行标YC 144-2008中规定三乙酸甘油酯纯度测定时,气相进样口温度为250℃。但我查到的三乙酸甘油酯沸点为:258-260 °C(lit.)℃。进样口的温度设置原则不应该是要高于被分析物的沸点,确保所有分析物经过进样口进样后能够完全气化吗?想寻求大家的解答
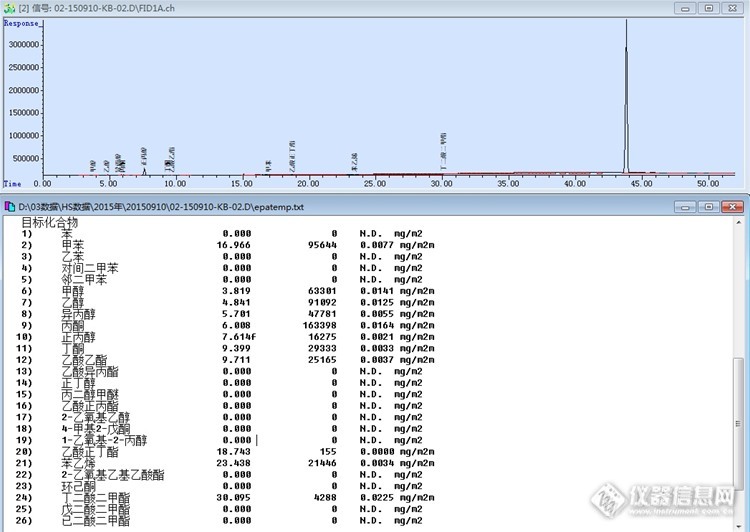
安谱三乙酸甘油酯的使用心得 我们实验室一直在测烟标中的VOC,参照烟标YC/T 207-2014的方法来检测,YC/T207-2014同YC/T207-2006相比培加了10种管控物质,同时对溶剂杂质也有要求,基于方法的加严,为了达到更好的检测数据,对检测用的试剂自然而然的加严了,所以,就采购的安谱药典级的三乙酸甘油酯。以前我们使用的某品牌的三乙酸甘油酯,如下图:http://ng1.17img.cn/bbsfiles/images/2017/10/2015091819191794_01_2769262_3.jpg现在我们使用的三乙酸甘油酯,如下图:http://ng1.17img.cn/bbsfiles/images/2017/10/2015091819194869_01_2769262_3.jpg现就两种不同的三乙酸甘油酯,做个对比测试,选择用新的20ml顶空进样瓶,然后分别移取不同品牌的三乙酸甘油酯到顶空进样瓶中,封盖,然后用顶空加气相(带FID检测器)检测,检测结果如下:某品牌的三乙酸甘油酯检测结果如下图:http://ng1.17img.cn/bbsfiles/images/2017/10/2015091819223603_01_2769262_3.jpg安谱药典级的三乙酸甘油酯检测结果如下图:http://ng1.17img.cn/bbsfiles/images/2017/10/2015091819223175_01_2769262_3.jpg两种品牌试剂的检测结果汇总表:http://ng1.17img.cn/bbsfiles/images/2015/09/201509181923_566740_2769262_3.jpg综上,安谱药典级的三乙酸甘油酯优于某品牌的三乙酸甘油酯,安谱药典级的三乙酸甘油酯值得版友们拥有。
怎么分离亚氨基二乙酸和氨基三乙酸,样品中氨基三乙酸含量少
原子吸收测定三乙酸甘油酯、沒食子酸丙酯中铅、镉的含量?那位做过,我现在查的沒食子酸丙酯中铅的测定按照GB/T5009.75测定,请问哪里有标准下载。三乙酸甘油酯、沒食子酸丙酯中铅镉的含量都是微量
请问EDTA中氨基三乙酸的含量,不合格的批次多吗?
通常液相色谱分析中,在流动相中加乙酸和三乙胺,大家来谈谈,在什么情况下需要加乙酸,什么情况下加三乙胺,加多少是怎么控制的?
有这么一种流动相,甲醇:水:三乙胺:乙酸=600:400:1:1那么加三乙胺和乙酸的作用是什么?
原子荧光测定三乙酸甘油酯、沒食子酸丙酯中砷、汞的含量?含量是微量的,样品是采用湿法消解还是采用微波消解?
氮川三乙酸,又名氨三乙酸,求其分析方法,化分、仪分均可,方法不限,特别是在混合体系中的分析方法,谢过先 补充:特别是氮川三乙酸与甘氨酸,亚氨基二乙酸,羟基乙酸等多组分的混合体系,换句话说,如果这几个东西混在一起,有什么方法能够将他们分别分析出来,一步也行,多步也行,任何方法都不限,欢迎大家讨论跟上次的问题一样,上次结贴太匆忙了,再次征集。感谢上次renture/jiangyunjun3/melu的帮助,找到一个分光法测痕量氮川的方法,测量范围是0.01~0.12ug/mL,但我们的样品是含量大概有0~10%左右的氮川,是否能够通过稀释来使用上面的分光方法还不知道。现在想征集一个能够分析常量氮川的方法,希望大家踊跃发言。
氮川三乙酸,又名氨三乙酸,求其分析方法,化分、仪分均可,方法不限,特别是在混合体系中的分析方法,谢过先[em0813] [em0813] [em0813]补充:特别是氮川三乙酸与甘氨酸,亚氨基二乙酸,羟基乙酸等多组分的混合体系,换句话说,如果这几个东西混在一起,有什么方法能够将他们分别分析出来,一步也行,多步也行,任何方法都不限,欢迎大家讨论
三乙酸甘油酯+硝酸,放在电热板上100度加热,会有危险吗?可能的反应产物是什么,谢谢大家
今天用液相色谱仪检测依地酸钙钠中的氨基三乙酸,用的是安捷伦的C8柱子,流动相为0.01mol/L的氢氧化四丁基铵:甲醇=90:10,溶剂为硝酸铜,可是压力很不稳定,好不容易平衡后,一进样,压力瞬间飘很高,在走样过程中逐渐下降,走一针过后,基线很不稳定,感觉柱子里面冲出来很多东西,有没有做过这个品种的前辈们啊,知道我一下吧,怎么改进?
GC,我用色谱柱TG-624 检测三乙胺、乙酸异丙酯和庚烷的混合样,分离总是不好。该如何调节?
[color=#444444]各位大神给点建议,[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质联用[/color][/url]所用的流动相是三氟乙酸和三乙胺配制的缓冲液,来检测发酵液中的有机酸,能不能给点建议,具体怎样配制此缓冲液?[/color]
我实验要在负离子模式下定量测定一种代谢物,但买到的对照品是三氟乙酸盐结合的形式,在论坛里看到说,三氟乙酸不能再负离子模式下使用,会抑制响应,那我的对照品就不能用了吗?应该怎么办呢?
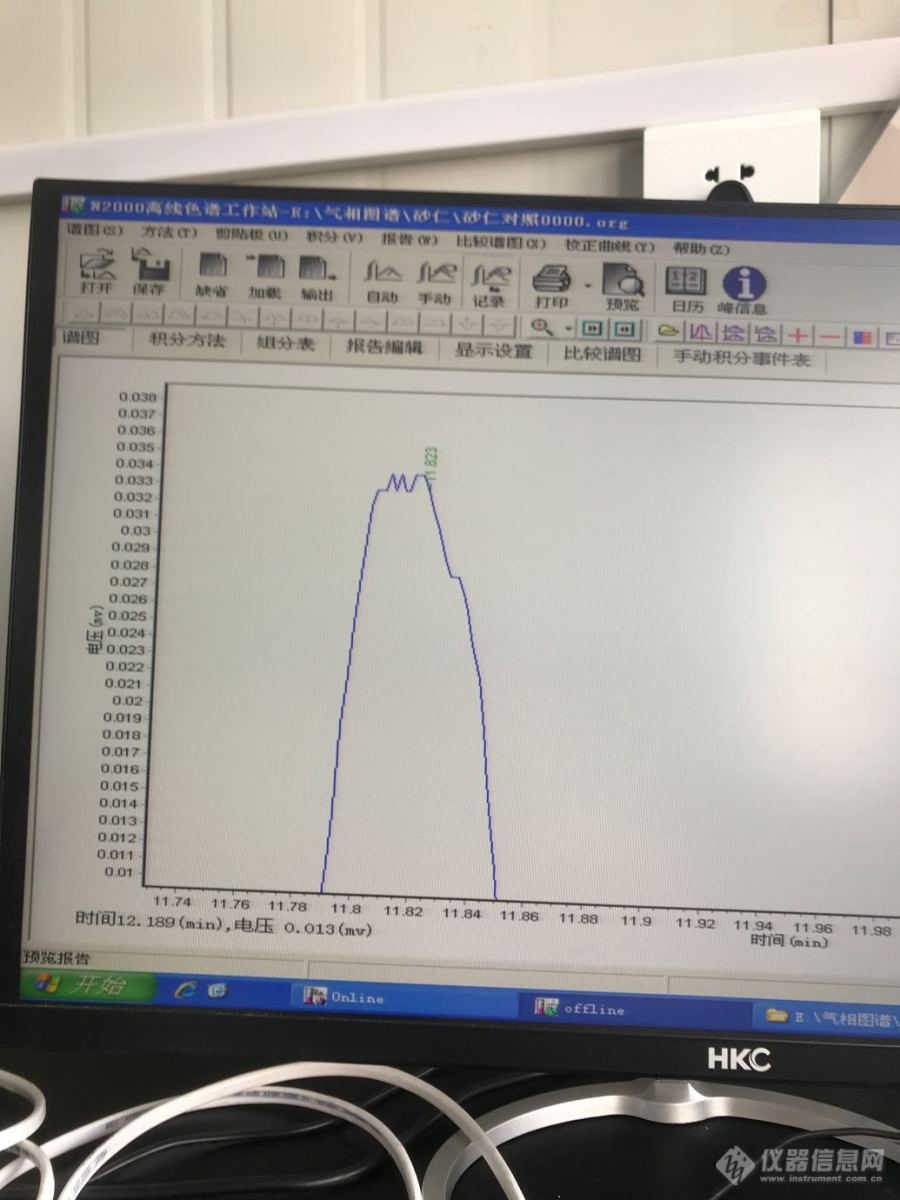
砂仁含量测定,乙酸龙脑脂对照品的峰是这样的,什么原因?[img=,690,920]https://ng1.17img.cn/bbsfiles/images/2019/09/201909291048233492_354_4008962_3.png[/img][img=,690,920]https://ng1.17img.cn/bbsfiles/images/2019/09/201909291048362627_1375_4008962_3.png[/img][img=,690,920]https://ng1.17img.cn/bbsfiles/images/2019/09/201909291048546892_2814_4008962_3.png[/img]
今天做乙酸乙酯残留,对照面积分别是702.2,722.5,738.6,790.4,821.2,rsd为6.5%,第一针和最后一针面积相差120左右,但rsd在10%内,这5个对照能采用吗?
在用液相色谱分析某些酸性药品时,有的时候会在流动相中加入三氟乙酸,加入三氟乙酸的目的是什么啊?有时候还会在流动相中加入二乙胺,目的是什么啊?仅仅是要调节pH值么?还有就是,二乙胺可以用三乙胺代替么?哪位高手给解答一下啊,具体点的。
在对黄连进行薄层色谱的时候,用环己烷:乙酸乙酯:异丙醇:甲醇:水:三乙胺(3:3.5;1:1.5:0.5:1)进行展开,但是没有展开完,就感觉有二次展开的情况,想问问出现这种情况的原因,和解决方法
我要做氨基三乙酸和亚氨基二乙酸的分离,但是在C18柱上试了好几种方法都没有保留,请专家帮忙
1.3 对照品混合溶液和样品的衍生取对照品混合溶液或样品200micron置于离心管中,加入0.1mol/l异硫氰酸苯酯乙腈溶液100 micron ,1.0 mol/l 三乙胺乙腈溶液100 micron,混匀,室温下放置1h后加入400 micron正己烷,漩涡混合器振荡1 min,静置10min 。用[url=https://insevent.instrument.com.cn/t/9p][color=#3333ff][url=https://insevent.instrument.com.cn/t/9p][color=#3333ff]移液器[/color][/url][/color][/url]吸取下层溶液,经0.22微米滤膜过滤后进样进行色谱分析。PITC-乙腈:36微升异硫氰酸苯酯+2.164毫升PITC,混合。三乙胺乙腈溶液:417微升三乙胺+2.583毫升乙腈,混合。1、三乙胺是用来调节PH值吗,或者还有其它作用吗?2、正己烷用来萃取,乙腈是干什么用呢?3、那个下面的PITC-乙腈配制也不是很懂,这样配怎么就成了上面需要的浓度呢?
[color=#444444]本人最近按2015版药典做了一个药用辅料-醋酸羟丙甲纤维素琥珀酸酯的的游离乙酸的含量测定实验。实验过程如下:[/color][color=#444444] 游离乙酸、琥珀酸 取本品0.102g,精密称定,置锥形瓶中,精密加入磷酸盐溶液(取0.02mol/L磷酸二氢钾溶液,用1mol/L氢氧化钠溶液调pH值至7.5)4.0ml,搅拌2小时,加磷酸溶液(取1.25mol/L磷酸1ml,置50ml量瓶中,加水稀释至刻度,摇匀)4.0ml,强力振摇,离心,上清液作为供试品溶液;精密称取琥珀酸0.13g,置100ml量瓶中,加水适量,振摇使完全溶解,加水至刻度,摇匀,作为琥珀酸贮备液;取加有水20ml的100ml量瓶,称重,精密加入冰乙酸2ml,再称重,用水稀释至刻度,摇匀,精密量取6ml,置100ml量瓶中,用水稀释至刻度,摇匀,作为乙酸贮备溶液;精密量取乙酸贮备液和琥珀酸贮备液各4.0ml,置同一25ml量瓶中,用流动相稀释至刻度,摇匀,作为对照溶液。照高效液相色谱法(中国药典2015年版四部通则0512)试验。以十八烷基硅烷键合硅胶为填充剂,以0.02moI/L磷酸二氢钾溶液(用6mol/L磷酸溶液调pH值至2.8)为流动相,流速每分钟1ml,检测波长为215nm。取对照溶液10μl, 注入液相色谱仪,按琥珀酸峰计算,理论板数不得少于8000。取供试品溶液与对照溶液各10μl,注入液相色谱仪,按干燥品计算,游离乙酸和琥珀酸总量不得过1.0%。[/color][color=#444444]计算公式: 游离乙酸含量=0.0768(WA/(W(1-干燥失重)))(γUA/γSA)[/color][color=#444444] 式中 WA为乙酸贮备溶液中冰乙酸量,mg;[/color][color=#444444] W为供试品的取样量,mg;[/color][color=#444444] γUA、γSA为供试品溶液、对照溶液中乙酸的峰面积。[/color][color=#444444] 游离琥珀酸含量=1.28(WS/(WUS(1-干燥失重)))(γUS/γSS)[/color][color=#444444] 式中 WS为琥珀酸贮备液中琥珀酸量,mg;[/color][color=#444444] WUS为供试品取样量,mg;[/color][color=#444444] γUS、γSS为供试品溶液、对照溶液琥珀酸的峰面积。[/color][color=#444444]我的问题是,根据“干燥品计算,游离乙酸和琥珀酸总量不得过1.0%”这句话,游离乙酸含量的最后计算的结果要不要乘以100%,比如我最后计算结果是0.0139,如果这个结果再乘以100%,就变为1.39%,从而超过限度,那么就需要重新做实验复核一遍。[/color]
我现在在做某物质的[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]残留溶剂,标准规定乙酸丁酯≤1000ppm,我配置对照品溶液时可以配置的比标准规定的低吗(比如对照品溶液配成500ppm)?样品中乙酸丁酯的残留大概是80ppm
请问液质联用做一种代谢物的定量分析时,买到的对照品是代谢物的三氟乙酸盐,如何在Q1扫描时将盐去掉,得到代谢物的母离子质核比?还是需要将对照品进行前处理?
[color=#333333]求助各位高手帮帮忙,最近在做醋酸纤维素琥珀酸酯乙酸的检测,按照2015版药典方法检测,出峰的时候发现琥珀酸有两个峰出现,不知道是什么原因,琥珀酸的对照买的是麦克林公司的试剂,纯度也都在99.5%。[/color][color=#333333][/color]
[align=right][b]SGLC-GC-039[/b][/align][b]摘要:[/b]本文建立了砂仁中乙酸龙脑酯含量测定的GC 方法。结果表明,参照2020版《中国药典》中色谱条件并对升温程序进行优化,采用色谱柱SH-1 分析砂仁中乙酸龙脑酯,乙酸龙脑酯峰形对称,理论塔板数按乙酸龙脑酯峰计算远高于10000,满足《中国药典》要求。此方法可为砂仁中乙酸龙脑酯含量测定提供参考。。[b]关键词:[/b]砂仁 乙酸龙脑酯 SH-1 GC[b]1. 实验部分1.1 实验仪器及耗材[/b]Shimadzu GC-2030[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱仪[/color][/url];色谱柱:SH-50(30 m,0.25 mm × 0.25 μm;P/N:227-36162-01;S/N:1553669);SHIMSEN Arc Disc HPTFE针式过滤器(P/N:380-00341-05);[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]GC-MS[/color][/url]认证样品瓶LabTotal Vial(P/N:227-34002-01);SHIMSEN Pipet[url=https://insevent.instrument.com.cn/t/9p][color=#3333ff]移液枪[/color][/url]:SHIMSEN Pipet PMII-10(P/N:380-00751-02);SHIMSEN Pipet PMII-100(P/N:380-00751-04);SHIMSEN Pipet PMII-1000(P/N:380-00751-06)。[b]1.2 对照品溶液的制备[/b]取芳樟醇对照品适量,精密称定,加乙酸乙酯制成每1 mL含0.1 mg的溶液,即得。[b]1.3 供试品溶液的制备[/b]取本品粉末(过三号筛)约1 g,精密称定,置具塞锥形瓶中,精密加入无水乙醇25 mL,密塞,称定重量,超声处理(功率300W,频率40 kHz)30 分钟,放冷,用无水乙醇补足减失的重量,摇匀,滤过,取续滤液,即得。。[b]1.4 分析条件[/b]色谱柱:SH-1 (30 m, 0.25 mm × 0.25 μm P/N:221-75719-30;S/N:1541069 )升温程序:初始温度80 ℃,保持1分钟,以每分钟2 ℃的速率升温至100 ℃,保持5分钟载气:N2进样口温度:230 ℃分流模式:分流(10:1)控制模式:恒线速度(30 cm/s)初始流速:1.09 mL/min检测器:FID,温度:250 ℃进样量:1 μL[b]2. 实验结果[/b]按照上述色谱条件(1.4)进行采集,对照品溶液和供试品溶液色谱图如下:[b]对照品溶液[/b][img=砂仁中乙酸龙脑酯含量测定]https://img.shimadzumall.com/Storage//userfiles/images/Img_articles/SGLC-GC-039_1.png[/img][font=arial, &][size=12px][/size][/font][b]供试品溶液[/b][img=砂仁中乙酸龙脑酯含量测定]https://img.shimadzumall.com/Storage//userfiles/images/Img_articles/SGLC-GC-039_2.png[/img][font=arial, &][size=12px][/size][/font][b]重现性[/b][img=砂仁中乙酸龙脑酯含量测定]https://img.shimadzumall.com/Storage//userfiles/images/Img_articles/SGLC-GC-039_3.png[/img][font=arial, &][size=12px][/size][/font][b]3. 结论[/b]本文建立了砂仁中乙酸龙脑酯含量测定的GC 方法。结果表明,参照2020版《中国药典》中色谱条件,采用色谱柱SH-1 分析砂仁中乙酸龙脑酯,乙酸龙脑酯峰形对称,理论塔板数按乙酸龙脑酯峰计算远高于10000,满足《中国药典》要求。此方法可为砂仁中乙酸龙脑酯含量测定提供参考。
据广东出入境检验检疫机构称,在日本进口的日式酱油、芥末酱中检测出了甲苯和乙酸乙酯。有关食品产自三家日本生产企业。其中甲苯的最高检出值为0.0053mg/kg,乙酸乙酯最高检出值为0.537mg/kg。此前,有日本媒体报道,日本有人食用了检出甲苯和乙酸乙酯的食品出现过不适症状。中国国家质检总局称,进口上述产品的中国进口企业,已开始对这三家企业生产的同类产品采取下架和批批检验措施,以确保中国消费者安全。甲苯 健康危害:对皮肤、粘膜有刺激性,对中枢神经系统有麻醉作用。 慢性中毒:长期接触可发生神经衰弱综合征,肝肿大,女工月经异常等。皮肤干燥、皲裂、皮炎。乙酸乙酯用作清漆稀释剂,人造革、硝酸纤维素塑料等的溶剂,也用作染料、药物、香精等的原料。作为增香剂用于食品和饲料中。
做氨基酸用的药品三乙胺,醋酸钠方法上说需要优级纯,可是优级纯的很难买到,用分析纯的可以不?
三乙基砷酸酯是无机物吗?
目前我们实验室用的维生素A醋酸酯的对照品的供应商断货了,求问一下大家都用的是哪些供应商的对照品?我们也可以去买。我们试用过Sigma的和USP的发现都不行。Sigma的是实际含量和COA上的含量出入较大。USP的是一个混合物有全反式的和CIS的,由于我们不是用的中国药典附录上测定维生素A的方法,所以我们的液相分不开这2种物质,所以也不能用。







 我要推广仪器
我要推广仪器
 下载APP
下载APP