm)时,可按7.1.1、7.1.2 判定结果。7.1.1(A)的抑菌圈小于(B)的抑菌圈差异在3 mm以上(含3 mm),且重复性良好,应判定该试样添加有β- 内酰胺酶,报告β- 内酰胺酶类药物检验结果阳性。7.1.2(A)的抑菌圈同(B)的抑菌圈差异小于3 mm,且重复性良好,应判定该试样未添加有β- 内酰胺酶,报告β- 内酰胺酶类药物检验结果阴性。7.2 如果(A)和(B)均不产生抑菌圈,应将样品稀释后再进行检测。附 录 A(规范性附录)培 养 基A.1 磷酸盐缓冲溶液(pH6.0)无水磷酸二氢钾8.0 g无水磷酸氢二钾2.0 g蒸馏水加至1000 mLA.2 生理盐水(8.5 g/L)氯化钠8.5 g蒸馏水1000 mL121℃高压灭菌15 min。A.3 青霉素标准溶液准确称取适量青霉素标准物质,用磷酸盐缓冲溶液溶解并定容为0.1mg/mL的标准溶液。当天配制,当天使用。A.4 β-内酰胺酶标准溶液准确量取或称取适量β-内酰胺酶标准物质,用磷酸盐缓冲溶液溶解并定容为16000 U/mL的标准溶液。当天配制,当天使用。A.5 舒巴坦标准溶液准确称取适量舒巴坦标准物质,用磷酸盐缓冲溶液溶解并定容为1 mg/mL的标准溶液,分装后-20 ℃保存备用,不可反复冻融使用。A.6 营养琼脂蛋白胨10 g牛肉膏3 g氯化钠5 g琼脂15-20 g蒸馏水1000 mL将上述成分加入蒸馏水中,搅混均匀,分装试管每管约5~8 mL,120℃高压灭菌15 min,灭菌后摆放斜面。A.7 抗生素检测培养基Ⅱ蛋白胨10 g牛肉浸膏3 g氯化钠5 g酵母膏3 g葡萄糖1 g琼脂14 g蒸馏水1000mL将上述成分加入蒸馏水中,搅混均匀,120 ℃高压灭菌15 min,其最终pH 值约为6.6。
大家好,一直在尝试做这两种物质,采用ESI源负离子模式,叔丁基对苯醌(TBBQ)会形成【M+H】(-)的特殊准分子离子,m/z=165,和叔丁基对苯酚(TBHQ)的准分子离子一样,m/z=165,导致他们的MRM通道完全一致,只能在色谱上区分,但是他们的保留时间测出来一模一样,改了不少次条件了都不太好。 我想请问 1.是不是两种物质已经发生了某种转化呢,实际已经是同一种物质,怎么验证呢? 2.如果没发生转化,怎么样在色谱上分开他们两个,应该怎么调整?实验室有T3 和C18 柱子,各种流动相都有
大家好,我想在[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]上分离叔丁基对苯酚和叔丁基对苯醌,但尝试了很多次始终做不到,他们两个的峰几乎完全在同一时间,我想请问一下大家,应该从哪方面尝试呢?实验室有T3和C18两种色谱柱,各种流动相和药品几乎都有。
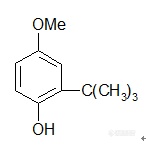
【中文名称】3-叔丁基-4-羟基茴香醚;3-叔丁基-4-羟基苯甲醚;2-叔丁基-4-甲氧基苯酚【英文名称】3-t-butyl-4-hydroxy anisole【结构或分子式】 http://ng1.17img.cn/bbsfiles/images/2012/03/201203241952_357143_1855403_3.jpg【相对分子量或原子量】180.25【熔点(℃)】48~55【沸点(℃)】264~270℃(9.77E4Pa)【毒性LD50(mg/kg)】 混合物大鼠经口2200【性状】 与3-叔丁基-4-甲氧基苯酚混合为无色至微黄色蜡状固体或粉末,略有刺激性气味。【溶解情况】 不溶于水。【用途】 市售品为3-叔丁基-4-甲氧基苯酚和2-叔丁基-4-甲氧基苯酚的混合物,用于食品用抗氧剂,抗氧能力3-叔丁基-4-甲氧基苯酚强,两者同时使用,起协同作用,可用于油脂、油炸食品、干鱼制品、饼干、方便面、罐头及腊肉制品等。除单独使用外,亦可与抗坏血酸、异抗坏血酸、柠檬酸等配合使用。【制备或来源】 (1)以对羟基苯甲醚为原料,在硫酸或磷酸催化剂存在下,在80℃下与叔丁醇反应而得混合物;(2)以对苯二酚和叔丁醇为原料,反应生成叔丁基对苯二酚,再以锌粉为催化剂,与硫酸二甲酯进行甲基化反应而得混合物。【其他】 遇铁离子会着色。与BHT、没食子酸丙酯、对苯二酚、蛋氨酸、卵磷脂、硫代二丙酸等有协同作用。【生产单位】略
请问有没有测过生活饮用水中的丁基黄元酸,不知道大家的标准品是在哪里买的?是买的丁基黄元酸还是丁基黄元酸钾?我们这里只能买到丁基黄元酸钾不知道能不能做出来!
我们稀释标准品的时候,有的标准上说乙腈,有的方法是用甲醇,有的方法是用叔丁基甲醚,这些试剂有什么不同和共同点吗?
[color=#00008B][size=4][em0808][/size][/color]求助各位,谁知道在哪可以找到抗氧化剂叔丁基对苯二酚(TBHQ)的检测标准。急盼,麻烦,谢谢各位[em0805]
问题描述:本人新买了有机锡的标准品一氯三丁基锡 CISn(C4H9)3,配制成工作液后加内标衍生,衍生产物为乙基三丁基锡C2H5Sn(C4H9)3 (TBT),走GC-MS,发现会出二乙基二丁基锡 (C2H5)2Sn(C4H9)2 (DBT).疑问:1:这个标准是不是存在分解,如果存在,分解发生在那个步骤,是标准配制还是要衍生或是进样口。2:因为多出的目标物(DBT)也是我们的检测目标物,这样就存在定量的问题,TBT偏小。DBT偏高。我把谱图发上来大家看下
买了sigma家的北塔-内酰胺酶标准品,规格为1KU,试剂条检出限为2U,要验收试剂条。因为是冻干粉,所以往里面加了10mL稀释液,然后再原奶到稀释1U,结果还是检出,然后看证书“lyophilized powder, 1,500-3,000 units/mg protein (using benzylpenicillin)” 里面为2800左右,然后按三倍稀释,还是做不出,是不是哪一个步骤错误了?
乳及乳制品中舒巴坦敏感β-内酰胺酶类药物检验方法杯碟法1、范围本标准规定了乳及乳制品中舒巴坦敏感β-内酰胺酶类药物的检验方法。本标准适用于乳及乳制品中舒巴坦敏感β-内酰胺酶类物质的检验。本方法的检出限为4U/mL。2、原理该方法采用对青霉素类药物绝对敏感的标准菌株,利用舒巴坦特异性抑制β-内酰胺酶的活性,并加入青霉素作为对照,通过比对加入β-内酰胺酶抑制剂与未加入抑制剂的样品所产生的抑制圈的大小来间接测定样品是否含有β-内酰胺酶类药物。3、设备和材料除微生物实验室常规灭菌及培养设备外,其他设备和材料如下:3.1 抑菌圈测量仪或测量尺。3.2恒温培养箱:36℃±1℃。3.3 高压灭菌器。3.4 无菌培养皿:内径90 mm,底部平整光滑的玻璃皿,具陶瓦盖。3.5 无菌牛津杯:外径(8.0士0.1) mm,内径(6.0士0.1) mm,高度(10.0士0.1) mm。3.6 麦氏比浊仪或标准比浊管。3.7 pH计。3.8 无菌吸管:1mL(0.01mL刻度值),10mL(0.1mL刻度值)。3.9 加样器:5μL~20μL,20μL -200μL及配套吸头。4、培养基和试剂 除另有规定外,所用试剂均为分析纯,水为GB/T6682中规定的三级水。4.1 试验菌种:藤黄微球菌(Micrococcus luteus) CMCC(B) 28001,传代次数不得超过14次。4.2 磷酸盐缓冲溶液:按附录A中A.1规定。4.3生理盐水(8.5 g/L):按附录A中A.2规定。4.4 青霉素标准溶液:按附录A中A.3规定。[size=1
本标准代替GB 1916-1980本标准规定了食品添加剂叔丁基-4-羟基茴香醚的技术要求、试验方法、检验规则、标志、包装、运输、贮存及保质期。
公司有一产品:樟脑磺内酰胺现测比旋度,用三氯甲烷做溶剂,C=1.0,每次数字显示很不稳定,大概一分钟才能显示数字,每次复测的结果相差很大仪器肯定没问题(标准旋光管校验过)是什么原因导致的呢?
大家好,我想请教下糖精钠中甲苯磺酰胺的检测方法,大家有没有做过该检测的,告知下仪器及参数设置是怎么样的?另:色谱用硅藻土有现货的吗?还是要按照药典方法自己制备的?急
最近要测n甲基甲酰胺标准品但是找了一些标准品厂家都没这个标,要证书的,所以也不能用溶剂代替。有买过的或者有货号的请提供一下给我
[size=2][font=宋体]2-氨基苯酚-4-磺酰胺的国标或行标[/font][/size][size=2][font=宋体][/font][/size]
谁有维生素E,磺胺嘧啶银(N-2-嘧啶基 -4-氨基苯磺酰胺银盐)的红外光谱图
各位老师,求一份做叔丁基甲基醚的翻译标准,U.SEPA8260D-2018这个标准的翻译版标准,看不懂英文。
PONY谱尼测试最新了解到,厚生劳动省医药食品局发布食安发0928第2号:部分修改食品、添加剂等的规格标准(2009年厚生劳动省告示第422号),设定农药氯虫酰胺、氰氟虫腙以及甲基碘在食品中的残留标准。根据此通知,厚生劳动省将如下记修改部分食品、添加剂等的规格标准(昭和34年厚生省告示第370号)。第1 修改的摘要根据食品卫生法(昭和22年法律第233号。以下简称“法”。)第11条第1项的规定,设定农药氯虫酰胺、氰氟虫腙以及甲基碘等在食品中的残留标准。第2 实施• 适用日期由公布之日起开始实施第3 应用须知1、此次,在设定了氯虫酰胺标准值的食品中,桃、西瓜以及香瓜是包括果皮的。2、此次,设定了标准值的氰氟虫腙是指:将氰氟虫腙(E-同分异构体)、氰氟虫腙(Z-同分异构体)以及作为氰氟虫腙代谢物的p-[m-(三氟甲基) 苯甲酰甲基] 苯甲腈换算为氰氟虫腙之后的和。第4 其它以残留标准值(根据“法”设定)以及农药取缔法(昭和23年法律第82号)为依据,在农林水产省将氯虫酰胺、氰氟虫腙以及甲基碘注册为农药。关于氯虫酰胺、氰氟虫腙以及甲基碘的检验法将在日后通知。PONY谱尼测试在农药残留方面具有丰富的经验,可以依据日本肯定列表进行检测。对于这三种农药,企业应该引起重视,PONY谱尼测试将鼎力帮助企业进行检测,顺利出口。
问题描述:本人新买了有机锡的标准品一氯三丁基锡 CISn(C4H9)3,配制成工作液后加内标衍生,衍生产物为乙基三丁基锡C2H5Sn(C4H9)3 (TBT),走GC-MS,发现会出二乙基二丁基锡 (C2H5)2Sn(C4H9)2 (DBT).疑问:1:这个标准是不是存在分解,如果存在,分解发生在那个步骤,是标准配制还是要衍生或是进样口。2:因为多出的目标物(DBT)也是我们的检测目标物,这样就存在定量的问题,TBT偏小。DBT偏高。我把谱图发上来大家看下







 我要推广仪器
我要推广仪器
 下载APP
下载APP