各路大侠,有做过甲苄喹啉的吗?标准品用什么试剂溶解的啊?参考“SN/T 2444-2010 进出口动物源食品中甲苄喹啉和癸氧喹酯残留量的测定 液相色谱-质谱∕质谱法”用的N,N-二甲基甲酰胺,还是不能完全溶解……
食品安全国家标准 食品添加剂 喹啉黄
[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]分析脂肪酸甲酯标准品,怎样处理标准品?用什么溶剂稀释?
徐建强(南京气象学院环境科学系,南京 210044)分光光度法是获得物质光吸收特性及定性、定量分析的重要手段,在冶金、地质、生物、医学、农业、环境监测、食品卫生等部门得到极其广泛的应用。分光光度法的发展不仅依赖于电子学、激光和计算机技术的发展和应用,而且还依赖于高灵敏度、高选择性有机试剂的合成和应用。喹啉类试剂作为光度分析的有机试剂,可以分为两类,一类是喹啉及其衍生物,如8-羟喹啉、8-巯基喹啉、8-氨基喹啉等,这类试剂可用作金属离子光度分析的显色剂,具有一定的灵敏度和选择性 另一类是喹啉偶氮化合物,喹啉偶氮化合物作为一大类显色剂已有多种试剂被合成和研究。其中8-氨基喹啉的8位偶氮衍生物前苏联学者研究得较早 我国的李亚文等也进行了系统深入的研究,近年来不断有一系列新的衍生物合成,这些化合物因其特有的灵敏度和选择性而备受化学工作者的关注。自从1955年程广禄等[3]首次提出PAN(即1-(2-吡啶偶氮)-2-萘酚)作为分析试剂后,越来越多的偶氮试剂相继被化学工作者合成并且用于光度分析研究。实践证明,偶氮化合物(azo-compounds)具有性质稳定、显色反应灵敏度高、选择性好、对比度大等优点,仍然是目前应用最广泛的一类显色剂。本文选用6-甲氧基-8-氨基喹啉作为原料,首先对其进行重氮化,得到重氮盐,然后与间苯二酚进行偶联反应,合成了新有机试\剂:4-(6-甲氧基-8-喹啉偶氮)-间苯二酚(简称MQAR)。对其进行了结构分析和鉴定。实验研究表明,MQAR能与Co、Cu、Fe、Ni等金属离子发生灵敏的显色反应,该试剂可用于试样中微量金属离子的测定(另文介绍),方法简便、快速、准确可靠,是一种比较理想的新有机试剂。1 合成方法1.1 试剂和仪器设备6-甲氧基-8-氨基喹啉(由工业品提纯所得),亚硝酸钠(AR),间苯二酚(AR),N,N-二甲基甲酰胺(AR),浓硫酸(AR),甲酸(AR)。中量有机化学制备仪、真空干燥箱、差热分析仪、Perkin-Elmer元素分析仪、IR-408型红外分光光度计、756MC型紫外-可见分光光度计、BRUKERARX300M核磁共振谱仪。1.2 合 成1.2.1 合成线路MQAR的合成线路如图1所示。[img]http://ng1.17img.cn/bbsfiles/images/2006/05/200605221937_18814_1634962_3.gif[/img]1.2.2 合成步骤(1)重氮化 在250mL三口烧瓶中,加入8.7g6-甲氧基-8-氨基喹啉,10mL甲酸,同时加入由15mL浓硫酸和10mL水配制而成的溶液,搅拌溶解,置于冰浴中冷却。在0~5℃时边搅拌边滴加3.5g亚硝酸钠与10mL水配制而成的溶液,控制在1h内加完,使反应充分完全,最后得到深红色的重氮盐溶液,用于下一步的反应。(2)偶联 取5.5g间苯二酚溶于75mL无水乙醇中,置于冰浴中冷却。在0~5℃时,边搅拌边加入上述重氮盐溶液,控制重氮盐溶液于1h内加完,继续搅拌2h,静置过夜得砖红色沉淀,抽滤,依次用水、无水乙醇洗涤,抽干。干燥后得棕黄色粗品。(3)精制 将偶联得到的粗品用N,N-二甲基甲酰胺重结晶两次,于真空干燥箱干燥,得到深红色的MQAR纯品。经测定产品熔点为194℃。2 结构分析2.1 薄层色谱分析(1)试剂和仪器设备 展开剂正丁醇∶无水乙醇∶2molL-1氨水=3∶1∶1 支持剂硅胶HF254+0.3%CMC 8×10cm2薄层板(自制) 层析缸。(2)结果和讨论 在不同极性的溶剂体系中进行展开,在薄层色谱板上只发现一个斑点,在紫外灯光下未发现其他斑点,(1)中所列展开剂效果最好,比移值Rf=0.66(图2)。结果表明,产品中只含有一种物质。2.2 元素分析用Perkin-Elmer元素分析仪对产品进行了元素分析,产品MQAR元素分析结果与理论计算值基本一致。2.3 紫外-可见吸收光谱分析用756MC型紫外-可见分光光度计测得2×10-5molL-1MQAR的10%DMF水溶液(pH=8.3时)的紫外-可见吸收光谱(图3)。Kmax=450nm,E=2.98×104Lmol-1cm-1。结果表明,产品分子是一个大的共轭体系。2.4 红外吸收光谱分析用IR-408型红外分光光度计对产品MQAR进行了红外吸收光谱分析(KBr压片法),产品的红外光谱解析结果见表1。解析结果表明,产品分子中含有酚羟基、芳环、偶氮基等官能团,还具有1,2,4-三取代苯结构。[img]http://ng1.17img.cn/bbsfiles/images/2006/05/200605221954_18815_1634962_3.gif[/img][img]http://ng1.17img.cn/bbsfiles/images/2006/05/200605221955_18816_1634962_3.gif[/img]2.5 核磁共振谱分析用BRUKERARX300M核磁共振谱仪(DMSO-d6溶剂,TMS内标)对产品进行核磁共振谱分析,得到化学位移、峰面积等(表2)。解析结果表明,产品分子中除了羟基质子以外,还含有甲氧基质子以及苯环和杂环质子。[img]http://ng1.17img.cn/bbsfiles/images/2006/05/200605221955_18817_1634962_3.gif[/img]2.6 产品的结构根据实验条件以及结构鉴定和分析,可以确定产品4-(6-甲氧基-8-喹啉偶氮)-间苯二酚的分子结构为[img]http://ng1.17img.cn/bbsfiles/images/2006/05/200605221956_18818_1634962_3.gif[/img]3 结 论研究了新有机试剂4-(6-甲氧基-8-喹啉偶氮)-间苯二酚的合成方法、实验条件和精制方法,通过薄层色谱法、元素分析、紫外-可见吸收光谱法、红外吸收光谱法和核磁共振谱法等分析手段,对合成的产品进行了分析和结构鉴定,实验结果确证合成了4-(6-甲氧基-8-喹啉偶氮)-间苯二酚纯品。
苄嘧磺隆二氯喹啉酸的图谱,二者出峰顺序我做的和标准怎么相反?请说说你们的意见,谢谢!
本人现急需氯亚铂酸钾和二(乙酰丙酮)铂的分析方法或行业标准,那位大虾有相关资料,请上传或发到我的信箱[color=#DC143C]zhangfy03@126.com[/color],不胜感激!!!!
我需要测定喹啉的浓度,但是不知道色谱的分析条件,主要包括柱子的选择,标准溶液的配制,测定条件,由于以前怎么用过,请高手多多指教!另外我还想问一下测定水中氨氮能不能用色谱,[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]行吗,还有具体条件,谢谢!
6-氨基喹啉基-N-羟基琥珀酰亚胺基甲酸酯(AQC)为衍生试剂,准备利用AQC做氨基酸分析?有做过的吗?是不是仅Waters有?其他公司有类似替代品吗?补充一下:大概300个样品。紫外检测
1、广泛用于金属的测定和分离。沉淀和分离金属离子的沉淀剂和萃取剂,能与下列金属离子络合:Cu?+2、Be?+2、Mg?+2、Ca?+2、Sr?+2、Ba?+2、Zn?+2、Cd?+2、Al?+3、Ga?+3、In?+3、Tl?+3、Yt?+3、La +3、Pb?+2、B?+3、Sb?+3、Cr?+3、MoO?+22、Mn?+2、Fe?+3、Co?+2、Ni?+2、Pd?+2、Ce?+3 1.用作医药中间体,是合成克泻痢宁、氯碘喹啉、扑喘息敏的原料,也是染料、农药中间体。该品是卤化喹啉类抗阿米巴药物的中间体,包括喹碘仿、氯碘喹啉、双碘喹啉等。这类药物通过抑制肠内共生菌而发挥抗阿米巴作用,对阿米巴痢疾有效,对肠道外阿米巴原虫无影响。国外报道本类药物能引起亚急性脊髓视神经病,故该药在日本和美国已禁用,双碘喹啉引起此病比氯碘喹啉较少见。8-羟基喹啉也是染料、农药的中间体。其硫酸盐和铜盐是优良的防腐剂、消毒剂和防霉剂。该品是化学分析的络合滴定指示剂。 2.用作沉淀和分离金属离子的络合剂和萃取剂,能与Cu+2、Be+2、Mg+2、Ca+2、Sr+2、Ba+2、Zn?+2、Cd+2、Al+3、Ga+3、In+3、Tl+3、Yt+3、La +3、Pb+2、B+3、Sb?+3、Cr+3、MoO?+22、Mn+2、Fe+3、Co+2、Ni+2、Pd+2、Ce+3、等多种金属离子络合。有机微量分析测定杂环氮的标准,有机合成。也是染料、农药及卤化喹啉类抗阿米巴药物的中间体。其硫酸盐和铜盐是优良的防腐剂。 3.加入环氧树脂胶黏剂中可提高对金属(尤其是不锈钢)的粘接强度和耐热老化性,用量一般为0.5~3份。是卤化喹啉类抗阿米巴药物的中间体,也是农药、染料的中间体。可作为防霉剂、工业防腐剂以及聚酯树脂、酚醛树脂和双氧水的稳定剂,还是化学分析的络合滴定指示剂。 4.本品是卤化喹啉类药物的中间体,也是染料、农药的中间体。其硫酸盐和铜盐是优良的防腐剂、消毒剂和防霉剂。化妆品中最大允许含量(质量分数)为0.3%,防晒产品和3岁以下儿童用品 ( 如爽身粉)禁用,并应在产品标签上注明 “ 3岁以下儿童禁用” 。在处理病菌感染的皮肤和细菌性传染湿疹时,乳液中8-羟基喹啉的质量分数为0.001%~0.02%。它也用作消毒剂、防腐剂和杀菌剂,其防霉菌作用强。8-羟基喹啉硫酸钾用于护肤膏霜和乳液中含量(质量分数)为0.05%~0.5%。
2011年6月9日,日本农林水产省发布G/SPS/N/JPN/279号通报:食品卫生法实施条例及食品和食品添加剂和标准规范修订案。该修正案批准异喹啉和吡咯作为食品添加剂使用并规定此类物质的标准和规范。该草案评议截止期为2011年8月8日,此后会尽快批准和生效。更多详情参见:http://members.wto.org/crnattachments/2011/sps/JPN/11_1820_00_e.pdf
求助:原乙酸三甲酯分析方法或分析标准
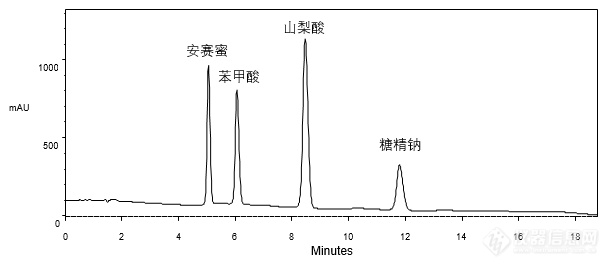
[align=center][b]GB 5009.28-2016食品安全国家标准 食品中苯甲酸、山梨酸和糖精钠的测定[/b][/align][align=center][b] ——标准品与乳品实际样品的分析[/b][/align][align=center][/align][align=left]本实验按照《GB5009.28-2016 食品安全国家标准食品中苯甲酸、山梨酸和糖精钠的测定》方法,分别对安赛蜜、苯甲酸、山梨酸、糖精钠的标准品混合溶液及加标乳品样品进行了分析。首先,使用CAPCELL PAK C[sub]18[/sub] MG S5 4.6 mm i.d. × 150mm色谱柱,对标准品混合溶液进行分析,如图1,安赛蜜、苯甲酸、山梨酸、糖精钠标准品均得到了良好的分析结果。[/align][align=left][/align][align=center][img=,611,268]http://ng1.17img.cn/bbsfiles/images/2018/03/201803221532276656_9890_2222981_3.png!w611x268.jpg[/img][/align][align=center]图1 标准品混合溶液分析色谱图[/align][img=,400,200]http://ng1.17img.cn/bbsfiles/images/2018/03/201803221532280132_6863_2222981_3.png!w400x200.jpg[/img][align=left][/align][align=left]其次,对乳品加标样品进行分析,如图2,糖精钠(Rt 12 min)与其后杂质峰之间未能取得基线分离,分离度仅为1.02。[/align][align=left][/align][align=center][img=,668,335]http://ng1.17img.cn/bbsfiles/images/2018/03/201803221533054905_2223_2222981_3.png!w668x335.jpg[/img][/align][align=center]图2 加标乳品样品分析色谱图[/align][align=left][img=,406,203]http://ng1.17img.cn/bbsfiles/images/2018/03/201803221533317202_2333_2222981_3.png!w406x203.jpg[/img][/align][align=left][/align][align=left]为改善糖精钠与杂质间的分离,在国标方法基础上,将流动相由[b]乙酸铵 / 甲醇 = 95 / 5[/b]调整为[b][b]乙酸铵 / 甲醇[/b][color=red]([/color][color=red]2 mmol/L [/color][color=red]甲酸)[/color]= 92 / 8[/b],再次对混合标准溶液和加标样品进行分析,结果如图3所示。[/align][align=left][/align][align=center][img=,690,545]http://ng1.17img.cn/bbsfiles/images/2018/03/201803221534141056_4073_2222981_3.png!w690x545.jpg[/img][/align][align=center]图3 混标与加标乳品样品分析色谱图[/align][align=left][img=,464,171]http://ng1.17img.cn/bbsfiles/images/2018/03/201803221535548985_7176_2222981_3.png!w464x171.jpg[/img][/align][align=left][/align][align=left]如图3,在酸性条件下,出峰顺序发生了变化,安赛蜜保留时间略有缩短,糖精钠保留时间明显缩短,由12 min缩短至8 min,苯甲酸和山梨酸保留时间分别延长至2 min和6 min;在分离度方面,糖精钠与苯甲酸之间分离度为2.79,苯甲酸与峰后杂质间分离度为2.04,所有色谱峰之间都达到了基线分离。[/align][align=left][/align][align=left]为使客户有更多选择,实验室又在国标原方法条件下继续筛选色谱柱,最终使用SUPERIOREX ODS S5 4.6 mm i.d. × 250 mm色谱柱时,仅微调有机相比例即可实现加标乳品样品的良好分析结果。如图4,杂质峰与糖精钠之间分离度达到2.48,达到基线分离要求。[/align][align=left][/align][align=center][img=,580,332]http://ng1.17img.cn/bbsfiles/images/2018/03/201803221537130173_1058_2222981_3.png!w580x332.jpg[/img][/align][align=center]图4 加标乳品样品分析色谱图[/align][align=left]*注:峰上标所示数字由下至上依次为分离度与不对称因子。[/align][align=left][img=,326,177]http://ng1.17img.cn/bbsfiles/images/2018/03/201803221537540634_9437_2222981_3.png!w326x177.jpg[/img][/align][align=left][/align][align=left]综上所述,按照国标《GB 5009.28-2016 食品安全国家标准食品中苯甲酸、山梨酸和糖精钠的测定》方法进行分析,使用CAPCELL PAK C[sub]18 [/sub]MG色谱柱对标准品混合溶液能得到良好分析结果,但在对加标乳品样品进行分析时,糖精钠与样品中的杂质未能实现基线分离,通过在流动相中添加甲酸可实现安赛蜜、糖精钠、苯甲酸、山梨酸及杂质的基线分离;另一方面,使用SUPERIOREX ODS色谱柱,在原条件基础上微调即可实现乳品中安赛蜜、苯甲酸、山梨酸、糖精钠及杂质间的良好分离。[/align]
手上有棕榈酸甲酯和硬脂酸甲酯的标准品各50mg,请问该怎么处理进气相会比较好?另外,关于脂肪酸的定量分析是用内标法比较好吗?外标法行吗?那这样的话稀释浓度梯度该为多少比较合适?拜托各位老师教教我..先.谢谢了!!
做农残实验时,提取水稻植株中的二氯喹啉酸时加二氯甲烷?为什么还要加饱和碳酸氢钠溶液?碳酸氢钠溶液有什么作用?
[color=#444444]关于批准发布《钢铁及合金 钙和镁含量的测定 电感耦合等离子体原子发射光谱法》等374项国家标准和3项国家标准修改单的公告中( [/color]2019年第7号 [color=#444444]), [/color]有多项涉及分析仪器分析方法,包括电感耦合等离子体原子发射光谱法、火焰[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收光谱[/color][/url]法、原子发射光谱法、[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法、分光光度法、X射线荧光光谱法、高效液相色谱法、液相色谱-串联质谱法、[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]-质谱法、超高效液相色谱串联质谱法、[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]质谱法等等。 [img]http://www.spcctech.com/FileUpLoad/jsFile/636984539272079206_1.jpg[/img] 行业范围包括工业、化妆品、塑料、电子等多种行业,谱标科技汇总与分析仪器分析方法相关的32项标准如下:[table][tr][td]序号[/td][td]标准编号[/td][td]标准名称[/td][td]代替标准号[/td][td]实施日期[/td][/tr][tr][td]1[/td][td]GB/T 223.88-2019[/td][td]钢铁及合金 钙和镁含量的测定 电感耦合等离子体原子发射光谱法[/td][td] [/td][td]2020/5/1[/td][/tr][tr][td]23[/td][td]GB/T 4698.17-2019[/td][td]海绵钛、钛及钛合金化学分析方法 第17部分: 镁量的测定 火焰[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收光谱[/color][/url]法[/td][td]GB/T 4698.17-1996[/td][td]2020/5/1[/td][/tr][tr][td]24[/td][td]GB/T 4698.21-2019[/td][td]海绵钛、钛及钛合金化学分析方法 第21部分:锰、铬、镍、铝、钼、锡、钒、钇、铜、锆量的测定 原子发射光谱法[/td][td]GB/T 4698.21-1996[/td][td]2020/5/1[/td][/tr][tr][td]31[/td][td]GB/T 6040-2019[/td][td]红外光谱分析方法通则[/td][td]GB/T 6040-2002[/td][td]2020/5/1[/td][/tr][tr][td]36[/td][td]GB/T 7739.13-2019[/td][td]金精矿化学分析方法 第13部分: 铅、锌、铋、镉、铬、砷和汞量的测定 电感耦合等离子体原子发射光谱法[/td][td] [/td][td]2020/5/1[/td][/tr][tr][td]50[/td][td]GB/T 12688.1-2019[/td][td]工业用苯乙烯试验方法 第1部分:纯度及烃类杂质的测定 [url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法[/td][td]GB/T 12688.1-2011[/td][td]2020/5/1[/td][/tr][tr][td]56[/td][td]GB/T 13747.5-2019[/td][td]锆及锆合金化学分析方法 第5部分:铝量的测定 铬天青S-氯化十四烷基吡啶分光光度法[/td][td]GB/T 13747.5-1992[/td][td]2020/5/1[/td][/tr][tr][td]57[/td][td]GB/T 13747.6-2019[/td][td]锆及锆合金化学分析方法 第6部分:铜量的测定 2,9-二甲基-1,10-二氮杂菲分光光度法[/td][td]GB/T 13747.6-1992[/td][td]2020/1/1[/td][/tr][tr][td]63[/td][td]GB/T 15076.3-2019[/td][td]钽铌化学分析方法 第3部分: 铜量的测定 火焰[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收光谱[/color][/url]法[/td][td]GB/T 15076.3-1994[/td][td]2020/1/1[/td][/tr][tr][td]66[/td][td]GB/T 16597-2019[/td][td]冶金产品分析方法 X射线荧光光谱法通则[/td][td]GB/T 16597-1996[/td][td]2020/5/1[/td][/tr][tr][td]94[/td][td]GB/T 20975.28-2019[/td][td]铝及铝合金化学分析方法 第28部分:钴含量的测定 火焰[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收光谱[/color][/url]法[/td][td] [/td][td]2020/5/1[/td][/tr][tr][td]95[/td][td]GB/T 20975.29-2019[/td][td]铝及铝合金化学分析方法 第29部分:钼含量的测定 硫氰酸盐分光光度法[/td][td] [/td][td]2020/5/1[/td][/tr][tr][td]96[/td][td]GB/T 20975.30-2019[/td][td]铝及铝合金化学分析方法 第30部分:氢含量的测定 加热提取热导法[/td][td] [/td][td]2020/5/1[/td][/tr][tr][td]97[/td][td]GB/T 20975.31-2019[/td][td]铝及铝合金化学分析方法 第31部分:磷含量的测定 钼蓝分光光度法[/td][td] [/td][td]2020/5/1[/td][/tr][tr][td]98[/td][td]GB/T 21114-2019[/td][td]耐火材料 X射线荧光光谱化学分析 熔铸玻璃片法[/td][td]GB/T 21114-2007[/td][td]2020/5/1[/td][/tr][tr][td]111[/td][td]GB/T 23524-2019[/td][td]石油化工废铂催化剂化学分析方法 铂含量的测定 电感耦合等离子体原子发射光谱法[/td][td]GB/T 23524-2009[/td][td]2020/5/1[/td][/tr][tr][td]123[/td][td]GB/T 24583.8-2019[/td][td]钒氮合金 硅、锰、磷、铝含量的测定 电感耦合等离子体原子发射光谱法[/td][td]GB/T 24583.8-2009[/td][td]2020/5/1[/td][/tr][tr][td]191[/td][td]GB/T 37544-2019[/td][td]化妆品中邻伞花烃-5-醇等6种酚类抗菌剂的测定 高效液相色谱法[/td][td] [/td][td]2020/1/1[/td][/tr][tr][td]192[/td][td]GB/T 37545-2019[/td][td]化妆品中38种准用着色剂的测定 高效液相色谱法[/td][td] [/td][td]2020/1/1[/td][/tr][tr][td]268[/td][td]GB/T 37626-2019[/td][td]化妆品中阿莫西林等9种禁用青霉素类抗生素的测定 液相色谱-串联质谱法[/td][td] [/td][td]2020/1/1[/td][/tr][tr][td]271[/td][td]GB/T 37628-2019[/td][td]化妆品中黄芪甲苷、芍药苷、连翘苷和连翘酯苷A的测定 高效液相色谱法[/td][td] [/td][td]2020/1/1[/td][/tr][tr][td]281[/td][td]GB/T 37638-2019[/td][td]塑料制品中多溴联苯和多溴二苯醚的测定 高效液相色谱法[/td][td] [/td][td]2020/1/1[/td][/tr][tr][td]282[/td][td]GB/T 37639-2019[/td][td]塑料制品中多溴联苯和多溴二苯醚的测定 [url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]-质谱法[/td][td] [/td][td]2020/1/1[/td][/tr][tr][td]283[/td][td]GB/T 37640-2019[/td][td]化妆品中氯乙醛、2,4-二羟基-3-甲基苯甲醛、巴豆醛、苯乙酮、2-亚戊基环己酮、戊二醛含量的测定 高效液相色谱法[/td][td] [/td][td]2020/1/1[/td][/tr][tr][td]284[/td][td]GB/T 37641-2019[/td][td]化妆品中2,3,5,4'-四羟基二苯乙烯-2-O-β-D-葡萄糖苷的测定 高效液相色谱法[/td][td] [/td][td]2020/1/1[/td][/tr][tr][td]287[/td][td]GB/T 37644-2019[/td][td]化妆品中8-羟基喹啉和硝羟喹啉的测定 高效液相色谱法[/td][td] [/td][td]2020/1/1[/td][/tr][tr][td]292[/td][td]GB/T 37649-2019[/td][td]化妆品中硫柳汞和苯基汞的测定 高效液相色谱-[url=https://insevent.instrument.com.cn/t/yp][color=#3333ff]电感耦合等离子体质谱[/color][/url]法[/td][td] [/td][td]2020/1/1[/td][/tr][tr][td]312[/td][td]GB/T 37667-2019[/td][td]煤灰中铁、钙、镁、钾、钠、锰、磷、铝、钛、钡和锶的测定 电感耦合等离子体原子发射光谱法[/td][td] [/td][td]2020/1/1[/td][/tr][tr][td]316[/td][td]GB/T 37673-2019[/td][td]煤灰中硅、铝、铁、钙、镁、钠、钾、磷、钛、锰、钡、锶的测定 X射线荧光光谱法[/td][td] [/td][td]2020/1/1[/td][/tr][tr][td]359[/td][td]GB/T 37757-2019[/td][td]电子电气产品用材料和零部件中挥发性有机物释放速率的测定 释放测试舱-[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]质谱法[/td][td] [/td][td]2020/1/1[/td][/tr][tr][td]362[/td][td]GB/T 37760-2019[/td][td]电子电气产品中全氟辛酸和全氟辛烷磺酸的测定 超高效液相色谱串联质谱法[/td][td] [/td][td]2020/1/1[/td][/tr][tr][td]375[/td][td]GB/T 8381.7-2009[/td][td]饲料中喹乙醇的测定 高效液相色谱法《第1号修改单》[/td][td]GB/T 8381.7-2005[/td][td]2019/6/4[/td][/tr][/table]
[font='Times New Roman']急需以下三个标准[/font][font='Times New Roman']GB/T 22661.1-2008 氟硼酸钾化学分析方法 第1部分:试样的制备和贮存[/font]GB/T 22661.3-2008 氟硼酸钾化学分析方法 第3部分:氟硼酸钾含量的测定 氢氧化钠容量法[font='Times New Roman']GB/T 22662.3-2008 氟钛酸钾化学分析方法 第3部分:氟钛酸钾含量的测定 硫酸高铁铵容量法[/font][font='Times New Roman'][/font]
[align=center][b]GB 22255-2014 食品安全国家标准 食品中三氯蔗糖(蔗糖素)的测定——三氯蔗糖标准品分析-RI[/b][/align]三氯蔗糖(TGS),是唯一以蔗糖为原料的功能性甜味剂,甜度可达蔗糖600倍。这种[url=http://baike.sogou.com/v130009.htm][color=windowtext]甜味剂[/color][/url]具有无能量,甜度高,甜味纯正,高度安全等特点,是最优秀的功能性甜味剂之一。[align=center][img=,170,99]http://ng1.17img.cn/bbsfiles/images/2018/03/201803080920210187_4197_2222981_3.png!w170x99.jpg[/img][/align][align=center]三氯蔗糖结构式[/align]实验室前期按照《GB 22255-2014 食品安全国家标准食品中三氯蔗糖(蔗糖素)的测定》方法,使用高灵敏度气溶胶型检测器——纳克级水凝粒子计数检测器(NQAD),得到了三氯蔗糖标准品的良好分析结果。本实验按照相同条件,使用示差折光检测器(RI)对三氯蔗糖标准品进行分析。色谱柱同样选择中等极性的普适型色谱柱CAPCELL PAK C[sub]18 [/sub]MGII S5 4.6 mm i.d. × 150 mm,得到结果如图1所示。三氯蔗糖保留时间为12.400min,与标准谱图保留时间基本一致,理论塔板数为12350,不对称因子为0.95,峰形良好。[align=center][img=,690,489]http://ng1.17img.cn/bbsfiles/images/2018/03/201803080945469257_8172_2222981_3.png!w690x489.jpg[/img][/align][align=center]图1 三氯蔗糖标准品分析色谱图(0.4 mg/mL)[/align]*注:峰上标数字由下至上依次为保留时间、理论塔板数及不对称因子。[img=,472,187]http://ng1.17img.cn/bbsfiles/images/2018/03/201803080945471937_6640_2222981_3.png!w472x187.jpg[/img][align=center][img=,690,435]http://ng1.17img.cn/bbsfiles/images/2018/03/201803080946205953_7240_2222981_3.png!w690x435.jpg[/img][/align][align=center]附图:GB方法中标准色谱图[/align]接下来,按照国标要求配制三氯蔗糖工作液,0.02 mg/mL、0.05 mg/mL、0.1 mg/mL、0.2 mg/mL、0.4 mg/mL,进行线性考察实验。线性实验结果如图2所示,R[sup]2[/sup]=0.9939,得到良好线性结果。同时,由于低浓度0.02 mg/mL、0.05 mg/ mL标准品溶液均未检出色谱峰,因此根据标准曲线最高浓度的信噪比计算出检出限(以S/N=3计)约为0.17 mg/ mL。[align=center][img=,650,398]http://ng1.17img.cn/bbsfiles/images/2018/03/201803080947051037_4812_2222981_3.png!w650x398.jpg[/img][/align][align=center]图2 三氯蔗糖标准曲线图[/align]综上,按照《GB 22255-2014 食品安全国家标准食品中三氯蔗糖(蔗糖素)的测定》方法,使用示差检测器(RI)进行检测,以及CAPCELL PAK C[sub]18[/sub] MGII S5 4.6 mm i.d. ×150 mm色谱柱进行分析,可得到三氯蔗糖标准品的良好线性分析结果;但RI检测器的检测灵敏度较低。
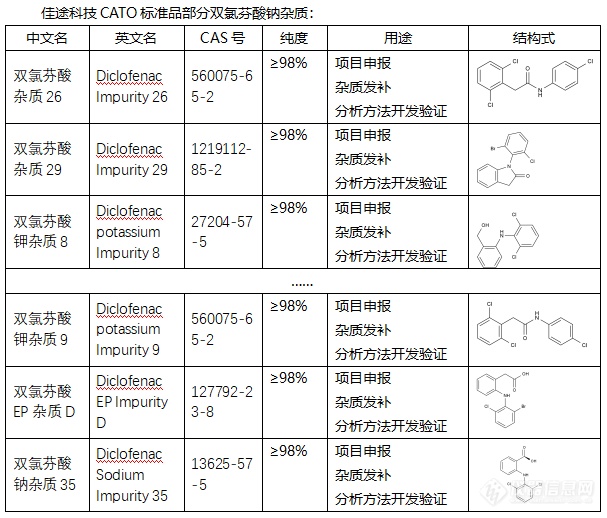
◇双氯芬酸钠杂质在双氯芬酸钠的生产和储存过程中,可能会产生一些杂质,双氯芬酸钠的杂质有多种,包括但不限于以下几种:双氯芬酸钠杂质A:这是一种具有特定CAS号(15362-40-0)和分子式(C14H9Cl2NO2)的杂质。其分子量为278.13,密度为1.4±0.1 g/cm3,沸点为488.6±45.0°C at 760 mmhg,熔点为115-119°C;双氯芬酸钠杂质(1-(2,6-DICHLOROPHENYL)INDOLIN-2,3-DIONE):这是一种具有CAS号的杂质,其化学式为C14H7Cl2NO2。双氯芬酸钠的其他杂质:除了上述两种杂质外,双氯芬酸钠还可能存在其他杂质,如乙酰氯芬酸杂质、醋氯芬酸杂质等。CATO标准品提供的双氯芬酸钠全套的杂质,这些杂质对于药物的纯度和稳定性研究至关重要,也是药物研发过程中不可或缺的一部分。[img=,607,518]https://ng1.17img.cn/bbsfiles/images/2024/02/202402192056045756_8062_6381607_3.png!w607x518.jpg[/img]广州佳途科技股份有限公司深知药物研发与质量控制的重要性,CATO标准品厂家,提供双氯芬酸钠全套的杂质,为客户提供更加精准、可靠的分析标准品,助力药物研发事业的快速发展,以满足客户在药物研发和质量控制方面的需求。
[align=center][b]GB 22255-2014 食品安全国家标准 食品中三氯蔗糖(蔗糖素)的测定——三氯蔗糖标准品分析-NQAD检测器[/b][/align]三氯蔗糖(TGS),是唯一以蔗糖为原料的功能性甜味剂,甜度可达蔗糖600倍。这种甜味剂具有无能量,甜度高,甜味纯正,高度安全等特点,是最优秀的功能性甜味剂之一。[align=center][img=,170,99]http://ng1.17img.cn/bbsfiles/images/2018/03/201803011004470313_2453_2222981_3.png!w170x99.jpg[/img][/align][align=center]三氯蔗糖结构式[/align]本实验按照[b]《GB 22255-2014 食品安全国家标准 食品中三氯蔗糖(蔗糖素)的测定》[/b]方法,使用[b][color=#ff0000]高灵敏度气溶胶型检测器——纳克级水凝粒子计数检测器(NQAD)[/color][/b]对三氯蔗糖标准品进行了分析。色谱柱选择中等极性普适型[color=#3333ff][b]CAPCELL PAK C18 MGII S5 4.6 mm i.d. × 150 mm[/b][/color],得到结果如图1所示。三氯蔗糖保留时间为12.709min,[b]与标准谱图保留时间基本一致,理论塔板数为9992,不对称因子为1.06,峰形良好。[/b][align=center][b][img=,690,497]http://ng1.17img.cn/bbsfiles/images/2018/03/201803011006155125_1559_2222981_3.png!w690x497.jpg[/img][/b][/align][align=center]图1 三氯蔗糖标准品分析色谱图[/align]*注:峰上标数字由下至上依次为保留时间、理论塔板数及不对称因子。[b][img=,633,176]http://ng1.17img.cn/bbsfiles/images/2018/03/201803011006367633_3986_2222981_3.png!w633x176.jpg[/img]附图:GB方法中标准色谱图[/b][align=center][b][img=,690,448]http://ng1.17img.cn/bbsfiles/images/2018/03/201803011007162573_9264_2222981_3.png!w690x448.jpg[/img][/b][/align][b][/b]接下来,按照国标要求配制三氯蔗糖工作液,浓度分别为0.02 mg/mL、0.05 mg/mL、0.1 mg/mL、0.2 mg/mL、0.4 mg/mL,进行线性考察实验。[b][color=#3333ff]由于NQAD检测器原理与常规蒸发光散射检测器ELSD不同,能够直接得到线性回归结果,不需要做对数方程,更加简单快捷。[/color][/b]线性结果如图2所示,R[sup]2[/sup]=0.996,得到良好线性结果。同时,我们根据标准曲线最低浓度的信噪比计算出定量限(以S/N=10计)约为3 μg/mL,[b][color=#ff0000]能够实现三氯蔗糖的高灵敏度检出[/color][/b]。[align=center][img=,658,399]http://ng1.17img.cn/bbsfiles/images/2018/03/201803011008425185_5014_2222981_3.png!w658x399.jpg[/img][/align][align=center]图2 三氯蔗糖标准曲线图[/align]
[color=#333333]食品中磷化铝的标准分析方法[/color]
我们实验室是检测饲料和原料含量的,以前别人介绍用过中国农业科技院分析检测中心研制的氨基酸分析用校核标准品(参比物),名字我忘记了,只记得有代号,1#、2#、3#、4#、5# 这5种。有知道的老师们请帮帮忙!
急需:HG\T3886-2006苄嘧磺隆二氯喹啉酸可湿性粉剂 标准,请大家帮帮忙!谢谢
成分分析中硫酸法测定棉和聚酯纤维的含量时,标准要求1克样品200ML硫酸,这量比较大,大家有用100ML硫酸做过实验吗?
8-羟基喹啉重量法(GB/T 5059.1-1985)1.方法提要试样以硝酸-氯酸钾饱和溶解分解,钼成钼酸析出,用EDTA络合铁等杂质,加氨水溶解钼酸按,过滤。取部分溶液,pH约4.8醋酸-醋酸按缓冲溶液中,以8-羟基喹啉沉淀钼用玻璃坩埚过滤,在130~140℃电热干燥箱内干燥称重,借此测得钼的含量。本方法适用于钼精矿中钼量的测定。测定范围:大于10%2.试剂氨水(比重0.9)硝酸-氯酸钾饱和溶液:加氯酸钾在硝酸(比重1.42)中,成饱和溶液。乙二胺四乙酸二钠ETDA(C10H12FeN2NaO8• 3H2O)(5 %)。甲基橙(0.1%)。盐酸(1+1)。醋酸-醋酸铵缓冲溶液(pH4.8):280克醋酸铵,用水溶解后,加220毫升冰醋酸,再用水稀释至1000毫升,混匀。8-羟基喹啉(3%):30克8-羟基喹啉溶解在1000毫升2mol/L醋酸中(冰醋酸118毫升)。3.分析步骤称取0.2305克试样,置于250毫升烧杯中,加15~20毫升硝酸-氯酸钾饱和溶液,待剧烈作用后,低温加热分解并蒸发至溶液约剩5毫升,取下,冷却。用少量水吹洗表面皿及烧杯内壁,加20毫升5 % ETDA溶液,摇匀,用氨水(比重0.9)中和至钼酸沉淀全部溶解并过量2毫升,冷却至室温,移入100毫升容量瓶中,用慢速滤纸干过滤,移取50.00毫升试液,置于250毫升烧杯中,加1滴0.1%甲基橙指示剂,用盐酸(1+1)中和至溶液颜色刚好变红,加5毫升醋酸-醋酸按缓冲溶液,用水稀释至溶液约120毫升,煮沸,取下。在不断搅拌下,徐徐加10毫升3%8-羟基喹啉溶液,置于低温电炉上静置2~3分钟。取下,用已在130~140℃干燥过并称重的3~4#玻璃坩埚过滤,用热水洗涤烧杯3~4次,洗涤沉淀8~10次,将坩埚连同沉淀置于130~140℃电电热干燥箱中干燥1小时,取出,置于干燥器内冷却至室温,称重。并反复千燥至恒重。钼的百分含量按下式计算; 式中;W1——玻璃坩埚与8-羟基喹啉钼的重量(克);,W2——玻璃坩埚的重量(克);V——试液总体积(毫升);V1——分取试液体积(毫升);W——称样量(克);0.2305——8-羟基喹啉钼换算成钼的系数。4.允许差含钼量(%)允许差(%)≤200.2520.01~3000.3030.000.405.注意事项①加硝酸-氯酸钾饱和溶液时应防止加入固体氯酸钾。如试样分解不完全应重复加硝酸-氯酸钾和溶液至试样分解完全。②此时不宜将溶液煮沸,否则沉淀会溅跳粘附在烧杯内壁上不易洗净。
刚刚看到国标GB/T 22110-2008 食品中反式脂肪酸的测定-气相色谱法 中用到反-9、12、15十八碳三烯酸甲酯标准品(trans-9,trans-12,trans-15-Octadecatrienoic acid methyl ester),有谁买过,哪个品牌可以提供?谢谢!
[color=#444444]我用辛酸甲酯methyloctanoate (C9H18O2) 做[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]的内标物,绘制不同分析物的标准曲线,各个分析物的相对校正因子差别很大。实验室人告诉我,如果分析物和内标结构差不多,那么校正因子越接近1。这是对的,不过有的化合物化学式差不多,结构却相差很多,这样校正因子差别也很大,我要如何判断我做出来的标准曲线和相对校正因子是对的呢?[/color][color=#444444]比如,我用辛酸甲酯做内标,测了两个化合物,苯乙酸(C8H8O2,含苯环和羧酸)和香兰素(C8H8O3,含苯环,羰基,甲氧基和羟基)。其中苯乙酸相对辛酸甲酯差别不是很大,而香兰素差别就大了。所以他们的标准曲线分别是y=0.7311x-0.0525 R2=0.99998,y-1.1526x-0.1764 R2=0.9982。不知道它们的校正因子对不对?有大神帮忙分析一下吗?[/color]
1 绿色食品添加剂使用准则中华人民共和国农业行业标准NY/T 392--2000绿色食品添加剂使用准则,4 .2 .7规定:“在任何情况下,绿色食品中不得使用下列食品添加剂”。其中包括生产绿色食品粉条不使用食品漂白剂…硫磺。(硫磺对人体的不良作用……)不得使用硫磺,但在生产控制与检测监督工作中,怎样才能检查出所检粉条中是否含有并使用过硫磺,对申报产品是否进行绿色食品认可的重要基础之一。但恰恰在实际工作中因缺乏相应的检测方法,而使这一工作陷人困境。2 生产控制与检测监督工作存在的问题例如,有一甘薯淀粉制品…甘薯粉条产品,申报绿色食品产品认可,绿色食品授权检测中心对其进行质量品质检测与评价工作,依据NY/T 392-200绿色食品添加剂使用准则规定,硫磺不得使用于此类产品,但该准则并未规定禁止使用亚硫酸盐等添加剂 而且,目前可以采用的国家检测方法标准主要是GB/T5009 .34一 1996食品中亚硫酸盐的测定方法 其他产品中亚硫酸盐的测定方法还有多种,但这些方法都共同存在着一个问题,即最终结果对硫磺、亚硫酸盐等添加剂无法予以区别,都是在最后结果时通过计算以二氧化硫或亚硫酸盐计。如此以来,当依据现行国家标准检测方法进行粉条产品含硫磺检测时,会出现这样一种无法结论的情况,如该样品未检测出二氧化硫,则该样品可下结论未使用硫磺 但如检测出二氧化硫时,该样品却无法下结论是否是硫磺使用所致,即可能是使用硫磺所致,也可能不是硫磺而是其他亚硫酸盐所致。在这种情况下检测中心检测后无法明确进行被检样品是否使用硫磺的判定。至此,绿色食品申报程序与检测工作过程因检测结果无法下结论而中断。也就是绿色食品添加剂禁用的漂白剂硫磺,因无明确专一的检测方法讲行监督控制.形成限量标准禁用与检测方法标准无法明确检测之间的矛盾。没有监督的禁用本质上是无效的,在实际工作中会出现2个极端情况:(l)严格要求时,实际是只要检出硫元素存在,结果就可以换算为二氧化硫,此时,因不能判明是否是硫磺所致,粉条产品就不能判定未使用硫磺,即该产品不能成为绿色食品 (2)第一种检验结论方式对许多未使用硫磺的粉条类产品显失公平,但如果不这样判定,当检出样品中硫时,是不是加有硫碘检测中心和认证管理机构只有相信申报受检者自己的承诺保证了,如此,禁止使用的规定本质上成为一种无效的要求。同样的问题在绿色食品添加剂标准中仍然存在,如明矾禁止使用,但现行的标准方法亦不能检测粉条中的明矾,而是检测铝离子 检铝离子就无法判断铝是粉条中本身具有的,还是工艺过程中加人的明矾,如此,又同硫磺的检测一样进人一个不能下结论状态,从而使检测方法和限量标准之间存在矛盾。无法落实检查的要求等于没做要求,这种要求只会给实际工作造成麻烦,使检测中心无所适从,导致检测与判定工作无法作出科学明确的结论。3 建议标准制定单位对标准进行进一步完善,如果提出的禁用指标在现条件下还无法从检测技术上予以解决,到不如将此类指标给出最低限量指标要求(如铝),指标高于背景值即可,如此,似更为科学实用。象粉条中使用硫磺的问题,现阶段禁止使用一切含硫化合物进入绿色食品粉条加工过程,才能实现绿色食品二氧化硫不得检出的要求,绿色食品申报检测工作中部分粉条生产企业的产品证明是可以做到的.
《食品安全国家标准 食品中农药最大残留限量》2016版正式颁布实施,这一农药残留的新国标,在标准数量和覆盖率上都有了较大突破,规定了433种农药在13大类农产品中4140个残留限量,较2014版增加490项,基本涵盖了我国已批准使用的常用农药和居民日常消费的主要农产品。 食品伙伴网标法中心结合2016版标准前言部分内容与2014版进行相应的对比分析,供参考: 1、对原标准中氟唑磺隆、甲咪唑烟酸、氟吡菌胺、三唑酮和三唑醇等5种农药残留物定义,敌草快等5种农药每日允许摄入量等信息进行了核实,修订了敌草快、三环锡等5种农药的ADI值。 2、增加了2,4-滴异辛酯等46种农药;增加了490项农药最大残留限量标准 2014版规定了食品中2,4-滴等387种农药3650项最大残留限量,2016版规定了433种2,4-滴等农药4140项最大残留限量。增加了46种农药:2,4-滴异辛酯、2甲4氯异辛酯、苯嘧磺草胺、苯嗪草酮、吡唑草胺、丙硫多菌灵、除虫菊素、毒草胺、多抗霉素、呋虫胺、氟吡菌酰胺、复硝酚钠、甲磺草胺、井冈霉素、抗倒酯、苦参碱、醚苯磺隆、嘧啶肟草醚、扑草净、嗪草酸甲酯、氰氟虫腙、氰烯菌酯、炔苯酰草胺、噻虫胺、三苯基乙酸锡、三氯吡氧乙酸、杀螺胺乙醇胺盐、莎稗磷、虱螨脲、特丁津、调环酸钙、五氟磺草胺、烯丙苯噻唑、烯肟菌酯、烯效唑、辛菌胺、辛酰溴苯腈、溴氰虫酰胺、唑胺菌酯、唑啉草酯、啶菌噁唑、丁吡吗啉、噁唑酰草胺、甲哌鎓、丁酰肼、唑嘧菌胺。 3、增加 12 项检测方法标准,删除1项检测方法标准 增加了SN/T 0162、SN 0198、SN/T 0931、SN/T 1624、SN/T 1989、SN/T 2229、SN/T 2231、SN/T 2237、SN/T 2323、SN/T 2387、SN/T 2795、SN/T 2807,删除了SN/T 0711,其中SN 0198标准已于2015年12月31日被认监委废止,废止依据为《国家认监委办公室关于公布2015年检验检疫行业标准复审结论的通知》。 4、修改了丙环唑等8种农药的英文通用名 修改了丙环唑、六六六、烯肟菌胺、氯啶菌酯、杀虫双、四氯苯酞、氯氟吡氧乙酸和氯氟吡氧乙酸异辛酯 5、将苯噻酰草胺和灭锈胺的限量值由临时限量修改为正式限量;对资料性附录 A 进行了修订,增加了干制蔬菜等3种食品名称,修改1项作物名 食品伙伴网对附录A部分内容的对比发现如下变化: 1)水果(核果类)的类别说明增加了青梅,枣修改为枣(鲜)。 2)水果(浆果和其他小型水果)的类别说明中露莓增加了备注:包括波森莓和罗甘莓。 3)水果(热带和亚热带水果)的类别说明中将大型果的木瓜修改为番木瓜。 4)干制水果的类别说明中增加了枣(干)等。 5)食品类别名称修改:将饮料修改为饮料类。 6、食品伙伴网在对比过程中发现,2016版标准除了以上所列变化外,还修正了其他一些内容: 1)引用的标准名称的修正,如GB/T 19648、GB/T 19469等部分标准的名称中 “兽”字已删除。 2)引用的作废标准的修正,如2014版标准中引用的是2006版GB/T 20770的标准名称,2016版标准已经修正为2008版GB/T 20770的标准名称。 3)农药中文名称修改:2014版标准中的2甲4氯(钠)修改为2甲4氯钠。 4)附录A中动物源食品部分类别的测定部位描述进行了修正。
问:请问乙酰氯的GC分析方法极其标准?我们是是化工企业,生产高纯乙酰氯,分析量很大,我们目前用GC-FID毛细管柱子,没有标准品,含量大约99。5%,请问这样分析的缺陷,用HPLC行不行?假如有其他微量杂质,是否还要用质谱仪?
标定盐酸标准滴定溶液的不确定度分析 作者:吴文英 张春雨 唐惠兰 来源:中华医学研究杂志 在理化分析过程中,一切测量结果都不可避免地具有不确定度。盐酸标准溶液是常用化学定量参比物质,其标定值的准确性直接影响常规分析质量。笔者以GB/T601《滴定分析(容量分析)用标准液的制备》为依据配制并标定盐酸根据JJF1059-1999《测定不确定度评定与表示》分析其测量不确定度。简述由标定过程中得到的不确定度。 1 实验部分 1.1 测定方法[1,2] 准确称量270℃~300℃干燥至恒重的基准碳酸钠(99.95%~100.05%)约0.2g左右,电子分析天平(精度为0.1mg),置于三角瓶中,加入50ml水使之溶解,加指示剂,用盐酸标准液滴定至终点同时作试剂空白实验。 1.2 主要计量仪器与试剂 电了分析天平:AG204;酸式滴定管:50ml A级。 1.3 建立数学模型 C=m (V1-V2)×0.05300 式中 C:盐酸标准滴定溶液的浓度(mol/L);m:基准无水碳酸钠的质量(g);V1:盐酸标准滴定溶液用量(ml);V2:试剂空白实验中盐酸标准滴定溶液用量(ml);0.05300:与1.00ml盐酸标准溶液[C(HCl)=1.000mol/L]相当于以克表示的无水碳酸钠的质量。 1.4 盐酸标准滴定溶液的标定结果 为获得标准溶液重复测量的不确定度分量,对同一标准溶液进行8次独立的标定。测定数据见表1。 表1 盐酸标准滴定溶液的标定结果 略 2 测量不确定度来源 从检测过程和数学模型分析,标定盐酸标准溶液的不确定度主要来源,由四个方面所引起。(1)测量的重复性(A类不确定度);(2)基准无水碳酸钠的纯度;(3)测量使用的电子分析天平及量具;(4)其他相关常数。 3 测量不确定度分析 3.1 A类不确定度的分析 利用表1中的测量结果,按照A类评定测量重复性的标准不确定度。具体计算过程:重复测量的平均值计算式:=1 n∑8 i=1xi=0.09951mol/L 单次测量的标准差按贝塞尔公式计算s(x)为 s(x)=∑8 i=1(xi-)2 n-1=0.0001555mol/L 的标准差s()为 s()=s(x) n=0.000155 8=0.0000548mol/L=5.48×10-5mol/L 由测量重复性引起的相对标准不确定度为U(x):0.0000548/0.09951=0.055%。 3.2 B类不确定度分析 3.2.1 基准碳酸钠的纯度 基准碳酸钠的纯度为1.0000±0.0005,视为矩形分布0.00053=0.00029,则标准不确定度为:由基准碳酸钠的纯度引入的相对不确定度u(p)为:0.029%。 3.2.2 天平称量所引入的标准不确定度 干燥器与天平称量仓内均放置同质硅胶,视为相同湿度,称量时无吸潮。电子天平检定证书标出线性为上0.2mg;可视为矩形分布,则标准不确定度为:因为称量采用的是减量法,故称量的标准不确定度为0.2mg /3=0.12mg:因为称量采用的是减量法,故称量的标准不确定度为:2×0.122=0.17mg,则由称量引入的相对标准不确定度u(m)为:0.17mg/0.2018g=0.084%。 3.2.3 标定体积的不确定度 (1)滴定管的校准:滴定使用50ml酸式滴定管(A级),按照检定规程,其最大允许误差为±0.05ml,相对允许误差为±0.1%,按照矩形分布,则滴定体积的相对标准不确定度u(V)为:u(V)=0.1%/3=0.0577%。(2)环境温度:实验环境在空调条件下,室温近似20℃。温度在20℃左右,标准溶液的温度补正值非常小,对实验结果影响可忽略不计,所以在不确定度分析中不把一温度影响引起的不确定度列入考虑范围。(3)滴定终点的判断:终点时的误差±0.05ml(1滴的体积),两点分布,现由终点分布判断引入的标准不确定度为0.05ml:相对标准不确定度为0.05ml/38.32ml=0.13%标定体积的影响引入相对标准不确定度U(V)为0.0572+0.132=0.142%。 3.2.4 其他常数 基准无水碳酸钠摩尔质量引起的标准不确定度很小,可以忽略。 4 合成标准不确定度 测量重复性、基准无水碳酸钠的纯度、天平称量、标定体积等的不确定度相互独立,故将上述数据合成得盐酸的相对合成标准不确定度U(C)为0.0552+0.0292+0.0842+0.1422=0.176%。 5 扩展不确定度 实验测得盐酸标准溶液浓度为0.09951mol/L,则测量结果的合成标准不确定度U(C)=0.09951mol/L×0.176%=0.000175mol/L。若取包含因子K=2,得测量结果的扩展不确定度U=2U(C)=0.00035mol/L。 6 测量结果的表示 盐酸标准滴定溶液的浓度可表示为:(0.09951±0.00035mol/L,K=2)。 【参考文献】 1 姚正堂,将已峰.奶制品中蛋白质测定的不确定度分析.中华医学研究杂志,2005,5(6):6. 2 国家技术监督局.JJF1059-1999测量不确定度与表示.北京:中国计量出版社,1997,81. 作者单位: 214171 江苏无锡,无锡市惠山区疾病预防控制中心







 我要推广仪器
我要推广仪器
 下载APP
下载APP