请问各位高手有没有叔丁醇锂的国标,行业标准也行,谢谢!
分析二甲基丁醇,三甲基丁醇,样品浓度大约为99%。请教分析方法及使用何种色谱柱?谢谢了!![em58]
哪位知道国产的异丁醇,异戊醇标准品哪有?

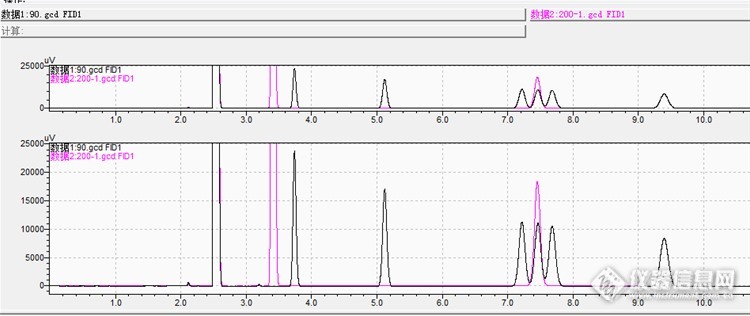
目前,许多国家规定,凡结构中具有不对称因素的药物,即“手性药物”,必须拆分其相应的立体异构体,并分别研究其药理、毒理和药物代谢性质。对已上市的消旋体药物,要重新评价其光学异构体的性质。对新申报的药品,一开始就要合成其光学异构体。这就要求我们在产品质量控制过程中,开发相应的光学异构体分离方案。 现大家常用HPLC来进行分离,色谱柱是一部分因素,但是色谱条件的选择,对分离的影响非常大。通常会首先正相条件来进行分离,使用的流动相含有烷烃(正己烷、正戊烷)以及醇类(异丙醇、乙醇、甲醇、叔丁醇),现通过一个案例看叔丁醇在手性分离中的应用。 项目名称是:[b][i]替格瑞洛[/i][/b],结构式分别如下:[img=,416,254]http://ng1.17img.cn/bbsfiles/images/2017/08/201708222022_01_1708019_3.png[/img][img=,374,252]http://ng1.17img.cn/bbsfiles/images/2017/08/201708222023_01_1708019_3.png[/img][img=,388,272]http://ng1.17img.cn/bbsfiles/images/2017/08/201708222023_02_1708019_3.png[/img] 接下来的就是选柱子、选方法了: 当然,开始从正相条件入手,选择了常规的两款多糖涂覆型手性柱:[b]月旭Ultimate Cellu-D(4.6×250mm,5μm)与月旭Ultimate Amy-D(4.6×250mm,5μm)[/b]开始测试。流动相选择了:[b]正己烷:乙醇=90:10[/b],测试效果如下: [img=,690,299]http://ng1.17img.cn/bbsfiles/images/2017/08/201708222030_01_1708019_3.png[/img] [img=,690,301]http://ng1.17img.cn/bbsfiles/images/2017/08/201708222031_01_1708019_3.png[/img] 结果分离效果均不理想,接下来用这两款柱子,醇类选择异丙醇和乙醇按不同比例来测试,均达不到分离要求,但是明显在[b] [/b]Amy-D的分离效果更好,所以后续方法的调整均在Amy-D柱上进行测试; 接下来怎么办呢,试试两种醇混合使用,看看效果: [img=,690,289]http://ng1.17img.cn/bbsfiles/images/2017/08/201708222036_01_1708019_3.png[/img] 上图是使用流动相:[b]正己烷:甲醇:乙醇=90:10:10[/b]来进行分离,结果三个物质均露面了,但是但是还是达不到分离要求,而且降低醇类的比例,也无法实现分离; 哎,看来正相体系是没有办法了,只能再试试模式了。现在[b]极性溶剂模式[/b]也是常用的一种模式,这种模式是使用100%的醇类或两种醇类混合使用,通常用于常规模式分离不理想的时候的一种选择吧!!! 先用[b]甲醇:异丙醇=80:20[/b]开始,出图先: [img=,690,192]http://ng1.17img.cn/bbsfiles/images/2017/08/201708222043_02_1708019_3.png[/img] 分离不开,不行再来,用[b]甲醇:乙醇=90:10:[/b] [img=,690,241]http://ng1.17img.cn/bbsfiles/images/2017/08/201708222045_01_1708019_3.png[/img] 结果三个物质均出现,两个杂质分别在主峰的前后,也达不到基线分离。 继续:100%甲醇、100%乙醇、100%乙腈。。。。。。还是分离不开,咋办。唉这时候已经想选择放弃,通过两个方法,杂质A与杂质B分别控制。 听说有些极端情况下,可以试试叔丁醇呢,那加点叔丁醇试试吧,结果在[b]甲醇:叔丁醇 = 78 : 22[/b]条件下,使用[b]Amy-D(4.6×250mm,5μm)[/b]分析,各个物质均达到基线分离,且杂质A与杂质B均在主峰之前出峰,是不是都是我们希望的,按照要求配置一个系统适应性溶液测试: [img=,690,321]http://ng1.17img.cn/bbsfiles/images/2017/08/201708222048_01_1708019_3.png[/img] 分析条件初步完成,接下来么,就是方法验证了,就不再一一列举了。 所以在进行手性分析时,当遇到分离度不合适,或者样品在烷烃中的溶解性不好以及难以洗脱的样品时,可以试试极性溶剂模式来进行分离,同时因为手性分离,不同醇类,会出现不同的分离效果,而甲醇、叔丁醇也可以当做添加剂的形式,配合其他醇类用于手性分离,有时候会出现意想不到的效果。
叔丁醇锂游离碱分析方法我要分析叔丁醇锂游离碱含量,请问有谁知道,请赐教
各位大大,本人现在正在做一个冻干制剂的叔丁醇残留检测,标准上规定用PEG20000为固定液的柱子,但未指定品牌和型号,我这里刚好有一根HP-INNOWAX的柱子,就试着做了一下,结果发现出峰时间在1分半钟左右,而且进的几个不同叔丁醇标准液的浓度和它们的峰面积基本呈线性。但是我觉得保留时间这么短,又有些担心,比如死时间出峰什么的。所以想问下各位,有没有人做过类似叔丁醇残留测定的,柱子是HP-INNOWAX或者DB-WAX的,这么短的保留时间能不能用。还有个问题,标准上规定的载气流量是以“毫升每分钟”记的,但是我们这的岛津GC上面只有载气压力显示,要是想换算成流量,应该怎么弄啊。 (本人是新手,还请各位多多帮忙,谢谢)
[color=#444444]本人现在正在做一个冻干制剂的叔丁醇残留检测,标准上规定用PEG20000为固定液的柱子,但未指定品牌和型号,我这里刚好有一根HP-INNOWAX的柱子,就试着做了一下,结果发现出峰时间在1分半钟左右,而且进的几个不同叔丁醇标准液的浓度和它们的峰面积基本呈线性。但是我觉得保留时间这么短,又有些担心,比如死时间出峰什么的。所以想问下各位,有没有人做过类似叔丁醇残留测定的,柱子是HP-INNOWAX或者DB-WAX的,这么短的保留时间能不能用。还有个问题,标准上规定的载气流量是以“毫升每分钟”记的,但是我们这的岛津GC上面只有载气压力显示,要是想换算成流量,应该怎么弄啊。[/color]
【求助】GC测定叔丁醇残留用什么气相色谱柱各位大大,本人现在正在做一个冻干制剂的叔丁醇残留检测,标准上规定用PEG20000为固定液的柱子,但未指定品牌和型号,我这里刚好有一根HP-INNOWAX的柱子,就试着做了一下,结果发现出峰时间在1分半钟左右,而且进的几个不同叔丁醇标准液的浓度和它们的峰面积基本呈线性。但是我觉得保留时间这么短,又有些担心,比如死时间出峰什么的。所以想问下各位,有没有人做过类似叔丁醇残留测定的,柱子是HP-INNOWAX或者DB-WAX的,这么短的保留时间能不能用。还有个问题,标准上规定的载气流量是以“毫升每分钟”记的,但是我们这的岛津GC上面只有载气压力显示,要是想换算成流量,应该怎么弄啊。 (本人是新手,还请各位多多帮忙,谢谢)
测四氟丙醇中的杂质叔丁醇的含量。因为现有两台[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]GC979/GC1690,手动进样0.8UL,都是FID。柱子都是兰化所的1701毛细柱。分析同一个样品,总峰面积1690为1080W,9790的为998W,结果1690那台叔丁醇的含量是9790的两倍,做了3次试验都是一样情况。两台色谱的载气,空气,氢气的流速都一样。不知道怎样才能确定哪个是准确的。是不是用内标?本人接触色谱不久对内标不是很懂,对叔丁醇选什么内标物好?内标物是不是要特别买?我看试剂的含量都是个范围。
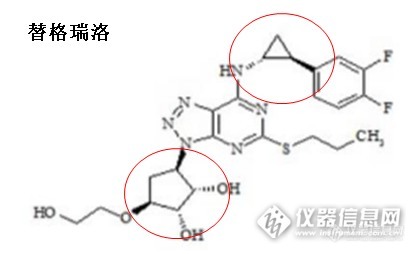
目前,许多国家规定,凡结构中具有不对称因素的药物,即“手性药物”,必须拆分其相应的立体异构体,并分别研究其药理、毒理和药物代谢性质。对已上市的消旋体药物,要重新评价其光学异构体的性质。对新申报的药品,一开始就要合成其光学异构体。这就要求我们在产品质量控制过程中,开发相应的光学异构体分离方案。 现大家常用HPLC来进行分离,色谱柱是一部分因素,但是色谱条件的选择,对分离的影响非常大。通常会首先选择正相条件来进行分离,使用的流动相含有烷烃(正己烷、正戊烷)以及醇类(异丙醇、乙醇、甲醇、乙腈),现通过一个案例看叔丁醇在手性分离中的应用。 项目名称是:替格瑞洛,结构式分别如下: [img=,286,223]http://ng1.17img.cn/bbsfiles/images/2017/08/201708250816_01_1708019_3.png[/img] [img=,297,222]http://ng1.17img.cn/bbsfiles/images/2017/08/201708250817_01_1708019_3.png[/img] [img=,310,222]http://ng1.17img.cn/bbsfiles/images/2017/08/201708250818_01_1708019_3.png[/img] 接下来就是选柱子,选方法了: 当然,开始先从正相条件入手,选择了常规的两款多糖涂覆型手性柱:月旭Ultimate Cellu-D(4.6×250mm,5μm)与月旭Ultimate[b] [/b]Amy-D(4.6×250mm,5μm)开始测试。流动相选择了:正己烷:乙醇=90:10,测试效果如下:[img]http://bbs.instrument.com.cn/xheditor/xheditor_skin/blank.gif[/img] [img=,690,299]http://ng1.17img.cn/bbsfiles/images/2017/08/201708250820_01_1708019_3.png[/img] [img=,690,301]http://ng1.17img.cn/bbsfiles/images/2017/08/201708250820_02_1708019_3.png[/img] 结果分离效果均不理想,接下来用这两款柱子,醇类选择异丙醇和乙醇按不同比例来测试,均达不到分离效果,但是明显在Amy-D柱上的分离效果更好,所以后续方法的调整均在Amy-D柱上进行测试; 再接下来怎么办呢,试试两种醇混合使用,看看效果: [img=,690,289]http://ng1.17img.cn/bbsfiles/images/2017/08/201708250821_01_1708019_3.png[/img] 上图是使用流动相:正己烷:甲醇:乙醇=90:10:10来进行分离,结果三个物质均露面了,但是但是还是达不到分离的要求,而且降低醇类的比例,也无法实现完全分离。 哎,看来正相体系是没有办法了,只能再试试别模式了。现在极性溶剂模式也是常用的一种模式,这种模式是使用100%的醇类或两种醇类混合使用,也算是常规模式分离不理想情况下的一种选择吧!!! 先用甲醇:异丙醇=80:20开始,出图先: [img=,690,192]http://ng1.17img.cn/bbsfiles/images/2017/08/201708250822_01_1708019_3.png[/img][img]http://bbs.instrument.com.cn/xheditor/xheditor_skin/blank.gif[/img] 分离不开,不行再来,试试甲醇:乙醇=90:10: [img=,690,241]http://ng1.17img.cn/bbsfiles/images/2017/08/201708250823_01_1708019_3.png[/img][img]http://bbs.instrument.com.cn/xheditor/xheditor_skin/blank.gif[/img] 三个物质均出现,两个杂质分别在主峰前后,也达不到基线分离。 继续:100%甲醇、100%乙醇、100%乙腈。。。。。。还是分离不开,咋办。唉,这时候已经想选择放弃,通过两个方法来,分别监控杂质A与杂质B的含量。 嗯,听说有些极端情况下,可以试试叔丁醇呢,那加点叔丁醇试试吧,结果在甲醇:叔丁醇 = 78 : 22条件下,使用Amy-D(4.6×250mm,5μm)分析,各个物质均达到基线分离,且杂质A与杂质B均在主峰之前出峰,是不是都是我们希望的,按照要求配置一个系统适应性溶液测试: [img=,690,321]http://ng1.17img.cn/bbsfiles/images/2017/08/201708250823_02_1708019_3.png[/img][img]http://bbs.instrument.com.cn/xheditor/xheditor_skin/blank.gif[/img] 分析条件的筛选初步完成,接下来么,就是方法验证了,就不再一一列举了。 所以在进行手性分析时,当遇到分离度不合适,或者样品在烷烃中的溶解性不好以及难以洗脱时,可以试试极性溶剂模式来进行分离,同时因为手性分离,不同醇类,会出现不同的分离效果,而甲醇、叔丁醇也可以当作添加剂的形式,配合其他醇类用于手性分离,有时候会出现意想不到的效果。
本人要测定叔丁醇中的纯度,找了很久都有找到如题标准,那位好心人支持一下,万分感谢!
如题TCD检测器 PEG填充柱。在分析乙醇的时候很正常,换作分析丁醇就会出现许多杂峰 而且基线要走很久才能平 有时候甚至一直出现像心电图一样的峰 很崩溃是丁醇样品的问题是样品不纯 还是柱子或者色谱的问题但是为什么分析乙醇时候峰就很正常!!!!!!!!!!!!!!
三氧化二铝柱能不能进醇类?我想知道丁醇在三氧化二铝柱中的出峰位置,怎么实现?
应用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url]做白酒成分分析时,如何在保证甲醇出峰较好时异丁醇和正丁醇能够分离(恒温条件)
问题描述:本人新买了有机锡的标准品一氯三丁基锡 CISn(C4H9)3,配制成工作液后加内标衍生,衍生产物为乙基三丁基锡C2H5Sn(C4H9)3 (TBT),走GC-MS,发现会出二乙基二丁基锡 (C2H5)2Sn(C4H9)2 (DBT).疑问:1:这个标准是不是存在分解,如果存在,分解发生在那个步骤,是标准配制还是要衍生或是进样口。2:因为多出的目标物(DBT)也是我们的检测目标物,这样就存在定量的问题,TBT偏小。DBT偏高。我把谱图发上来大家看下
[em09] 路过的大哥大姐?有知道异丁醇分析方法的帮忙指点一下!不甚感激!!我用的是戴安公司的HPLC反相柱Diomamoilsil C18主要用什么流动相?ahead35@163.com
DMA做溶剂,却有醇峰,定位炔丁醇和混合溶剂跑出来的峰,混合溶剂里炔丁醇附近有两个峰,是反应了吗
空气与废气样品中挥发性有机物常见的分析方式是活性炭管采样二硫化碳解吸。比如苯系物,丙酮,丁酮,环己酮,正己烷,乙酸甲酯,乙酸乙酯,乙酸丁酯,二氯甲烷,1,2-二氯乙烷,异丙醇,正丁醇。参考的依据有 环境空气苯系物的测定 活性炭吸附/二硫化碳解吸-[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法HJ584-2010,《合成革与人造革工业污染物排放标准》VOCs监测技术导则 GB 21902-2008 附录C, 还有工作场所空气有毒物质测定 GB/T160 系列。[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]多组分分析时尽量使用相同的条件,这样可以尽可能的减少进样而减少分析时间。这次遇到活性炭管样品要求检测甲苯,二甲苯,正丁醇,实验中发现平常的分析条件正丁醇与对二甲苯出峰时间重叠,于是通过摸索分析条件最终成功摸索出低温恒温,高温恒温,程序升温三种分离方法。 平常苯系物分析色谱条件:岛津GC-2010plus带AOC-20i自动进样器,DB-WAX(30m*0.53mm*1.0um),进样口200℃,检测器200℃,柱温恒温80℃,线速度25cm/s,分流比20,进样1ul。正丁醇采用相同的条件。苯系物与正丁醇比较图:黑色为苯系物其中三连峰是乙苯,对二甲苯,间二甲苯,红色为正丁醇与对二甲苯几乎完全重合。[img=,690,357]http://ng1.17img.cn/bbsfiles/images/2017/09/201709281523_01_2103464_3.png[/img] 这个重叠峰分离有些特殊,因为正丁醇前面连着乙苯,后面连着间二甲苯。想分离前移或后移正丁醇峰比较大的距离才有用,之前我用极性柱DB-FFAP分离过二硫化碳中1,2-二氯乙烷与乙酸丁酯,分离异常艰难,所以刚开始我就想这个难度不小,有人说得用60m的DB-WAX,可惜我没有!当然这种分离可以换非极性柱来做,不过我不想换非极性柱,因为平常大多用这根柱子,这次想要发挥这根柱子的潜力。 虽然样品没要求测乙苯,但根据经验乙苯与二甲苯经常共存,于是配制苯,甲苯,乙苯,对二甲苯,间二甲苯,邻二甲苯 ,正丁醇约30ug/ml 混合二硫化碳溶液,另配制正丁醇二硫化碳溶液单标做定性。以下图谱因为节省分析时间只要 乙苯,对二甲苯,间二甲苯,正丁醇出峰 就停止采样,烘烤色谱柱至邻二甲苯出峰再进行下次分析。首先想到降低柱温,降低柱流速来实现分离。分析条件1:柱温改成65℃,柱流速改成17cm/s(以下各条件都是这个流速),发现正丁醇后移与间二甲苯重叠:[img=,690,287]http://ng1.17img.cn/bbsfiles/images/2017/09/201709281443_01_2103464_3.png[/img]这说明这招有效,于是分析条件2:继续减低柱温至55℃:发现正丁醇跑到间二甲苯后面完全分离![img=,690,281]http://ng1.17img.cn/bbsfiles/images/2017/09/201709281448_01_2103464_3.png[/img]峰是分离开了,可是正丁醇出峰时间达26min感觉太慢了,于是想加快分离速度,分析条件3:柱温改成60℃[img=,690,319]http://ng1.17img.cn/bbsfiles/images/2017/09/201709281454_01_2103464_3.png[/img]可见正丁醇与乙苯,对二甲苯,间二甲苯完全分离,正丁醇出峰时间减少至21.5min。这个条件为低温恒温。至此分析条件摸索好了,可是再琢磨下:既然正丁醇降温可以后移,那么升温应该会前移,前移比较多的话可以跑到乙苯前面,这样出峰时间更少,岂不是更好!条件4:柱温改成90℃,这次正丁醇确实是前移了不过和乙苯重叠了四连峰变成了三连峰:[img=,561,355]http://ng1.17img.cn/bbsfiles/images/2017/09/201709281459_01_2103464_3.png[/img]分析条件5:柱温升至100℃,正丁醇跑到乙苯前面完全分离,分析时间大大缩短!此条件为高温恒温。[img=,633,381]http://ng1.17img.cn/bbsfiles/images/2017/09/201709281501_01_2103464_3.png[/img]想到[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]大多使用程序升温那就用程序升温再试试。分析条件6:初温50℃以3℃/min至80℃保持5min,正丁醇与间二甲苯重叠:[img=,690,338]http://ng1.17img.cn/bbsfiles/images/2017/09/201709281507_01_2103464_3.png[/img]分析条件7:初温40℃保持2min以2℃/min升至70℃保持17min,正丁醇跑到间二甲苯后面完全分离!此条件为程序升温。[img=,690,329]http://ng1.17img.cn/bbsfiles/images/2017/09/201709281511_01_2103464_3.png[/img]至此摸索出三种条件都可以分开乙苯,对二甲苯,间二甲苯,正丁醇,分别是低温恒温,高温恒温,程序升温三者的比较:[img=,690,309]http://ng1.17img.cn/bbsfiles/images/2017/09/201709281515_01_2103464_3.png[/img]可见灵敏度是:高温恒温>程序升温>低温恒温 分析时间是:程序升温>低温恒温>高温恒温样品分析:因为样品可能有别的有机物,分析时间不宜太快于是采用程序升温方法,三个峰分别是乙苯,对二甲苯,间二甲苯,而未检出正丁醇:[img=,680,362]http://ng1.17img.cn/bbsfiles/images/2017/09/201709281519_01_2103464_3.png[/img]总结:1.原因分析是通过降低柱温使得DB-WAX与正丁醇相互作用力(主要是偶极力)加强的更显著,所以与乙苯,对二甲苯,间二甲苯比较是后移,反之亦然。 2.摸索出三种方法:低温恒温,高温恒温,程序升温,灵敏度是:高温恒温>程序升温>低温恒温 分析时间是:程序升温>低温恒温>高温恒温(比较到四连峰最后一个峰的出峰时间为止)。低温恒温的好处是基线平整,高温恒温的好处是分析时间非常快,如果样品比较干净是比较适合的,而程序升温适合的样品复杂些。[u][/u]
同为极性柱子,为什么乙醇叔丁醇在PEG20M的柱子上乙醇出峰在前,FFAP的柱子顺序相反呢?
问题描述:本人新买了有机锡的标准品一氯三丁基锡 CISn(C4H9)3,配制成工作液后加内标衍生,衍生产物为乙基三丁基锡C2H5Sn(C4H9)3 (TBT),走GC-MS,发现会出二乙基二丁基锡 (C2H5)2Sn(C4H9)2 (DBT).疑问:1:这个标准是不是存在分解,如果存在,分解发生在那个步骤,是标准配制还是要衍生或是进样口。2:因为多出的目标物(DBT)也是我们的检测目标物,这样就存在定量的问题,TBT偏小。DBT偏高。我把谱图发上来大家看下
正丁醇部位的分离是天然药物化学专业分离较难的部分,但分到有价值的东西几率较大,所以值得我们为之一搏。 一般来说,正丁醇部位常含有单糖、二糖等小分子糖,多种酚类化合物,以及苷类化合物,极性较大,在硅胶柱上吸附较多,成点性差,分离效果不好。因此,一般说来,对于正丁醇部位可采取以下方法: 1. 大孔树脂柱砍段。 样品水溶解后上样,依次用水,30%、60%、95%乙醇溶液洗脱,分别合并、收集。一般说来目标化合物多在60%段,水,30%段多为一些水溶性单糖、二糖及多酚类化合物,可以考虑弃去。而95%部分多和乙酸乙酯部位大极性段重叠,可以乙酸乙酯部位合并处理。 2. 反相柱分段。 如果正丁醇部位量不是太大,或者课题组有大反相柱,可以考虑用反相柱砍段。本课题组就有一根500g的反相柱,专门用于砍段,效果比正相柱好了许多。唯一需要提醒注意的是,由于我们一般是用正相硅胶板检测化合物分离情况,所以反相柱 砍段后往往各组份在硅胶板上看起来比较混乱,不如硅胶柱砍段后那么直观,一定要小心对比,否则会越分越乱。 3.正相柱分配色谱层析。 如果正丁醇部位量太大,或者课题组没有大的相柱用来分段,那么可以考虑氯仿:甲醇:水 体系来砍段。我一般用8:2:1、7:3:1以及6.5:3.5:1依次洗脱,效果不会很好,但两到三次反复上柱后各部分还是可以看得到点的。这时再会反相柱细分,拿十到二十个点应该没问题的。 总体来说,这部分较难,但等你分到好东西了,难也值得,小小经验和大家分享,祝大家实验顺利!
帮忙推荐下:能检测醇类(甲醇、丁醇、丙醇等)、氯代烃、丙烯酸、甲基丙烯酸的柱子?
我想咨询我要用热导一根柱子来分析水中的甲醇或水中的叔丁醇含量的话,我用什么样的柱子呢 什么型号呢?
[em0808] 我做了个酯化反应,正丁醇和乙酸的,然后想用GC-MS检测它们反应进行的程度我用的是外标法,然后刚才结果出来了,连续三个小时内,正丁醇峰面积几乎没有变化,但是产物确实越来越多,怎么会这样?理论上正丁醇峰也该减少才是。另外,我在做标准样品时,用的配比就是反应初始配比5ml:32ml=乙酸:正丁醇。但是这个标样的峰面积也会变化,上周测的都在4,000,000左右,这周测的几个都在4,650,000左右。。。这是为什么呢?
有机化工产品精馏过程副产有丁醇水溶液,取样发现取样瓶内很快就分层了,除了水,丁醇外,还有少量甲醇杂质。[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]有啥办法准确定量其中的丁醇含量。
安捷伦7890A,顶空自动进样器,19091S-510色谱柱检测乙醇,叔丁醇內标,以前都正常乙醇保留时间为1.2分钟,叔丁醇3.3分钟,现在突然叔丁醇0.6分钟就出峰了,请问一下是什么原因呢?
(应助版友在其他版面的标准求助)GB 10618-1989 食品添加剂 正丁醇1990-02-01实施,改号调整为HG 2926-1989。HG 2926-1989(1997年确认) 食品添加剂 正丁醇1990-02-01实施,现行有效。[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=118515]GB 10618-1989 食品添加剂 正丁醇(调整为HG 2926-1989)[/url]GB 11962-1989 食品添加剂 丁酸1990-09-01实施。改号调整为QB/T 2796-2006。QB/T 2796-2006 食品添加剂 丁酸2006-10-11实施,现行有效。[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=118516]GB 11962-1989 食品添加剂 丁酸(调整为QB/T 2796-2006)[/url]
[align=center][b]GB 22255-2014 食品安全国家标准 食品中三氯蔗糖(蔗糖素)的测定——三氯蔗糖标准品分析-RI[/b][/align]三氯蔗糖(TGS),是唯一以蔗糖为原料的功能性甜味剂,甜度可达蔗糖600倍。这种[url=http://baike.sogou.com/v130009.htm][color=windowtext]甜味剂[/color][/url]具有无能量,甜度高,甜味纯正,高度安全等特点,是最优秀的功能性甜味剂之一。[align=center][img=,170,99]http://ng1.17img.cn/bbsfiles/images/2018/03/201803080920210187_4197_2222981_3.png!w170x99.jpg[/img][/align][align=center]三氯蔗糖结构式[/align]实验室前期按照《GB 22255-2014 食品安全国家标准食品中三氯蔗糖(蔗糖素)的测定》方法,使用高灵敏度气溶胶型检测器——纳克级水凝粒子计数检测器(NQAD),得到了三氯蔗糖标准品的良好分析结果。本实验按照相同条件,使用示差折光检测器(RI)对三氯蔗糖标准品进行分析。色谱柱同样选择中等极性的普适型色谱柱CAPCELL PAK C[sub]18 [/sub]MGII S5 4.6 mm i.d. × 150 mm,得到结果如图1所示。三氯蔗糖保留时间为12.400min,与标准谱图保留时间基本一致,理论塔板数为12350,不对称因子为0.95,峰形良好。[align=center][img=,690,489]http://ng1.17img.cn/bbsfiles/images/2018/03/201803080945469257_8172_2222981_3.png!w690x489.jpg[/img][/align][align=center]图1 三氯蔗糖标准品分析色谱图(0.4 mg/mL)[/align]*注:峰上标数字由下至上依次为保留时间、理论塔板数及不对称因子。[img=,472,187]http://ng1.17img.cn/bbsfiles/images/2018/03/201803080945471937_6640_2222981_3.png!w472x187.jpg[/img][align=center][img=,690,435]http://ng1.17img.cn/bbsfiles/images/2018/03/201803080946205953_7240_2222981_3.png!w690x435.jpg[/img][/align][align=center]附图:GB方法中标准色谱图[/align]接下来,按照国标要求配制三氯蔗糖工作液,0.02 mg/mL、0.05 mg/mL、0.1 mg/mL、0.2 mg/mL、0.4 mg/mL,进行线性考察实验。线性实验结果如图2所示,R[sup]2[/sup]=0.9939,得到良好线性结果。同时,由于低浓度0.02 mg/mL、0.05 mg/ mL标准品溶液均未检出色谱峰,因此根据标准曲线最高浓度的信噪比计算出检出限(以S/N=3计)约为0.17 mg/ mL。[align=center][img=,650,398]http://ng1.17img.cn/bbsfiles/images/2018/03/201803080947051037_4812_2222981_3.png!w650x398.jpg[/img][/align][align=center]图2 三氯蔗糖标准曲线图[/align]综上,按照《GB 22255-2014 食品安全国家标准食品中三氯蔗糖(蔗糖素)的测定》方法,使用示差检测器(RI)进行检测,以及CAPCELL PAK C[sub]18[/sub] MGII S5 4.6 mm i.d. ×150 mm色谱柱进行分析,可得到三氯蔗糖标准品的良好线性分析结果;但RI检测器的检测灵敏度较低。
[align=center][b]GB 22255-2014 食品安全国家标准 食品中三氯蔗糖(蔗糖素)的测定——三氯蔗糖标准品分析-NQAD检测器[/b][/align]三氯蔗糖(TGS),是唯一以蔗糖为原料的功能性甜味剂,甜度可达蔗糖600倍。这种甜味剂具有无能量,甜度高,甜味纯正,高度安全等特点,是最优秀的功能性甜味剂之一。[align=center][img=,170,99]http://ng1.17img.cn/bbsfiles/images/2018/03/201803011004470313_2453_2222981_3.png!w170x99.jpg[/img][/align][align=center]三氯蔗糖结构式[/align]本实验按照[b]《GB 22255-2014 食品安全国家标准 食品中三氯蔗糖(蔗糖素)的测定》[/b]方法,使用[b][color=#ff0000]高灵敏度气溶胶型检测器——纳克级水凝粒子计数检测器(NQAD)[/color][/b]对三氯蔗糖标准品进行了分析。色谱柱选择中等极性普适型[color=#3333ff][b]CAPCELL PAK C18 MGII S5 4.6 mm i.d. × 150 mm[/b][/color],得到结果如图1所示。三氯蔗糖保留时间为12.709min,[b]与标准谱图保留时间基本一致,理论塔板数为9992,不对称因子为1.06,峰形良好。[/b][align=center][b][img=,690,497]http://ng1.17img.cn/bbsfiles/images/2018/03/201803011006155125_1559_2222981_3.png!w690x497.jpg[/img][/b][/align][align=center]图1 三氯蔗糖标准品分析色谱图[/align]*注:峰上标数字由下至上依次为保留时间、理论塔板数及不对称因子。[b][img=,633,176]http://ng1.17img.cn/bbsfiles/images/2018/03/201803011006367633_3986_2222981_3.png!w633x176.jpg[/img]附图:GB方法中标准色谱图[/b][align=center][b][img=,690,448]http://ng1.17img.cn/bbsfiles/images/2018/03/201803011007162573_9264_2222981_3.png!w690x448.jpg[/img][/b][/align][b][/b]接下来,按照国标要求配制三氯蔗糖工作液,浓度分别为0.02 mg/mL、0.05 mg/mL、0.1 mg/mL、0.2 mg/mL、0.4 mg/mL,进行线性考察实验。[b][color=#3333ff]由于NQAD检测器原理与常规蒸发光散射检测器ELSD不同,能够直接得到线性回归结果,不需要做对数方程,更加简单快捷。[/color][/b]线性结果如图2所示,R[sup]2[/sup]=0.996,得到良好线性结果。同时,我们根据标准曲线最低浓度的信噪比计算出定量限(以S/N=10计)约为3 μg/mL,[b][color=#ff0000]能够实现三氯蔗糖的高灵敏度检出[/color][/b]。[align=center][img=,658,399]http://ng1.17img.cn/bbsfiles/images/2018/03/201803011008425185_5014_2222981_3.png!w658x399.jpg[/img][/align][align=center]图2 三氯蔗糖标准曲线图[/align]
[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]校验时精密度太差是什么原因,FID检测器,PEG-20M柱,标准品异丁醇浓度1mg/ml







 我要推广仪器
我要推广仪器
 下载APP
下载APP