盐酸吗啡是否可以不用衍生化,用OV-17柱子直接进样检测?
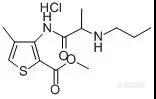
[color=black]复方盐酸阿替卡因注射液为复方制剂,是盐酸阿替卡因与肾上腺素的灭菌水溶液,作为口腔用局部麻醉剂,适用于涉及切骨术及粘膜切开的外科手术过程。[/color][color=black] [/color][img=,156,99]https://ng1.17img.cn/bbsfiles/images/2019/03/201903211036489419_4502_2297_3.jpg!w156x99.jpg[/img][align=center][/align][align=left][b][color=black]盐酸阿替卡因(Articaine hydrochloride M.W.:320.84)[/color][/b][/align][align=center][b][color=black] [/color][/b][/align][color=black]在现有国家药品标准(YBH17082004-2015Z)分析方法中,流动相添加了离子对试剂-庚烷磺酸钠,并在pH为2.0的强酸条件下进行相应分析,不利于色谱柱的使用寿命。大曹三耀实验室参考USP方法,以冰醋酸水溶液-乙腈作为流动相,选用CAPCELL PAK C18 MGII色谱柱,实现了复方盐酸阿替卡因注射液中盐酸阿替卡因的定量和有关物质的良好分析(复方盐酸阿替卡因注射液由客户提供)。[/color][color=black]CAPCELLPAK C18 MGII[/color][color=black]液相色谱柱,其采用高纯度硅胶作为基质,通过减少硅胶微细孔的数量来增大有效比表面积;并且采用新包被技术Ultimate Polymer Coating,实现了对硅醇基极大程度的封锁,兼具分离性能和普适性能,通用性非常好。[/color][align=left][b][color=#0070c0]实验方法[/color][/b][/align][align=left][img=,500,358]https://ng1.17img.cn/bbsfiles/images/2019/03/201903211037456969_5082_2297_3.jpg!w730x523.jpg[/img][/align][align=left]图1[color=black]盐酸阿替卡因[/color]对照品及供试品溶液[/align][align=left][img=,500,248]https://ng1.17img.cn/bbsfiles/images/2019/03/201903211038541919_2603_2297_3.jpg!w572x284.jpg[/img][/align][align=center][/align][align=center][/align][color=black]为进行有关物质分析,该实验将注射液样品以流动相稀释100倍,作为有关物质供试品溶液,再将该有关物质供试品溶液以流动相进一步稀释100倍,作为自身对照溶液。以冰醋酸水溶液-乙腈作为流动相,选用CAPCELL PAK C18 MGII色谱柱,通过调整流动相比例及柱温,最终在18%乙腈、柱温30℃条件下实现了盐酸阿替卡因供试品溶液及对照品的良好分析。[/color]如图2、3,使用CAPCELL PAK C18 MGII色谱柱进行分析,盐酸阿替卡因和有关物质均能得到良好分析结果,主峰与峰前杂质得到了良好分离,分离度为1.90(见表1)[img=,400,311]https://ng1.17img.cn/bbsfiles/images/2019/03/201903211045536855_9516_2297_3.jpg!w574x447.jpg[/img][img=,400,295]https://ng1.17img.cn/bbsfiles/images/2019/03/201903211045540875_8483_2297_3.jpg!w698x516.jpg[/img][align=left] 图2 [color=black]盐酸阿替卡因[/color]有关物质供试品溶液及空白 图3 自身对照溶液[/align][align=center][/align][align=left]表1 有关物质结果详表[/align][align=left][img=,600,323]https://ng1.17img.cn/bbsfiles/images/2019/03/201903211042513275_6690_2297_3.jpg!w786x424.jpg[/img][/align][align=center][/align]综上实验结果,使用CAPCELL PAK C18 MGII S5 4.6mm i.d.×250 mm色谱柱,以冰醋酸水溶液-乙腈为流动相体系,在30°C柱温条件下,能够实现复方盐酸阿替卡因注射液中盐酸阿替卡因的定量和有关物质的良好分析。[color=black] [/color]
请问哪有超纯的硫酸,盐酸,硝酸,乙酸等及光谱纯金属卖?
做个试验 想要盐酸贝那普利的对照品图,还有说两个光谱一致的依据是什么,全波数都对上? 包括指纹区?
单位考试题1杯是色谱纯的盐酸,1杯是分析纯的硝酸由于倒出来时候忘记了写标签,请问你用什么方法去辨认?
我现在在做中药,其中要检测吗啡和罂粟碱的含量,问题是流动相一直没有找到好的组合,另外对照品只有盐酸罂粟碱,不知道盐酸罂粟碱和罂粟碱的出峰时间差多大,希望各位高手i能帮忙给个好建议
[color=#444444]大家好,我最近做盐酸四环素的标准曲线,标品溶于甲醇中,用液相色谱分析,流动相为甲醇-乙腈-0.01mol/L草酸水溶液,配比有待优化。我参考相关文献对配比调试了多次,但是10mg/L浓度的盐酸四环素甲醇溶液的响应值最高峰高才11.5!很多配比下都是4或者5的响应值。按道理响应值应该是几十几百才对!根本问题出在哪?急!!!![/color]

10,抽取5个版友);中奖名单:zgx3025(注册ID:v2844608)吕梁山(注册ID:shih20j07)大川之子,纵横四海(注册ID:chuangu120)dahua1981(注册ID:dahua1981)捌道巴拉巴巴巴(注册ID:v3082413)http://ng1.17img.cn/bbsfiles/images/2016/12/201612071513_01_1610895_3.jpghttp://ng1.17img.cn/bbsfiles/images/2016/12/201612071513_02_1610895_3.jpg【注意事项】同样的答案,每人只能发一次PS:该贴浏览权限为“回贴仅作者和自己可见”,回复的版友仅能看到版主的题目及自己的回答内容,无法看到其他版友的回复内容。下午3点之后解除,即可看到正确答案、获奖情况及所有版友的回复内容。=======================================================================复方甘草片中的吗啡含量测定方法:SPE基质:药品应用编号:101793化合物:吗啡固定相:ProElut C18-U色谱柱/前处理小柱:ProElut C18-U 1000mg/6ml 30/pkg样品前处理:样品制备:取固相萃取柱(ProElut C18-U 1 g/6 mL Cat#:63506) 一支,依次用甲醇-水(3:1)15 ml及水5 ml冲洗,再用pH值约为9的氨水溶液(取水适量滴加氨试液至pH值约为9)冲洗至流出液pH值约为9(约10 mL),待用。取本品20片,精密称定,研细,精密称取约10片量,置磨口锥形瓶中,精密加水90ml,超声处理5分钟,精密加稀盐酸(6→10)10ml,摇匀,超声处理20分钟,使吗啡溶解,取出,放至室温,滤过,精密量取续滤液1.0ml,置上述固相柱上,滴加氨试液(300 μL)适量使柱内溶液的pH值约为9(上样前应取同体积续滤液预先调试,以确定滴加氨试液的量),摇匀,待溶剂滴尽后,用水约20ml冲洗,用含2%甲醇的5%醋酸溶液洗脱,用5ml量瓶收集洗脱液至刻度,摇匀,即得。对照品配制:另取吗啡对照品适量,精密称定,用含2%甲醇的5%醋酸溶液溶解并定量稀释制成每1ml中约含吗啡0.01mg的溶液,同法测定。色谱条件:分析条件 色谱柱:Inspire C8,250×4.6 mm,5 μm (Cat#:81106) 流动相:0.05mol/L KH2PO4:0.0025庚烷磺酸钠水溶液:乙腈=5:5:2 流 速:1.0 mL/min 柱 温:30 ℃ 检测器:UV 220 nm 进样量:10 μL文章出处:天津迪马实验室关键字:复方甘草片中,吗啡,2010药典,Inspire C8,81106,ProElut C18-U,63506摘要:复方甘草片中的吗啡含量测定谱图:http://www.dikma.com.cn/Public/Uploads/images/mafei(1).PNG图例:吗啡
借用xuanleer的帖子提一下几个疑问:不知道从何说起,我们在做某些碱性化合物检测时,购买的标准品通常是盐酸盐、硫酸盐、草酸盐之类的,如下面的糠氨酸(二盐酸盐)以及莱克多巴胺盐酸盐、四环素盐酸盐等等,疑问:(1)想问下大家这类目标化合物在进(HPLC、GC)色谱分析时,(色谱峰)是以游离碱的形式(糠氨酸、莱克多巴胺、四环素)存在还是以游离碱盐酸盐的形式存在?(2)这类化合物不少选用酸性环境下进行HPLC/LC-MS分析,其原因是否是让目标化合物以游离碱的形式(糠氨酸、莱克多巴胺、四环素)存在? (3) 如果第(2)个问题是对的,哪进GC分析时得到色谱峰是以何种形式存在的?也是游离碱吗?欢迎各位老师专家解答啊!顺祝大家节日快乐!参考资料如下:糠氨酸的鉴定适用于《NYT 939-2005 复原乳的鉴定》,具体见附件。色谱柱:LAEQ-462572 CNW Athena C18-WP 液相色谱柱,4.6*250mm,5um流动相:A=0.1%三氟乙酸水溶液;B=0.1%三氟乙酸乙腈平衡:A:B=99:1梯度:0min:99%A/1%B,25min:79%A/21%B检测波长:280nm流速:1ml/min进样浓度:2ppm柱温:室温标准品:CDDD-SC494-10MG,糠氨酸(二盐酸盐),品牌 NeoMPS,现货供应。参考:http://bbs.instrument.com.cn/shtml/20120903/4223471/
借用xuanleer的帖子提一下几个疑问:不知道从何说起,我们在做某些碱性化合物检测时,购买的标准品通常是盐酸盐、硫酸盐、草酸盐之类的,如下面的糠氨酸(二盐酸盐)以及莱克多巴胺盐酸盐、四环素盐酸盐等等,疑问:(1)想问下大家这类目标化合物在进(HPLC、GC)色谱分析时,(色谱峰)是以游离碱的形式(糠氨酸、莱克多巴胺、四环素)存在还是以游离碱盐酸盐的形式存在?(2)这类化合物不少选用选型环境下进行HPLC/LC-MS分析,其原因是否是让目标化合物以游离碱的形式(糠氨酸、莱克多巴胺、四环素)存在? (3) 如果第(2)个问题是对的,哪进GC分析时得到色谱峰是以何种形式存在的?也是游离碱吗?欢迎各位老师专家解答啊!顺祝大家节日快乐!参考资料如下:糠氨酸的鉴定适用于《NYT 939-2005 复原乳的鉴定》,具体见附件。色谱柱:LAEQ-462572 CNW Athena C18-WP 液相色谱柱,4.6*250mm,5um流动相:A=0.1%三氟乙酸水溶液;B=0.1%三氟乙酸乙腈平衡:A:B=99:1梯度:0min:99%A/1%B,25min:79%A/21%B检测波长:280nm流速:1ml/min进样浓度:2ppm柱温:室温标准品:CDDD-SC494-10MG,糠氨酸(二盐酸盐),品牌 NeoMPS,现货供应。参考:http://bbs.instrument.com.cn/shtml/20120903/4223471/
我的样品需要用盐酸进行处理再进样,可是发现盐酸竟然存在两个色谱峰(色谱柱是碳18柱,流动相为乙氰:水=10:90),正好严重干扰了我的样品峰。我配了好几种不同纯度的(分析纯,优级纯,工艺超纯)pH=1.5的盐酸,都有这两个峰。不知道这两个峰是什么物质?有没有纯度更好的盐酸。或者该怎样纯化盐酸?
采购光谱仪的参考标准有哪些?
参考美国药典液相色谱条件,用YMC-Triart C8色谱柱测定盐酸奥洛他定滴眼液的含量,系统适应性试验中理论塔板数、拖尾因子、相对标准偏差等均符合规定。
看到饲料中盐酸克伦特罗的检测,与大家分享! 饲料中盐酸克伦特罗的检测1.盐酸克伦特罗准确定性定量产品配置采用SPE-LC对饲料中盐酸克伦特罗准确定性定量,参考配置如下: 仪器配置:P1201等度系统基本配置 色谱柱:Hypersil BDS C18色谱柱(4.6×250mm,5µm) 固相萃取柱: Thermo HyperSep Retain CX(3ml,200mg)参考前处理设备 超声波清洗器 离心机 氮吹仪 酸度调节计2.样品处理准确称取饲料5g左右,准确加入0.5%偏磷酸50mL,超声提取15min,每5min取出振摇一次,超声结束后手摇10s,并取上清液以4000r/min离心10min,取上清液10mL置分液漏斗中滴加氢氧化钠溶液,充分振摇,调pH值至12左右,溶液用3-mL乙醚萃取两次,萃取液用无水硫酸钠干燥,于50℃下干燥,残渣用0.01mol/L盐酸溶液2mL溶解,待净化。3.样品净化依次用3mL甲醇、3mL超纯水和3mL 0.5mol/L高氯酸润洗固相萃取小柱,取待净化液注入SPE小柱中,依次用3mL超纯水、3mL甲醇淋洗,用3mL 5%的氨化甲醇进行洗脱,收集液用氮气吹干,残渣用40:60的甲醇和0.05%磷酸混合溶液溶解,过滤,即可。4.样品检测将净化的样品溶液注入液相色谱仪,在243nm下检测,谱图见图1。
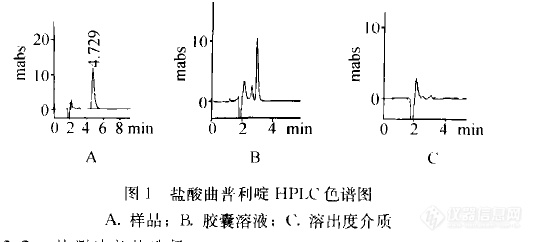
【作者中文名】黎志芳;【作者英文名】Li Zhi-fang (Guangzhou Institute for Drug Control; Guangzhou 510160);【作者单位】广州市药品检验所 广州;【摘要】目的 用HPLC法测定盐酸曲普利啶胶囊中盐酸曲普利啶的溶出量。方法 以Diamonsil C18柱(150 mm×4.6 mm,5μm)为固定相,流动相为甲醇-0.4%醋酸铵溶液(含0.15%三乙胺,用醋酸调pH 7.0)(70:30),流速为1.0 mL·min-1,检测波长为278 nm,进样量40μL,采用外标法定量。结果 盐酸曲普利啶线性范围为1.672-8.363μg·mL-1(r=0.999 9),回收率为100.0%。结论 本方法结果准确,重现性良好,操作简便。用 于测定盐酸曲普利啶胶囊中盐酸曲普利啶的溶出量,较原方法更合理、准确、简便。http://ng1.17img.cn/bbsfiles/images/2012/08/201208061427_381888_2379123_3.jpg
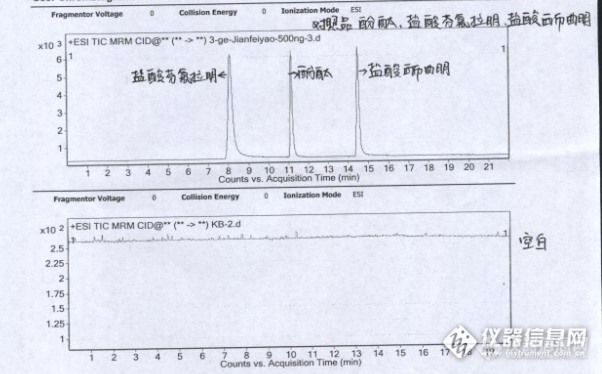
减肥产品中酚酞、盐酸西布曲明及盐酸芬氟拉明的检测盐酸西布曲明,盐酸芬氟拉明,酚酞已被国家食品药品监督管理总局明令禁止用于食品(含保健品)了,目前还有部分违规企业在减肥产品中违法添加。盐酸西布曲明曾为处方药,但目前已在全球大多数国家停止使用。盐酸西布曲明(Sibutramine Hydrochloride)是西布曲明(Sibutramine)的氯化物,是一种中枢神经抑制药物,曾用于肥胖症的治疗。酚酞是化学品和临床处方药,有严格的适应症,需在医生指导下应用,若长期过量服用可能引发严重的副作用。在制药上作为医药原料,其药品名称为酚酞片(Phenolphthalein Tablets),主要用于治疗习惯性、顽固性便秘。过量或长期滥用,可造成人体电解质代谢紊乱,严重时甚至可诱发心律失常。婴儿和哺乳期妇女禁用,幼儿和孕妇慎用。市场上抽检的三批次的减肥产品违法添加盐酸西布曲明,酚酞的检测:仪器型号及编号 Agilent 1200 LC/MS 6410B 天平型号及编号 BP211D,LD310-2 色谱条件:色谱柱:phenomenex C18柱(100x3.0 mm,2.6 μm) 预柱 Agilent 预柱流速(mL/min) 0.2 进样量(uL) 5 柱温(℃) 25℃ http://ng1.17img.cn/bbsfiles/images/2017/01/201701191701_668681_2166779_3.png质谱条件:电喷雾离子化源(ESI) 碰撞气压力(Mpa) 0.15 Nebulizerpressure(Psi) 15 drying GasFlow(L/min) 6 Dry Temp(℃) 350 电离源 ESI ,正离子模式 http://ng1.17img.cn/bbsfiles/images/2017/10/2016071708440193_01_0_3.pnghttp://ng1.17img.cn/bbsfiles/images/2017/10/2016071708440482_01_2166779_3.png将标准品分别配制成1mg/mL的酚酞,盐酸芬氟拉明,盐酸西布曲明标准储备液,分别吸取标准储备溶液进行稀释,得到100ng/mL,80ng/mL,50ng/mL,20ng/mL,10ng/mL,5ng/mL的标准工作溶液。2.标准曲线的制作取各标准工作溶液5 uL注入液质仪,采集数据。以峰面积为纵坐标(Y),以标准工作溶液浓度(X)为横坐标绘制标准曲线。酚酞,盐酸芬氟拉明,盐酸西布曲明标准品及空白的色谱图、质谱图及工作曲线:http://ng1.17img.cn/bbsfiles/images/2017/10/2016071717420205_01_0_3.pnghttp://ng1.17img.cn/bbsfiles/images/2017/10/2016071717421213_01_0_3.pnghttp://ng1.17img.cn/bbsfiles/images/2017/10/2016071717422190_01_2166779_3.pnghttp://ng1.17img.cn/bbsfiles/images/2017/10/2016071717433884_01_0_3.pnghttp://ng1.17img.cn/bbsfiles/images/2017/10/2016071717434225_01_2166779_3.png3.试样提取取各试样适量(约相当于一次用量),置50mL离心管中,精密加入甲醇20mL,超声处理15min,放冷至室温,10000r/min离心5min,取上清液用50%甲醇稀释。稀释过程:①0.2→2.0ml (稀释10倍);②0.1→2.0ml(共稀释200倍); ③0.1→2.0ml(共稀释4000倍)③0.1→2.0ml(共稀释80000倍)样品1、2、3号的酚酞,盐酸西布曲明,盐酸芬氟拉明色谱图http://ng1.17img.cn/bbsfiles/images/2016/07/201607171745_600844_2166779_3.png样品1号酚酞,盐酸西布曲明,盐酸芬氟拉明的质谱图http://ng1.17img.cn/bbsfiles/images/2016/07/201607171745_600845_2166779_3.pnghttp://ng1.17img.cn/bbsfiles/images/2016/07/201607171745_600846_2166779_3.pnghttp://ng1.17img.cn/bbsfiles/images/2016/07/201607171746_600847_2166779_3.png从图中可见1号未检出盐酸芬氟拉明;2、3号样品的质谱图略。定量分析 酚酞的含量(mg/粒)= C样×V样 ×样品稀释倍数×W平 /W样×10-6 供试品编号 10粒内容物装量(g) 平均装量(g) 取样量 (g) 检测结果(ng/mL) 含量 (mg/粒) 平均含量 (mg/粒) 1号 2.6327 0.2633 0.2692 16.5391 25.88 27.8 0.2623 18.5693 29.82 2号 2.4988 0.2499 0.2691 15.0804 22.41 [align=c

今天将为大家介绍两个使用资生堂色谱柱对盐酸小檗碱进行分析的文献数据,请参考。盐酸小檗碱(Berberine Hydrochloride)分子式:C20H18ClNO4·2H2O 分子量:407.85黄色结晶性粉末;无臭,味极苦。在热水中溶解,在水或乙醇中微溶,在三氯甲烷中极微溶解,在乙醚中不溶。对痢疾杆菌、大肠杆菌、肺炎双球菌、金葡菌、链球菌、伤寒杆菌及阿米巴原虫有抑制作用。临床主要用于肠道感染及菌痢等。还发现本品有抗心律失常的作用。 小檗碱有较强的体内外抗肿瘤活性并能诱导B16细胞分化;同盐酸阿糖胞苷在体外具有协同作用。(一)加味左金丸 http://ng1.17img.cn/bbsfiles/images/2017/03/201703021026_01_2222981_3.png【色谱条件】色谱柱:CAPCELL PAK C18 S5; 4.6mm i.d.×250 mm流动相:乙腈/0.05mol/L磷酸二氢钠溶液(含0.2%的三乙胺,用磷酸调pH值为3.0)=27/75流 速:1.0mL/min温 度:40℃检 测:UV263nm进样量:10μL*摘自:中国医药指南,2013年5月,第11卷,第14期,401-402 (二)三黄片 http://ng1.17img.cn/bbsfiles/images/2017/03/201703021026_02_2222981_3.png【色谱条件】色谱柱:CAPCELL PAK C18 MGII S5; 4.6 mm i.d.×150 mm流动相:乙腈/水(每1000ml流动相中含磷酸二氢钾3.4g,十二烷基硫酸钠1.7g)=1/1流 速:1.0mL/min检 测:UV265nm注:文献中所用液相方法与2015年版《中国药典》中三黄片检测方法一致。*摘自:药物研究,2013年,第7期,26-28
[color=#444444]最近在做含量分析的物质本身没有紫外吸收,于是用2-羟基-1-萘甲醛进行衍生化,目前还在摸条件。[/color][color=#444444]文献说要在酸性条件下反应,所以之前做的反应液中都是有盐酸的,最后溶液pH>2就没有再处理,进样之后找到了衍生剂峰和衍生产物峰。[/color][color=#444444]前两天做条件优化实验的时候,脑洞开开,把盐酸去掉了,结果发现色谱图上衍生剂的峰之前很近的位置多出了一个峰。[/color][color=#444444]一开始以为是发生了副反应,结果进了一针衍生剂发现本来就有两个峰,而溶液里加点盐酸,前一个峰就消失了。[/color][color=#444444]现在估计是衍生剂降解了,就是觉得挺神奇的,为什么加了盐酸会有这样的现象呢?一时也没有条件做质谱,暂不能确定出的峰都是什么情况,不知道有没有人遇到过类似的问题?跪求高人解答啊~[/color][color=#444444]色谱的话就是ODS柱,80%甲醇水做流动相,没有用盐,紫外检测(最近只有可变波长检测器available,之前用PDA找最大吸收波长的时候也没发现这个问题= =)。[/color][color=#444444]PS:想了一下,会不会是和羟基氢的解离有关啊?[/color]
我用盐酸邻菲罗啉显色测定污水中铁的含量数值和[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收光谱[/color][/url]测定的结果相差好大,有几十倍的区别,可见光测的是1.8%,原子测得是0.07%,个人认为简单的六价铬、二价铜、钴离子,镍离子的干扰都不会造成这样大的误差。而我的标准曲线做得还不错,试剂无混浊,显色也明显,就是不明原因。因为此事,我还被领导批评工作不认真,唉。请教各位高手!可能出现的原因都跟我说说,请教请教!感激不尽
我用盐酸邻菲罗啉显色测定污水中铁的含量数值和[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收光谱[/color][/url]测定的结果相差好大,有几十倍的区别,可见光测的是1.8%,原子测得是0.07%,个人认为简单的六价铬、二价铜、钴离子,镍离子的干扰都不会造成这样大的误差。而我的标准曲线做得还不错,试剂无混浊,显色也明显,就是不明原因。因为此事,我还被领导批评工作不认真,唉。请教各位高手!可能出现的原因都跟我说说,请教请教!感激不尽
直读光谱仪的检定规程参考哪个标准?是指仪器不是样品。
众所周知,目前新的光谱仪器层出不穷,而相关行业的国标却未与时俱进。采购光谱仪器之前,需要参考相关国标吗?检测机构,会不会因为采用了新的(国标中没提到的)检测仪器出据检测报告而惹上官司呢?
用的NY1110-2006标准水溶肥料汞砷铬铅铬的限量及其含量测定买了三种样品。是尿素、有机肥、复合肥1.是否可用这个标准来测定铬镉铅的含量?2.标准中配标液的时候要加4mL盐酸,用水定容,一开始没加盐酸,后来加入盐酸后吸光度前后有很大差异,是否需要加盐酸?3,在GB18877-2002有机-无机复混肥料中,标液是直接用盐酸定容的,会不会有很大的差异?4.加入4mL盐酸后测得标曲后,测空白,与样品的时候会有负值出现。空白样都是负值,测Cd,得出的浓度都是负的,有的比空白大有的小,是什么情况?谢谢!!!
建立盐酸非索非那定有关物质检查方法。方法: ODS C18柱为分离柱,0.01mol/L的KH2PO4 (pH3.5)∶甲醇(40∶60)为流动相,检测波长为220nm。结果 盐酸非索非那定与有关物质能有效地分离,有关物质的最小检出限是0.2μg/mL。结论:本法快捷、简便、准确,能很好地控制非索非那定的质量。
请问各位:如何得到氘代盐酸溶液的红外谱图数据?谢谢
有没有同仁做盐酸小檗碱的有关物质的?发图扑来看看啊?
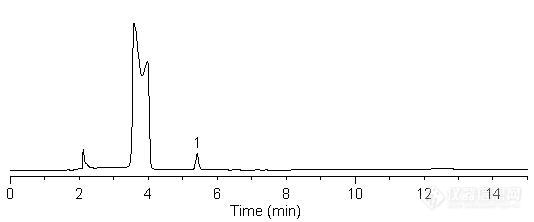
复方甘草片中的吗啡含量测定DM-93样品制备 制备方法: 吗啡含量测定:样品制备:取固相萃取柱(ProElut C18-U 1 g/6 mL) 一支,依次用甲醇-水(3:1)15 ml及水5 ml冲洗,再用pH值约为9的氨水溶液(取水适量滴加氨试液至pH值约为9)冲洗至流出液pH值约为9(约10 mL),待用。取本品20片,精密称定,研细,精密称取约10片量,置磨口锥形瓶中,精密加水90ml,超声处理5分钟,精密加稀盐酸(6→10)10ml,摇匀,超声处理20分钟,使吗啡溶解,取出,放至室温,滤过,精密量取续滤液1.0ml,置上述固相柱上,滴加氨试液(300 μL)适量使柱内溶液的pH值约为9(上样前应取同体积续滤液预先调试,以确定滴加氨试液的量),摇匀,待溶剂滴尽后,用水约20ml冲洗,用含2%甲醇的5%醋酸溶液洗脱,用5ml量瓶收集洗脱液至刻度,摇匀,即得。对照品配制:另取吗啡对照品适量,精密称定,用含2%甲醇的5%醋酸溶液溶解并定量稀释制成每1ml中约含吗啡0.01mg的溶液,同法测定。 分析条件 色谱柱:Inspire C8,250×4.6 mm,5 μm (Cat#:81106)流动相: 0.05mol/L KH2PO4:0.0025庚烷磺酸钠水溶液:乙腈=5:5:2 流速: 1.0 mL/min 柱温: 30 ℃ 检测器: UV 220 nm 进样量: 10 μL 吗啡对照: http://ng1.17img.cn/bbsfiles/images/2012/06/201206151459_372651_2370618_3.jpg1-吗啡 峰号 保留时间 min 峰面积μV*s 峰高mV 理论塔板数N USP拖尾因子 分离度 1 5.349 581405 94977 14928.574 1.141 复方甘草片样品: http://ng1.17img.cn/bbsfiles/images/2012/06/201206151459_372652_2370618_3.jpg1-吗啡 &#
买的是盐酸吗啡,只溶于水,咋整啊,还要做线性方程?
罂粟蒴果中可待因、吗啡、罂粟碱的GC-MS检验罂粟含有丰富的吗啡等吗啡型生物碱,为一年生草本,有乳状液体,茎自基部分枝,高60-120cm ,有白霜,无毛,下部叶有叶柄,上部叶抱茎,心脏形,有不对称粗齿,蒴果球形,直径2.5-5cm ,花期4-5月,果期6-8月。随着我国禁毒工作的不断深入,在铲除非法种植dupin原植物的同时,在法庭科学上,须对这类植物进行相关生物碱的检验。本文介绍了利用GC-MS对罂粟蒴果中可待因、吗啡、罂粟碱进行检测,方法简单。1 实验部分1.1仪器与试剂Varin3900 /2100GC-MS. 甲醇,丙酮,无水硫酸钠,均为分析纯。三种标准品均从公安部物证鉴定中心购得。1.2实验条件气相色谱条件:DB-5 30m X0.25mmX0.32 um 弹性石英毛细管柱;柱温:80℃(1min)20℃/min280℃(19min);进样口温度:250℃;分流比40:1;载气:高纯氦;进样量1ul.质谱条件:传输线温度240℃,离子阱温度150℃,EI源(70eV),扫描范围40-550amu.1.3检材的提取与净化取检材2-5克,加10克无水硫酸钠研磨,以丙酮提取,过滤,水浴或微波挥干,1ml甲醇溶解待检。2 结果与讨论该法中,罂粟蒴果中可待因、吗啡、罂粟碱的保留时间分别为12.33min、12.70min、16.26min,离子碎片峰依次为:可待因m/z299、162、124;吗啡285、162、215、124;罂粟碱339、324、308、292、266等。
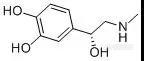
复方盐酸阿替卡因注射液为复方制剂,是盐酸阿替卡因与肾上腺素的灭菌水溶液,作为口腔用局部麻醉剂,适用于涉及切骨术及粘膜切开的外科手术过程。[img=,144,61]https://ng1.17img.cn/bbsfiles/images/2019/03/201903191545418661_4518_2222981_3.jpg!w144x61.jpg[/img][color=black] [/color][color=#3e3e3e]肾上腺素 L(-)-Epinephrine M.W. : 183.2 [/color][b]在国家药品标准(YBH17082004-2015Z)[/b]中,在对复方盐酸阿替卡因注射液中肾上腺素进行分析时,使用[b]甲醇-水[/b]进行梯度洗脱,但由于[b]肾上腺素极性较强[/b],即使初始梯度为纯水相条件,肾上腺素仍紧邻死时间出峰,[b]保留不佳,易受到溶剂峰干扰,无法进行准确定量。[/b]我们分别尝试使用反相柱CAPCELL PAK C18 MGII加离子对试剂,以及直接使用离子交换色谱柱CAPCELL PAK SCX UG80两种方式,对复方盐酸阿替卡因注射液中肾上腺素和硫酸肾上腺素进行保留分析(复方盐酸阿替卡因注射液由客户提供)。CAPCELL PAK C18 MGII液相色谱柱,其采用高纯度硅胶作为基质,通过减少硅胶微细孔的数量来增大有效比表面积;并且采用新包被技术Ultimate Polymer Coating,实现了对硅醇基极大程度的封锁,兼具分离性能和普适性能,通用性非常好。CAPCELL PAK SCX UG80是强阳离子交换柱,使用高纯度硅胶,填料中金属杂质很少,使配位化合物的吸附得到了极大程度抑制,兼具聚合物和硅胶填料的优点。[b][color=#0070c0]实验方法[/color][color=#0070c0]方法一[/color][color=#0070c0]使用[/color][color=#0070c0]CAPCELL PAK C18MGII[/color][color=#0070c0]色谱柱[/color][color=#0070c0]+[/color][color=#0070c0]离子对试剂[/color][/b]如图1,对肾上腺素对照品溶液进行分析,肾上腺素主峰保留时间为5.69 min,拖尾因子为1.19,理论塔板数为12538。在相同色谱条件下,尝试对亚硫酸肾上腺素标准品及注射液中的亚硫酸肾上腺素进行分析。如图3,亚硫酸肾上腺素标准品溶液能够得到良好分析结果,注射液(客户提供的样品)中未明显见亚硫酸肾上腺素出峰,保留时间为3.55min,拖尾因子为1.14,理论塔板数为14955。[b][color=#0070c0]方法二[/color][color=#0070c0] CAPCELL PAK SCX UG80[/color][color=#0070c0]色谱柱[/color][/b][color=#000000]考虑到使用离子对试剂的流动相条件具有流动相配制麻烦、有损色谱柱寿命、平衡时间长等缺点,我们也尝试使用键合磺酸基团的强阳离子交换柱 ——CAPCELL PAK SCX UG80进行分析。[/color][color=#000000]如图4,在流动相中添加磷酸二氢铵,通过对盐浓度进行调整,在5 mmol/L磷酸二氢铵(磷酸调pH=2.5)条件下,亚硫酸肾上腺素保留时间为3.32 min,然而出现峰形拖尾现象,拖尾因子为2.0,不如CAPCELL PAK C18 MGII色谱柱添加离子对试剂所得分析结果好。[/color][align=center][/align][align=left][img=,400,284]https://ng1.17img.cn/bbsfiles/images/2019/03/201903191547381421_926_2222981_3.jpg!w584x416.jpg[/img] [img=,400,276]https://ng1.17img.cn/bbsfiles/images/2019/03/201903191548310561_8067_2222981_3.jpg!w572x395.jpg[/img][/align][align=left][img=,400,166]https://ng1.17img.cn/bbsfiles/images/2019/03/201903191547576748_522_2222981_3.jpg!w612x254.jpg[/img] [img=,400,167]https://ng1.17img.cn/bbsfiles/images/2019/03/201903191548525761_4184_2222981_3.jpg!w624x262.jpg[/img][/align][align=left]图1 MGII分析肾上腺素对照品溶液结果(离子对条件) 图2 MGII分析注射液结果(离子对条件)[/align][align=center][/align][img=,400,258]https://ng1.17img.cn/bbsfiles/images/2019/03/201903191550354951_2539_2222981_3.jpg!w644x416.jpg[/img] [img=,400,250]https://ng1.17img.cn/bbsfiles/images/2019/03/201903191555486850_3396_2222981_3.jpg!w644x403.jpg[/img][img=,400,147]https://ng1.17img.cn/bbsfiles/images/2019/03/201903191551260881_9828_2222981_3.jpg!w696x256.jpg[/img] [img=,400,164]https://ng1.17img.cn/bbsfiles/images/2019/03/201903191556163846_9739_2222981_3.jpg!w632x260.jpg[/img][align=left]图3 MGII分析亚硫酸肾上腺素标准品和注射液结果(离子对条件) 图4 SCX UG80分析亚硫酸肾上腺素对照品溶液和供试品溶液[/align][align=left][/align][align=left]综上实验结果,使用中等极性色谱柱CAPCELL PAK C18 MGII S5 4.6 mm i.d. × 250 mm,在流动相中添加5 mM辛烷磺酸钠、30°C柱温条件下进行梯度洗脱,能够实现复方盐酸阿替卡因注射液中肾上腺素和亚硫酸肾上腺素的良好保留与分析。[/align][b][color=#0070c0][/color][/b][align=left][b][color=#0070c0] [/color][color=#0070c0] [/color][/b][/align]







 我要推广仪器
我要推广仪器
 下载APP
下载APP