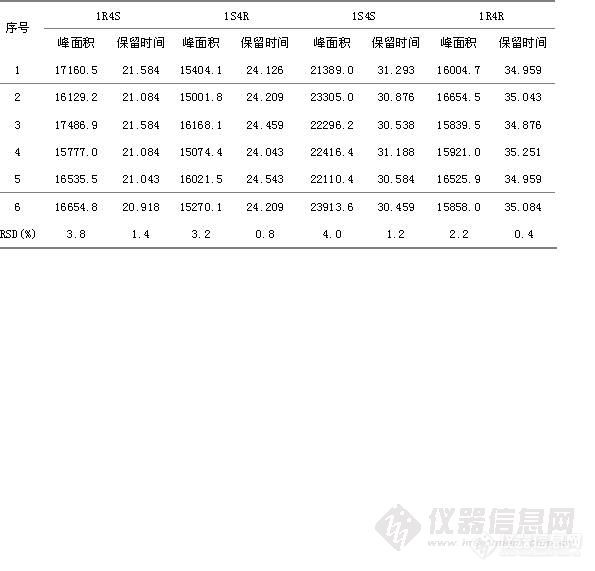
HPLC法测定盐酸舍曲林片的有关物质 摘要:目的:建立盐酸舍曲林片有关物质的测定方法,其外标法测定其四种光学异构体,自身对照法测定其它有关杂质。方法:采用依利特Hypersil ODS2色谱柱(4.6mm×150mm,5μm);流动相为磷酸二氢钠缓冲液 β-环糊精12.8g,HP-β-环糊精17.3g,加水至1000ml]-乙腈[font=Times New Roman]-[font=宋体]三乙胺([font=Times New Roman]800[font=宋体]:[font=Times New Roman]200[font=宋体]:[font=Times New Roman]10[font=宋体])[font=Times New Roman],[font=宋体]用磷酸调节[font=Times New Roman]p H [font=宋体]为[font=Times New Roman]2.5±0.5为流动相,流速为每分钟1ml/min,检测波长为214nm。结果:建立了β-环糊精、HP-β-环糊精手性流动相拆分盐酸舍曲林光学异构体,的方法,结论:该方法专属性好,无干扰,盐酸舍曲林异构体与主峰分离良好。可作为控制盐酸舍曲林片质量的方法。关键词:HPLC外标法;HPLC自身对照法;盐酸舍曲林片;光学异构体; β-环糊精; HP-β-环糊精。盐酸舍曲林是一种选择性抑制[font=Times New Roman]5-[font=宋体]羟色胺再摄取的抗抑郁药([font=Times New Roman]SSRI[font=宋体]),为新一代抗抑郁药的代表药物,目前已跃成为抗抑郁治疗的一线药和首选药。盐酸舍曲林由美国辉瑞公司开发,并于[font=]1990[font=宋体]年[font=]12[font=宋体]月最初在英国上市,目前为止已经在[font=]30[font=宋体]个国家陆续上市。盐酸舍曲林(商品名:郁尔复片剂)于[font=Times New Roman]1996[font=宋体]年[font=Times New Roman]12[font=宋体]月获得中国药品行政保护,并于当年在中国以片剂形式(不含原料药)上市,用于治疗精神抑郁症。现在国内各大医院、药店均有销售。盐酸舍曲林在中国无专利保护。[font=Times New Roman]2004[font=宋体]年[font=Times New Roman]6[font=宋体]月该行政保护终止。由于盐酸舍曲林存在4种光学异构体,在合成工艺过程中可能会引入中间杂质盐酸舍曲林顺式左旋异构体,盐酸舍曲林反式左旋异构体,反式右旋异构体,因其均为手性异构体,眼前其异构体检测方法为毛细管电泳法,且只对其异构体顺式左旋异构体进行检测,其它杂质检测采用反相HPLC,且其盐酸舍曲林主峰不能很好的与其相邻异构体分开,参照相关文献,并经方法学研究,建立了盐酸舍曲林有关物质的检测方法,采用HPLC外标法测定[
怎样用液相测定舍曲林的含量?波长,流动相?
各位老师好!请问有没有人知道顺式氯氰菊酯和反式氯氰菊酯左右旋体的旋光度各是多少?
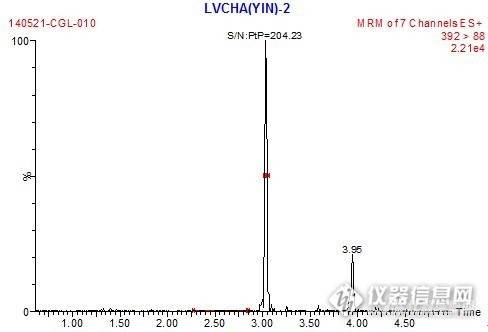
【生活中的仪器分析】样 品:蔬菜、水果、茶叶、茶粉等食品检测项目:草甘膦、草铵膦、氨甲基膦酸参考标准:SN/T 1923-2007检测仪器:a.WATERS液相色谱串联质谱仪:配有电喷雾(ESI)离子源(可用其他品牌作用等效的高效液相色谱质谱仪替代)b.Biotagevacmaster固相萃取仪c.IPRE Qclean PMG草甘膦专用固相萃取柱d.BiotageTurbovap LV 快速浓缩仪e.IKA MS3 涡旋混匀器g.TOMY-MX307离心机g.昆山超声波清洗器实验过程:1.提取及预处理称取2-5g(精确到0.001g)试样于50 mL聚丙烯离心管中,加入100μL内标液,加入20.0 mL水超声提取30min,于10000 r/min离心5min,取1.0 mL上清液于2mL子弹头离心管中,加入100μL酸度调节剂(注A),涡旋混匀,15000r/min离心5 min,待净化。注A:酸度调节剂配制方法:纯水+色谱纯甲醇+盐酸=160+40+13.4(V/V/V)2.固相萃取净化І将PMG-І柱(蓝柱)用2mL甲醇和2 mL 0.5%甲酸淋洗活化并自然滴干,将加入酸度调节剂处理的提取液(2)转移到小柱上,用5mL刻度试管收集流出液(1-2滴/秒),用1.0mL 0.5%甲酸洗柱并真空抽干,合并流出液,用移液枪吸取50%NaOH调pH7-9(用1-14pH试纸,根据样品不同约20-50μL),加水定容到3 mL刻度,混匀,待衍生。3.衍生步骤准确吸取600 μL净化液(3)于2mL子弹头离心管中,加入200 μL 5%硼砂溶液,边涡旋,边加入200 μL 25g/L FMOC-Cl乙腈溶液(注B),放置10min,加入50 μL甲酸,涡旋混匀,15000 r/min离心5min,吸取上清液准备过PMG-ІІ柱。注B:25 g/L FMOC-Cl乙腈溶液配制方法:称取0.25 gFMOC-Cl,溶解于10 mL色谱纯乙腈中。4.固相萃取净化ІІ 将PMG-ІІ柱(红柱)用2 mL甲醇和2 mL 0.5%甲酸淋洗并自然滴干,将上清液过PMG-ІІ柱,用3 mL水淋洗小柱,真空抽干5-10 min,再加入2 mL正己烷淋洗小柱,滴干后真空抽干5 min,最后用5 mL 5%氨水/甲醇洗脱小柱(1-2 mL/min)并用5 mL刻度试管收集流出液,45℃,氮气吹至近干,用20%乙腈定容1.0 mL,涡旋混匀,过0.2 μm PTFE膜后上机测试。5.测定5.1色谱条件a.色谱柱:Waters BEH-C18,1.7 μm,2.1 mm×100 mm;b.流动相:5mmol/L乙酸铵:乙腈梯度洗脱,梯度表见表1; 表1 流动相及梯度 时间(min)流速(mL/min)5mmol/L乙酸铵(%)乙腈(%)00.3901020.362384.40.362384.50.35956.50.35956.60.390109.00.39010c.检测器:串联四极杆质谱仪;d.柱温:35℃;e.进样量:10 μL。5.2质谱分析条件a)电离源:电喷雾正离子模式;b)毛细管电压:3.50KV;c)源温度:120℃;d)脱溶剂气温度:400℃;e)脱溶剂气流量:700L/h;f)碰撞室压力:2.7í10-3mbar;g)特征离子及参数见表2。 表 2 草甘膦和氨甲基膦酸的主要特征离子 化合物保留时间(min)母离子+(m/z)锥孔电压(V)子离子(m/z)碰撞能量(eV)草甘膦1.32392.215*88.02515214.01
旋光性物质的核磁与相应外消旋物质有什么不同?
目的:建立盐酸舍曲林片含量及有关物质测定的高效液相色谱方法。方法:色谱柱为Diamonsil C18(4.6mm×200mm,5μm);流动相为甲醇-0.2%三乙胺溶液(用磷酸调到pH4.0)(60∶40),流速1.5mL/min;检测波长225nm。结果:进样量在2.49~19.94μg范围内,与峰面积线性关系良好,平均回收率为99.55%,RSD为0.60%(n=9)。结论:该方法简单、快速、准确,重现性及专属性好,可用于盐酸舍曲林片的质量控制。
如果中间产物有一个手性碳,并不分离,是外消旋体,环合之后,又出现一个手性碳, 上面两个不同的氢,是会出现两个小峰吗?对其他的影响呢?谢谢!!
2-甲基苯并咪唑和邻硝基苯胺的的紫外吸光度事多少?
求助各位版友,如何用HPLC分析吗啉、甲基吗啉和nmmo?根据吗啉结构应该紫外吸收很弱吧,用紫外检测器能做么?谢谢
在珠三角,大大小小的工業區充斥這個城市的每一個角落,然而連接這一個個工業區的不是別的,就是為這些工業區的普通員工提供飲食的小攤,駐紮在街道兩旁,架鍋起灶,每逢下班高峰,整個街道就像是哪個飯店的廚房,鍋聲瓢聲吆喝聲伴隨著不斷經過車輛的喇叭聲,場面極其夸張。然而隱藏在喧囂下的是這些沒有受過多少教育的普工的健康危機,而這種危機就藏在這些小攤的鍋盆裡:在一次不經意中,我發現了這些小攤的炒菜用油,我驚奇的發現,基本大部分的攤位用的油都是一種外觀,渾濁不堪,色如機車潤滑油脂,在我看了幾家後,我心頭的疑惑更大了,這也不像是我之前了解的地溝油,那會是怎麼樣的油,而且如此廣泛的運用如此不明來歷的油,讓我不免對我們這些打工姐妹兄弟產生憐憫之心!
請問專家:ICP-AES在進行濃度曲線標定時,一般取几個濃度點(除空白點外),Linearity應為達到多少才可以正常使用。另外問一下,濃度點的選取是否與我們所測樣品的大概范圍有關,即如果待測元素是微量相應選取的濃度值可以大一些,如果待測元素是痕量相應選取的濃度值要小一些,謝謝!!
请问外消旋体在碳谱和氢谱上有什么特点,如何利用核磁来区分对映异构体和非对映异构体
求助求助:关于吗啉及甲基吗啉的液相紫外检测方法,各位大侠帮帮忙啊
长前胡【英文名】root of West szechwan Hogfennel【来源】药材基源:为伞形科植物长前胡的根。拉丁植物动物矿物名:Peucedanum turgeniifolium wolff.采收和储藏:秋季采挖根,去除茎叶,洗净,晒干。【原形态】多年生草本,高40-80cm。根圆柱形,长8-15cm,径0.5-1.5cm,下部常分枝,顶部残留众多棕色的叶鞘纤维。茎单一,具细长纵条纹,下部分枝,常带淡紫色,下部光滑,上部粗糙,有短毛。基生叶叶柄长1-7cm,基部具略带紫色的狭窄叶鞘,抱茎;叶片轮廓长卵形,二至三回羽状分裂,第一回羽片3-4对,末国顺裂片线形、倒披针形或倒卵形,基部楔形,先端裂片基部渐狭呈楔形,长1-2.5cm,宽0。5-1。5cm,边缘具2-3粗锯齿或呈浅齿状,叶柄及下表面常短糙毛,边缘具短睫毛;茎上部叶无柄,具叶鞘抱茎,叶片一回羽状分裂,裂片狭长,细小。复伞形花序顶生和侧生,花序梗粗壮,顶端多糙毛;无总苞片;伞辐5-12(-20),有短毛;小总苞片8-12,线状披针形,先端长渐尖,密生短柔毛;小伞花序有花10-20朵,花柄有毛;萼齿细小、不显着;花瓣近圆形,折色,外有稀疏柔毛;花柱基圆锥形,花柱向下弯曲。分生果卵状椭圆形,背部扁压,长3-3.5mm,宽2-3mm,有稀疏短毛,背棱和中材呈线形突起,侧棱狭翅状;每棱槽内有油管3-4,合生面油管6-8(-10)。花期7-9月,果期9-10月。【生境分布】生态环境:生于海拔2000-3600m的高山阳性山坡、草地、灌丛和河谷滩地上。资源分布:分布于甘肃、四川等地。【化学成份】全草含甘露醇(D-mannitol),长前胡甲素(tur-geniifolin A),长前胡乙(turgeniifolin B),长前胡丙素(turaeni-folin C),顺式消旋凯诺内酯(cis-khellactone),反式消旋凯诺内酯(trans-khellactone),7-羟基-8-(2′,3′-二羟基-3′-甲基-丁基)香豆精,异氧化前胡素(isooxypeucedanin),消旋美丽前胡素(peuformo-sin),消旋双异戊酰凯诺内酯(diisovalerylhellactone)。【性味】味苦;辛微寒【归经】归肺经【功能主治】宣散风热;降气祛痰。主风热感冒;咳喘痰黄;头痛;胸闷【用法用量】内服:3-9g。
急用亚甲基兰脱色方法请教亚甲基兰脱色方法的详细步骤,感激不尽,谢谢!!!
文献上用亚甲基蓝脱色力来衡量活性炭的能力,想问一下甲基蓝脱色力是不是就是标准上说的甲基蓝吸附量?如果不是,那它是什么?具体怎么算一直弄不明白谢谢了
求助大家一个问题,顺式环辛烯在紫外光后可以转化为逆式环辛烯,我需要测定溶液中顺式环辛烯和逆式环辛烯的浓度,但是市面上只搜到了顺-环辛烯,我怎么样才能确定逆式环辛烯的浓度呢?谢谢大家解答~
今天做氯仿中甲基对硫磷~FPD检测器~配的5,10,15,20,25的浓度曲线……分流比10曲线线性很不好~为什么??难道必须用不分流低浓度进样嘛?
现在应该说这一点是清楚的:氰戊菊酯其中一个峰一定会与顺式氰戊菊酯重合,无论采取什么分离方式。因为氰戊菊酯本身就包含顺式氰戊菊酯。这种说法应该没错吧?问题是,我现在就是需要计算同一个茶样中,氰戊菊酯和顺式氰戊菊酯2个项目的含量。氰戊菊酯的含量我一直有做,就是用氰戊菊酯的标曲来计算,每个浓度的氰戊菊酯标样都有2个峰,计算时用的是组校准,而非浓度合计。而我的疑问就是:不论是出几个峰,茶样中顺式氰戊菊酯的含量该如何计算?应该用顺式氰戊菊酯的标样作曲线,从而计算含量;而不应该是直接用氰戊菊酯的标曲计算,对吧?不好意思,脑袋愚笨,多有打扰!!!
今天主要讲解色谱峰拖尾的原因、提供相应的解决方法,并阐述色谱峰拖尾柱外效应的概念、原理及其在实际应用中的影响。 [b]一、色谱峰拖尾的原因[/b] 色谱峰拖尾是色谱分析中常见的问题,其原因复杂多样,主要包括以下几个方面: [list=1][*][font=-apple-system, BlinkMacSystemFont, &]仪器因素[/font]: [list][*]进样量过大:过多的样品进入色谱柱,导致峰形展宽和拖尾。 [*]色谱柱安装不正确:如柱子两端安装位置不当,泄漏或柱端切割不平整。 [*]色谱柱污染或流失:柱内填料污染或流失,影响分离效果。 [*]载气流速问题:载气流速过高或过低,都可能引起拖尾。 [*]进样器污染:进样针或汽化室衬管污染,影响样品进样。 [/list][*][font=-apple-system, BlinkMacSystemFont, &]样品因素[/font]: [list][*]样品性质:样品中的某些成分可能在柱填料上强烈吸附和解吸,导致拖尾。 [*]样品浓度:高浓度样品溶液中,溶质的吸附和解吸过程更加明显,增加拖尾现象。 [*]样品溶剂不合适:溶剂的洗脱强度过大或过小,都可能导致拖尾。 [/list][*][font=-apple-system, BlinkMacSystemFont, &]操作条件[/font]: [list][*]柱温过低:柱温不足会影响溶质在柱填料上的扩散速度,增加拖尾。 [*]流速控制不当:流速过高或过低都会影响分离效果,导致拖尾。 [/list][/list][b]二、色谱峰拖尾的解决方法[/b] 针对上述原因,可以采取以下措施解决色谱峰拖尾问题: [list=1][*][font=-apple-system, BlinkMacSystemFont, &]优化仪器条件[/font]: [list][*]检查并调整进样量,避免过大。 [*]确保色谱柱正确安装,无泄漏且柱端切割平整。 [*]定期维护和更换色谱柱,避免污染和流失。 [*]调整载气流速至适宜范围。 [*]清洁进样器和汽化室衬管,确保无污染。 [/list][*][font=-apple-system, BlinkMacSystemFont, &]改善样品制备[/font]: [list][*]优化样品前处理方法,减少杂质干扰。 [*]适当稀释高浓度样品,控制进样浓度。 [*]选择合适的样品溶剂,避免溶剂效应。 [/list][*][font=-apple-system, BlinkMacSystemFont, &]调整操作条件[/font]: [list][*]提高柱温,促进溶质扩散。 [*]优化流速设置,确保分离效果。 [*]必要时可更换更高效的色谱柱或调整流动相组成。[/list][/list][list][*][b]三、色谱峰拖尾柱外效应的概念、原理及影响[/b][/list]柱外效应是指色谱柱之外的因素导致的色谱峰展宽现象,主要由进样装置、检测池及它们与柱之间的连接管路等因素引起。这些因素会增加样品的扩散和传质阻力,从而导致色谱峰形展宽和拖尾。 原理:柱外效应通过增加样品的纵向扩散和传质阻力来恶化峰形。例如,管路过长或直径过粗、管路接头不匹配或有死体积等都会增加柱外效应。 影响:在实际应用中,柱外效应对色谱分析的准确性和重现性产生显著影响。它会导致色谱峰展宽、拖尾甚至分裂,降低分离度和灵敏度。因此,在色谱分析中应尽可能减小柱外效应的影响,以提高分析结果的准确性和可靠性。 为减小柱外效应的影响,可采取以下措施: [list][*]缩短连接管路长度,减小死体积。 [*]使用管径适中且内壁光滑的管路。 [*]确保管路接头匹配良好,无泄漏。 [*]优化进样装置和检测池的设计,减少样品在其中的滞留时间。 [*]综上所述,色谱峰拖尾是色谱分析中常见的问题,其原因复杂多样。通过优化仪器条件、改善样品制备和调整操作条件等措施可以有效解决这一问题。同时,应关注柱外效应对色谱分析的影响,并采取相应措施减小其影响以提高分析结果的准确性和可靠性。[/list]
用UV1902紫外分光光度计测水中硝态氮,双波长法220nm、275nm下侧吸光值,今天的标曲吸光值突然和前面几个月的相差很大,甚至出现很多负值,标曲已经做不出来,是不是仪器坏了,该如何检查?
各位老师:我做水中有机磷的扩项。用的5750.9-2006的方法。做敌敌畏,甲拌磷,对硫磷,甲基对硫磷。内吸磷,乐果,马拉硫磷,毒死蜱八种农药。水样250mL加50mL的二氯甲烷萃取。结果一下午做的三个加标。加标1,振荡时间和静置分层时间都没控制,收集萃取液后,用加无水硫酸钠的漏斗脱水。回收率除内吸磷外。其他在70%以上。加标2和加标3同时做的,振荡时间比较统一,均为2分钟左右。但是静置分层时间没有控制。结果加标2除内吸磷回收率为0以外。其他都还好。但是加标3除敌敌畏,甲基对硫磷以外都很差。全部三个加标都是,先加标再用氯化钠饱和,再做的。操作差别也不大。最后都是50度水浴,氮吹。怎么结果差那么大呢?各位老师,能不能帮忙查找下原因?能不能告诉下,这个实验的注意事项。先谢谢各位老师了!
[b]1 方法依据[/b]本方法依据GB 5009.188-2003《蔬菜、水果中甲基托布津、多菌灵的测定》。[b]2 方法原理[/b]用甲醇自试样中提取出甲基托布津,在pH 1~2时,用二氯甲烷提取,甲基托布津经闭环反应转变为多菌灵,提纯后,用紫外分光光度法进行定量测定。多菌灵经提取后可直接测定吸光度而进行定量。[b]3 试剂[/b]3.1 甲醇3.2 二氯甲烷3.3 三氯甲烷3.4 石油醚:沸程30℃~60℃3.5 乙酸-乙酸铜溶液:称取2g乙酸铜,加100ml冰乙酸,稍加热溶解,用水稀释至200ml3.6 盐酸(1+11):量取盐酸90ml,用水稀释至1000ml3.7 氢氧化钠溶液(80g/L):称取8g氢氧化钠,加水溶解并稀释至100ml3.8 氢氧化铵溶液(1+7):量取氨水10ml,加水稀释至80ml3.9 氯化钠溶液(100g/L)3.10 甲基托布津标准溶液:准确称取50.0mg甲基托布津,置于烧杯中,用三氯甲烷溶解并转移至50ml容量瓶中,稀释至刻度。此溶液每毫升相当于1.0mg甲基托布津。3.11 甲基托布津标准使用液:吸取10.0ml甲基托布津标准溶液置于100ml容量瓶中,加三氯甲烷稀释至刻度,此溶液每毫升相当于100.0ug甲基托布津。3.12 多菌灵标准溶液:准确称取50.0mg多菌灵置于烧杯中,用盐酸(1+11)溶解移入50ml容量瓶中,并稀释至刻度,此溶液每毫升相当于1.0mg多菌灵。3.13 多菌灵标准使用液:吸取10.0ml多菌灵标准溶液,置于100ml容量瓶中,加盐酸(1+11)稀释至刻度。此溶液每毫升相当于100.0ug多菌灵。[b]4仪器[/b]4.1 空气冷凝管4.2 紫外分光光度计[b]5分析步骤[/b]5.1 标准曲线的制备5.1.1甲基托布津标准曲线:吸取0、0.10、0.30、0.50ml甲基托布津标准使用液(相当于0、10、30、50ug甲基托布津),分别置于30ml圆底离心管中,挥干溶剂后,各加10ml乙酸-乙酸铜溶液及2粒玻璃珠,接上空气冷凝管,小火缓缓煮沸0.5h,取下,用20ml盐酸(1+11)从冷凝管顶端洗涤冷凝管和圆底离心管,并移入125ml分液漏斗中,用二氯甲烷提取二次,每次10ml,弃去二氯甲烷层,酸溶液中加25ml氢氧化钠溶液(80g/L)至pH6.0~6.5(pH试纸试),用二氯甲烷提取二次,每次20ml,合并二氯甲烷提取液,用10ml水洗涤一次,静置分层后,将二氯甲烷层分入另一个干的分液漏斗中,准确加入10ml盐酸(1+11),振摇5min,静置分层后,盐酸提取液用1cm石英比色杯,以盐酸(1+11)调节分光光度计零点,测读250nm~300nm的吸光度,以波长为横坐标,吸光度为纵坐标,绘制吸收图谱。将图谱上260nm和290nm吸光度读数点连成直线,设直线上282nm的吸光度A,两者之差为ΔA(ΔA=A-A’,为校正吸光度)。再以校正吸光度为纵坐标,甲基托布津的含量为横坐标,绘制各甲基托布津标准点ΔA值的标准曲线。5.1.2多菌灵标准曲线:吸取0、0.10、0.30、0.50ml多菌灵标准使用液(相当于0、10、30、50ug多菌灵),置于盛有20ml盐酸(1+11)的分液漏斗中,各用二氯甲烷提取二次,每次10ml,弃去二氯甲烷层,水溶液用氢氧化铵(1+7)中和到pH为6.0~6.5(pH试纸试),用二氯甲烷提取二次,每次20ml,提取液用10ml水洗涤一次,以下按甲基托布津自“将二氯甲烷层分入另一个干的分液漏斗中,准确加入10ml盐酸(1+11)”起依法操作,并绘制吸收图谱,计算出ΔA值后,绘制多菌灵的标准曲线。5.2 试样的提取和分离称取50.0g切碎、混匀的试样,加50ml甲醇振摇0.5h,用布什漏斗抽滤,容器和滤器用甲醇洗涤二次,每次15ml~20ml,抽干后,滤液移入烧杯中,抽滤瓶用约10ml水洗涤,洗液并入滤液内,在水浴上用空气流吹去部分甲醇后,移入分液漏斗中,加30ml氯化钠溶液(100g/L),用石油醚振摇提取二次,每次25ml,弃去石油醚,加盐酸酸化至pH为1~2(用pH试纸试),用二氯甲烷提取二次,每次25ml,合并二氯甲烷提取液,用25ml水洗涤一次分出二氯甲烷层留作甲基托布津测定用。水洗涤液合并入水层,留作多菌灵测定用。5.3 甲基托布津的测定二氯甲烷提取液自然挥干后,用10ml乙酸-乙酸铜溶液分次溶解残渣,并转入30ml圆底离心管中,加2粒玻璃珠,以下按5.1.1自“接上空气冷凝管”起依法操作,计算出试样的ΔA值,再与甲基托布津的标准曲线比较,计算试样中的含量。5.4 多菌灵的测定 取5.2中留作多菌灵测定的水溶液,再氢氧化铵(1+7)中和至pH6.0~6.5,然后按5.1.2多菌灵标准曲线“用二氯甲烷提取二次”起依法操作,计算出试样中ΔA值,再与多菌灵标准曲线比较,计算试样中的含量。[b]6结果计算[/b]试样中甲基托布津、多菌灵含量按下式计算: X=m′×1000/m×1000式中: X —试样中甲基托布津、多菌灵含量,单位为毫克每千克(mg/kg)m′[i]—[/i]样品中甲基托布津、多菌灵含量,单位为微克(ug);m —试样的质量,单位为克(g);计算结果表示到两位有效数字。[b]7试验结果报告[/b]7.1校准曲线及线性范围: [table][tr][td=1,3] [align=center]工作[/align] [align=center]曲线[/align] [/td][td] [align=center]顺序号[/align] [/td][td] [align=center]1[/align] [/td][td] [align=center]2[/align] [/td][td] [align=center]3[/align] [/td][td] [align=center]4[/align] [/td][/tr][tr][td] [align=center]质量( ug )[/align] [/td][td] [align=center]0.00[/align] [/td][td] [align=center]10.00[/align] [/td][td] [align=center]30.00[/align] [/td][td] [align=center]50.00[/align] [/td][/tr][tr][td] [align=center]吸光值(A)[/align] [/td][td] [align=center]0.000[/align] [/td][td] [align=center]0.035[/align] [/td][td] [align=center]0.093[/align] [/td][td] [align=center]0.163[/align] [/td][/tr][tr][td=2,1] [align=center]回归方程[/align] [/td][td=4,1] [align=center]y = 0.0032x +0.0004[/align] [/td][/tr][tr][td=2,1] [align=center]相关系数[/align] [/td][td=4,1] [align=center]0.9984[/align] [/td][/tr][/table]7.2 方法的精密度称取约50.0g样品,按照步骤5处理,平行制备9份样品,计算的含量并求出相对标准偏差,结果见下表所示。 [table=620][tr][td=2,1] [align=center]顺序号[/align] [/td][td] [align=center]1[/align] [/td][td] [align=center]2[/align] [/td][td] [align=center]3[/align] [/td][td] [align=center]4[/align] [/td][td] [align=center]5[/align] [/td][td] [align=center]6[/align] [/td][td] [align=center]7[/align] [/td][td] [align=center]8[/align] [/td][td] [align=center]9[/align] [/td][/tr][tr][td=1,4] [align=center]1[/align] [/td][td] [align=center]取样量(g)[/align] [/td][td] [align=center]50.0176[/align] [/td][td] [align=center]50.0229[/align] [/td][td] [align=center]50.0010[/align] [/td][td] [align=center]50.0098[/align] [/td][td] [align=center]50.0076[/align] [/td][td] [align=center]50.0089[/align] [/td][td] [align=center]50.0006[/align] [/td][td] [align=center]50.0097[/align] [/td][td] [align=center]50.0011[/align] [/td][/tr][tr][td] [align=center]吸光度(A)[/align] [/td][td] [align=center]0.041[/align] [/td][td] [align=center]0.041[/align] [/td][td] [align=center]0.043[/align] [/td][td] [align=center]0.042[/align] [/td][td] [align=center]0.041[/align] [/td][td] [align=center]0.04[/align] [/td][td] [align=center]0.042[/align] [/td][td] [align=center]0.041[/align] [/td][td] [align=center]0.040[/align] [/td][/tr][tr][td] [align=center]多菌灵含量(ug)[/align] [/td][td] [align=right]14.06 [/align] [/td][td] [align=right]14.06 [/align] [/td][td] [align=right]14.69 [/align] [/td][td] [align=right]14.38 [/align] [/td][td] [align=right]14.06 [/align] [/td][td] [align=right]13.75 [/align] [/td][td] [align=right]14.38 [/align] [/td][td] [align=right]14.06 [/align] [/td][td] [align=right]13.75 [/align] [/td][/tr][tr][td] [align=center]浓度 mg/kg[/align] [/td][td] [align=right]0.26 [/align] [/td][td] [align=right]0.26 [/align] [/td][td] [align=right]0.27 [/align] [/td][td] [align=right]0.26 [/align] [/td][td] [align=right]0.26 [/align] [/td][td] [align=right]0.25 [/align] [/td][td] [align=right]0.26 [/align] [/td][td] [align=right]0.26 [/align] [/td][td] [align=right]0.25 [/align] [/td][/tr][tr][td] [align=center]平均值[/align] [/td][td=10,1] [align=center]0.26 (S=0.0061)[/align] [/td][/tr][tr][td]相对标准偏差 [align=center]RSD(%)[/align] [/td][td=10,1] [align=center]2.34[/align] [/td][/tr][tr][td=2,1] [align=center]备注[/align] [/td][td=9,1] [align=center]结论:九个平行样品的相对标准偏差为2.34%,小于5%,符合要求。[/align] [/td][/tr][/table]7.3方法的中间精密度随机选取2名实验人员,按照步骤5测定砷的含量,每个实验者平行测定3份样品,求出两者实验结果的相对标准偏差,结果见下表所示。[align=center]表2 中间精密度实验结果[/align] [table][tr][td] [align=center]实验者[/align] [/td][td] [align=center]多菌灵含量(mg/kg)[/align] [/td][td] [align=center]平均结果(mg/kg)[/align] [/td][td] [align=center]相对标准偏差(%)[/align] [/td][/tr][tr][td=1,3] [align=center] [/align] [align=center]甲[/align] [/td][td] [align=center]0.26[/align] [/td][td=1,3] [align=center]0.26[/align] [/td][td=1,6] [align=center]2.99[/align] [/td][/tr][tr][td] [align=center]0.25[/align] [/td][/tr][tr][td] [align=center]0.26[/align] [/td][/tr][tr][td=1,3] [align=center] [/align] [align=center]乙[/align] [/td][td] [align=center]0.24[/align] [/td][td=1,3] [align=center]0.25[/align] [/td][/tr][tr][td] [align=center]0.25[/align] [/td][/tr][tr][td] [align=center]0.25[/align] [/td][/tr][tr][td=4,1] 接受标准:从精密度测试所得的6组数据中间精密度测试的相对标准偏差应小于5.0%。[/td][/tr][/table]7.4方法的准确度分别向试样中加入一定量的多菌灵,使其砷的加标浓度为10ug/L的样品2ml、分别制备3份样品,按照步骤5处理,同时测定未加标样品硫脲含量,分别计算回收率、结果见下表所示。 [table][tr][td=3,1] [align=center]样品[/align] [/td][td=3,1] [align=center]标准品[/align] [/td][td=3,1] [align=center]样品加标测量值[/align] [/td][td=1,2] [align=center]回收率(%)[/align] [/td][td=1,2] [align=center]平均[/align] [align=center]回收率(%)[/align] [/td][/tr][tr][td] [align=center]浓度(mg/kg)[/align] [align=center] [/align] [/td][td] [align=center]取样量(g)[/align] [/td][td] [align=center]多菌灵含量[/align] [align=center](ug)[/align] [align=center]M[sub]1[/sub][/align] [/td][td] [align=center]浓度(ug/ml)[/align] [/td][td] [align=center]取样量(ml)[/align] [/td][td] [align=center]M[sub]2[/sub][/align] [align=center](ug)[/align] [/td][td] [align=center]吸光度(A)[/align] [/td][td] [align=center]浓度[/align] [align=center](mg/kg)[/align] [/td][td] [align=center]质量[/align] [align=center]M[/align] [/td][/tr][tr][td] [align=right]0.252 [/align] [/td][td] [align=center]50.0305[/align] [/td][td] [align=center]12.6[/align] [/td][td] [align=center]10[/align] [/td][td] [align=center]2[/align] [/td][td] [align=center]20[/align] [/td][td] [align=center]0.331[/align] [/td][td] [align=center]0.63[/align] [/td][td] [align=right]32.81 [/align] [/td][td] [align=center]101.0[/align] [/td][td=1,3] [align=center]102.1[/align] [/td][/tr][tr][td] [align=right]0.248[/align] [/td][td] [align=center]50.0725[/align] [/td][td] [align=center]12.4[/align] [/td][td] [align=center]10[/align] [/td][td] [align=center]2[/align] [/td][td] [align=center]20[/align] [/td][td] [align=center]0.340[/align] [/td][td] [align=center]0.64[/align] [/td][td] [align=right]33.13 [/align] [/td][td] [align=center]103.6[/align] [/td][/tr][tr][td] [align=right]0.256 [/align] [/td][td] [align=center]50.0024[/align] [/td][td] [align=center]12.8[/align] [/td][td] [align=center]10[/align] [/td][td] [align=center]2[/align] [/td][td] [align=center]20[/align] [/td][td] [align=center]0.322[/align] [/td][td] [align=center]0.64[/align] [/td][td] [align=right]33.13 [/align] [/td][td] [align=center]101.6[/align] [/td][/tr][tr][td=3,1] [align=center]备注[/align] [/td][td=8,1] [align=center]△%=(M- M[sub]1[/sub])*100/ M[sub]2[/sub][/align] [/td][/tr][/table]7.5 检出限DL=0.02mg/kg取20次平行测定空白样的结果,按IUPAC规定DL=KSb/50a其中K=3Sb:空白多次测得信号的标准偏差:0.0008a: 校准曲线的斜率:0.0032 [table=595][tr][td] [align=center]顺序号[/align] [/td][td] [align=center]1[/align] [/td][td] [align=center]2[/align] [/td][td] [align=center]3[/align] [/td][td] [align=center]4[/align] [/td][td] [align=center]5[/align] [/td][td] [align=center]6[/align] [/td][td] [align=center]7[/align] [/td][td] [align=center]8[/align] [/td][td] [align=center]9[/align] [/td][td] [align=center]10[/align] [/td][/tr][tr][td] [align=center]吸光度(A)[/align] [/td][td] [align=center]0.007[/align] [/td][td] [align=center]0.008[/align] [/td][td] [align=center]0.007[/align] [/td][td] [align=center]0.006[/align] [/td][td] [align=center]0.007[/align] [/td][td] [align=center]0.006[/align] [/td][td] [align=center]0.008[/align] [/td][td] [align=center]0.007[/align] [/td][td] [align=center]0.008[/align] [/td][td] [align=center]0.007[/align] [/td][/tr][tr][td] [align=center]顺序号[/align] [/td][td] [align=center]11[/align] [/td][td] [align=center]12[/align] [/td][td] [align=center]13[/align] [/td][td] [align=center]14[/align] [/td][td] [align=center]15[/align] [/td][td] [align=center]16[/align] [/td][td] [align=center]17[/align] [/td][td] [align=center]18[/align] [/td][td] [align=center]19[/align] [/td][td] [align=center]20[/align] [/td][/tr][tr][td] [align=center]吸光度(A)[/align] [/td][td] [align=center]0.006[/align] [/td][td] [align=center]0.007[/align] [/td][td] [align=center]0.007[/align] [/td][td] [align=center]0.008[/align] [/td][td] [align=center]0.006[/align] [/td][td] [align=center]0.006[/align] [/td][td] [align=center]0.008[/align] [/td][td] [align=center]0.007[/align] [/td][td] [align=center]0.008[/align] [/td][td] [align=center]0.008[/align] [/td][/tr][tr][td] [align=center]Sb[/align] [/td][td=10,1] [align=center]0.0008[/align] [/td][/tr][tr][td] [align=center]检出限(μg/g )[/align] [/td][td=10,1] [align=center]0.02[/align] [/td][/tr][tr][td] [align=center]公式[/align] [/td][td=10,1] [align=center]DL=f*K*Sb/50a ( K=3, Sb:空白信号的标准偏差,a:曲线斜率)[/align] [/td][/tr][tr][td] [align=center]备注[/align] [/td][td=10,1] [align=center]DL=K*Sb/50a [/align] [/td][/tr][/table][b] [/b]8 是否对方法偏离?是□否■[b]9 结论[/b]我公司对[u]蔬菜、水果中甲基托布津、多菌灵的测定 [/u]的检测符合G[u]B 5009.188-2003[/u]要求。
最近检测样品收样要求检甲基托布津,查资料发现甲基托布津遇水就变成多菌灵了,那测甲基托布津有意义吗?请高手指点

10,抽取5个版友);中奖名单:翠湖园(注册ID:hhx050)牛一牛(注册ID:v2700892)mengzhaocheng(注册ID:mengzhaocheng)yifan1117(注册ID:yifan1117)sixingxing(注册ID:v2889187)http://ng1.17img.cn/bbsfiles/images/2016/12/201612081545_01_1610895_3.jpghttp://ng1.17img.cn/bbsfiles/images/2016/12/201612081545_02_1610895_3.jpg【注意事项】同样的答案,每人只能发一次PS:该贴浏览权限为“回贴仅作者和自己可见”,回复的版友仅能看到版主的题目及自己的回答内容,无法看到其他版友的回复内容。下午3点之后解除,即可看到正确答案、获奖情况及所有版友的回复内容。=======================================================================乳制品中农药残留的检测方法:GC-MS基质:乳制品应用编号:101803化合物:甲胺磷,敌敌畏,异丙威,氧乐果,乐果,林丹,甲基对硫磷,马拉硫磷,倍硫磷,双硫磷,三唑醇,硫丹,DDT,哒螨灵,顺式氰戊菊酯,反式氰戊菊酯,苯醚甲环唑,环氧七氯(内标)固定相:DM-5 MS色谱柱/前处理小柱:DM-5MS 30m x 0.25mm x 0.25um样品前处理:1 样品准备/提取 牛奶、原料乳等液态奶制品 (1)称取10 g样品,加入10 mL乙腈,涡旋混合1 min; (2)再依次加入10 mL乙腈,4 g 氯化钠,2 mL 200 g/L 乙酸铅溶液,涡旋振荡混合5 min,4000 rpm离心5 min; (3)取上清液10 mL,作为上样液待净化。 2 SPE柱净化 ProElut DPC(Cat#:65353) (1)活 化:加入10 mL乙腈,流出液弃去; (2)上 样:将上样液加入柱中,接收流出液; (3)洗 脱:10 mL乙腈洗脱,收集流出液。合并步骤(2)、(3)流出液; (4)浓 缩:在40 ℃下用减压蒸馏将流出液浓缩至大约2 mL,转移至氮吹管,用4 mL乙腈分两次润洗旋蒸瓶,并转移到氮吹管中; 在35 ℃水浴条件下,用氮气缓慢吹至低于1 mL(不能吹干),用乙腈定容至1 mL,供GS-MS检测。色谱条件:GC-MS 分析条件 色谱柱:DM-5MS 30 m×0.25 mm×0.25 μm(Cat.#: 8221) 进样口温度:290 ℃ 升温程序:初始温度40 ℃,保持0.5 min,以30 ℃/min升温至130 ℃,再以5 ℃/min升温至250 ℃,再以10 ℃/min升温至300 ℃,保持5 min 载气:氦气, 流速:1.2 mL/min 进样方式:不分流进样 进样量:1 μL 离子源温度:230 ℃ 接口温度:280 ℃ 溶剂延迟:5 min文章出处:天津迪马实验室关键字:乳制品,农药残留,DM-5MS,8221,ProElut DPC,65353,甲胺磷,敌敌畏,异丙威,氧乐果,乐果,林丹,甲基对硫磷,马拉硫磷,倍硫磷,双硫磷,三唑醇,硫丹,DDT,哒螨灵,顺式氰戊菊酯,反式氰戊菊酯,苯醚甲环唑,环氧七氯(内标)摘要:适用于牛奶等液态乳制品中农药残留的检测。谱图:http://www.dikma.com.cn/Public/Uploads/images/chayezhongnongcan1(1).PNGhttp://www.dikma.com.cn/Public/Uploads/images/chayezhongnongcan2(1).PNGhttp://www.dikma.com.cn/Public/Uploads/images/chayezhongnongcan3(1).PNG图例:1-甲胺磷,2-敌敌畏,3-异丙威,4-氧乐果,5-乐果,6-林丹,7-甲基对硫磷,8-马拉硫磷,9-倍硫磷,10-双硫磷,11-三唑醇,12-硫丹,13-DDT,14-哒螨灵,15-顺式氰戊菊酯,16-反式氰戊菊酯,17-苯醚甲环唑,18-环氧七氯(内标)

“超高效液相色谱-串联质谱法测定大豆中草甘膦及其代谢物氨甲基膦酸的残留”是本人去年开展大豆中草甘膦检测项目整个试验过程的总结,欢迎各位老师和同行批评指正,该文章还未在任何刊物上发表。[align=center][b]超高效液相色谱-串联质谱法测定大豆中草甘膦及其代谢物氨甲基膦酸的残留[/b][/align][align=center][/align][align=center]户江涛[/align][align=center](黑龙江省农垦科学院测试化验中心,黑龙江 佳木斯 154007 )[/align]摘要:采用超高效液相色谱-串联质谱法建立了快速检测大豆中草甘膦和氨甲基膦酸残留量的分析方法。试样经水超声提取,二氯甲烷去除脂肪,C[sub]18[/sub]固相萃取柱净化后,在硼酸钠缓冲溶液中与9-芴甲氧羰酰氯(FMOC-Cl)进行衍生反应,其衍生产物在C[sub]18[/sub]色谱柱上以 2 mmol/L 乙酸铵溶液和乙腈为流动相,进行液相色谱分离:质谱检测采用电喷雾正离子化模式和多反应监测模式(MRM)。结果表明,草甘膦和氨甲基膦酸在0.001~0.5 mg/L范围内线性关系良好,相关系数(R)分别为0.9996和0.9993,定量限(LOQ)均为0.01mg/kg。在空白大豆样品添加浓度为0.02、0.2、2 mg/kg 时,草甘膦和氨甲基膦酸的平均回收率分别为80.2%~91.5%和77.7%~89.3%,相对标准偏差(RSD)分别为3.37%~6.96%和4.11%~8.27%(n=6)。本方法快速、简便、灵敏,适用于大豆中草甘膦和氨甲基膦酸残留的同时检测。关键词:超高效液相色谱-串联质谱;大豆;草甘膦;氨甲基膦酸;衍生反应[align=center]Determination of glyphosate and its metabolite aminomethyl-phosphonic acid residues in soybean by ultra performance liquid chromatography-tandem mass spectrometry[/align][align=center]HU Jiangtao[/align][align=center]([i]Testing and Analysis Center of Heilongjiang Academy of Land Reclamation Sciences, Jiamusi 154007,China[/i])[/align][b]Abstract:[/b]A method[b] [/b]was developed for the determination of glyphosate(PMG) and aminomethyl-phosphonic acid(APMA) residues in soybean by ultra performance liquid chromatography-tandem mass spectrometry(UP[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url]/MS). After extracted with water under ultrasonication, the sample was defatted with dichloromethane and purified by C[sub]18 [/sub]solid phase extraction cartridge, and then PMG and APMA were derivatized using 9-fluorenylmethoxycarbonyl(FMOC-Cl) in borate buffer for 2 h.The derivatives of PMG and APMA were separated on a Waters BEH C[sub]18[/sub] column with gradient elution with the mobile phase of 2 mmol/L ammonium acetate and acetonitrile, and finally detected by positive eletrospray ionization-mass spectrometry(ESI[sup]+[/sup]-MS/MS) in multiple reaction monitoring(MRM) mode.The results showed the linearities of PMG and APMA were good in the concentration range of 0.001~0.5 mg/L ,and the correlation coefficients were 0.9996 and 0.9993 respectively. The limit of quantification(LOQ) of PMG and APMA was both 0.01mg/kg. At the spiked levels of 0.02、0.2、2 mg/kg in the blank soybean samples, the mean recoveries of PMG and APMA were 80.2%~91.5% and 77.7%~89.3% respectively, and the relative standard deviation(RSD) of PMG and APMA were 3.37%~6.96% and 4.11%~8.27% res-pectively(n=6).This method is fast,simple,sensitive, and suitable for simultaneous determination of PMG and APMA in soybean.[b]Key words: [/b]ultra performance liquid chromatography-tandem mass spectrometry (UPLC-MS/MS) soybean glyphosate(PMG) aminomethyl-phosphonic acid(APMA) derivatization草甘膦(Glyphosate,PMG)又名镇草宁、农达,分子式为C[sub]3[/sub]H[sub]8[/sub]NO[sub]5[/sub]P,是1971年美国孟山都公司研发的一种有机磷除草剂,因其兼具内吸、传导性、灭生性及非选择性,同时不易在生物体内累积,故广泛应用于农业生产中一年生和多年生杂草防除,是目前世界上应用最广、生产量最大的除草剂[sup][/sup]。草甘膦及其在植物中的主要代谢物氨甲基膦酸(Aminomethyl-phosphonic acid,APMA,分子式为CH[sub]6[/sub]NO[sub]3[/sub]P)均属于强极性、易溶于水的高沸点化合物,具有不易挥发、无紫外吸收等特性,因此用常规方法分析检测十分困难[sup][/sup]。 目前, PMG和APMA残留检测的方法主要有色谱法(GC[sup][/sup]、LC[sup][/sup]、IC[sup][/sup])、质谱法(GC/MS[sup][/sup]、ICP/MS[sup][/sup]、LC/MS/MS[sup][/sup])、光谱法[sup] [/sup]等。光谱法虽然操作简便,但其灵敏度不高,而[url=https://insevent.instrument.com.cn/t/3p][color=#3333ff]离子色谱[/color][/url]法[sup][/sup]只能适用于水样等简单基质,用于植物源样品检测时干扰太大;用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]和[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质联用[/color][/url]技术检测时,需要将PMG和APMA衍生转化为可气化物质,其引入试剂多、过程相对繁琐,效率较低[sup][/sup];用LC/MS/MS法直接检测时[sup][/sup],由于PMG和APMA分子量(分别为169、111)均较小,其主要碎片离子的质荷比多在100以下,检测实际样品时受基质干扰严重,灵敏度较低,因此柱前衍生——[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质联用[/color][/url]法成为近年来国内外检测PMG和APMA残留的主流方法[sup][/sup]。以9-芴甲氧羰酰氯(FMOC-Cl)做为衍生试剂,在硼酸盐缓冲溶液中与PMG和APMA水提取液相容性好,过程简单,其衍生产物在LC/MS/MS中响应信号高,碎片离子干扰小,适合定性定量分析。 目前,采用柱前衍生——LC/MS/MS法检测茶叶、稻米等基质中PMG和APMA残留的报道很多[sup][/sup],专门针对大豆基质的报道很少。行业标准[sup][/sup]的适用范围虽然包括了大豆基质,但该方法在实验过程中试剂用量大、操作繁琐(反复调pH值)、衍生时间长(需过夜),尤其是使用阳离子交换柱(CAX)洗脱时需要加入11 mL 1%的盐酸甲醇水(20/80,v/v),水分含量过高导致旋转蒸发时很难蒸干,容易造成PMG和APMA回收率不稳定。本文专门针对大豆这类高蛋白、高脂肪含量的特殊基质,采用纯水作为提取试剂,二氯甲烷去除脂溶性杂质,C[sub]18[/sub]固相萃取小柱净化后采用FMOC-Cl衍生,最后用UPLC/MS/MS测定。该方法前处理过程简便、快速、灵敏度高,适用于大豆中PMG和APMA的残留检测。[b]1 实验部分[/b]1.1 材料与试剂 草甘膦、氨甲基膦酸(纯度≥99%,德国Dr.Ehrensorfer公司);FMOC-Cl(纯度99%,Sigma公司),使用时配置成10g/L的丙酮溶液;乙腈、二氯甲烷、甲酸、乙酸铵(色谱纯,美国Fisher公司);十水四硼酸钠(优级纯,天津市科密欧化学试剂有限公司),使用时配置成50g/L的水溶液;实验用水为Millipore纯水仪制备;C[sub]18[/sub]固相萃取小柱(200mg/3ml,美国Agilent公司)。1.2 仪器与设备 Acquity UPLC型超高效液相色谱仪(Waters公司);XEVO TQ-S三重四级杆质谱仪(Waters公司);CR21GⅢ型高速离心机(HITACHI公司);KQ5200DB型台式超声波仪(昆山市超声仪器有限公司);涡旋混合器(IKA公司)。1.3 标准溶液的配置 分别称取草甘膦和氨甲基膦酸标准品10mg(精确到0.1mg),用水溶解并定容至10mL,配置成质量浓度为1.0 mg/mL标准储备液,于4℃冰箱保存待用;使用时用水逐级稀释成所需浓度的混合标准工作液。1.4 样品前处理 提取:称取粉碎均匀后的试样1.0g(精确到0.01g)于50mL聚乙烯离心管中,加入10.0mL超纯水,涡旋混合30 s并超声提取20 min后,以10000 r/min离心3 min,将上清液转移至另一离心管中,加入5 mL二氯甲烷涡旋混合30 s,以10000 r/min离心3 min,上清液待净化。 净化:取2.5 mL上清液加入到C[sub]18[/sub]固相萃取柱(使用前依次用3mL甲醇和3mL超纯水活化)中,弃去最初的几滴流出液(约0.5 mL),将剩余部分用5 mL玻璃管收集,待衍生。 衍生:取1.0 mL净化液于5 mL离心管中,依次加入1.0 mL 50g/L的硼酸钠溶液和 1.0 mL 10g/L的FMOC-Cl衍生液,混匀后室温下衍生2 h,以10000 r/min离心3 min,取上清液过0.22 mm有机系微孔滤膜后,供UPLC/MS/MS分析测定。1.5 液相色谱及质谱条件 液相色谱:色谱柱:Waters BEH C[sub]18[/sub](1.7 μm,50mm×2.1mm);柱温:30℃;流速:0.5 mL/min;进样量:2 μL;流动相A:乙腈;流动相B: 2 mmol /L 的乙酸铵水溶液。梯度洗脱程序:0~0.5min,10% A;0.5~3. 0 min,10%~100% A;3. 0 ~4. 0 min,100%A,4 ~4.1min,100% A~10% A,4.1 ~5.0min 10% A。 质谱:离子源:电喷雾离子源( ESI [sup]+[/sup] ) ;扫描方式:正离子扫描;检测方式:多反应监测( MRM);毛细管电压:3.2 kv;离子源温度:150℃;去溶剂气温度:500℃;去溶剂气流量:1000 L /h;定性、定量离子对及碰撞能量见表1。[align=center]表1 PMG-FMOC和 AMPA-FMOC的MRM质谱参数[/align][align=center]Table 1 MRM parameters of PMG-FMOC and AMPA-FMOC[/align][table][tr][td][align=center]Analyte[/align][/td][td][align=center]Cone/V[/align][/td][td][align=center]Parent ion/(m/z)[/align][/td][td][align=center]Daughter ion/(m/z) [/align][/td][td][align=center]Collision energy/V[/align][/td][/tr][tr][td][align=center]PMG-FMOC[/align][align=center] [/align][align=center]AMPA-FMOC[/align][/td][td][align=center]30[/align][align=center] [/align][align=center]30[/align][/td][td][align=center]392[/align][align=center][sup] [/sup][/align][align=center]334[/align][align=center][sup] [/sup][/align][/td][td][align=center]88[/align][align=center]214﹡[/align][align=center]112﹡[/align][align=center]179[/align][/td][td][align=center]14[/align][align=center]8[/align][align=center]11[/align][align=center]20[/align][/td][/tr][/table]﹡quantitative ion[b]2 结果与讨论[/b]2.1 色谱及质谱条件的优化 流动相的选择:对比了酸性体系(0.1%甲酸水溶液)与非酸性体系(乙酸铵水溶液)分别于甲醇、乙腈的流动相体系组合,结果发现两种分析物在酸性体系中分离效果欠佳,峰形拖尾严重,而在非酸性体系中其色谱分离效果得到明显改善,峰形对称;乙腈比甲醇体系洗脱能力更强,可以有效缩短分析时间。故本研究采用乙酸铵水溶液+乙腈流动相体系,并比较了1、2、5 mmol/L三种乙酸铵浓度与乙腈的组合,结果发现随着乙酸铵浓度的增加,目标物响应值虽略有提高但相差不大,而同时仪器背景值却显著升高,综合考虑目标物信号强度、信噪比、色谱分离效果以及分析时间等因素,本实验最终选择了2 mmol /L 乙酸铵水溶液+乙腈分析体系。质谱的选择:PMG、 AMPA对应的衍生物PMG-FMOC、AMPA-FMOC分子量分别为391、333。用超纯水配置10 mg/L 混合标准溶液直接注射到质谱中,在正负离子模式下分别进行母离子全扫描,发现正离子模式下392、334具有很好的响应,然后分别以392、334为母离子进行子离子全扫描,各得到两组丰度高、干扰小的子离子对进行MRM监测,最终确定的质谱条件见表1。2.2 前处理条件的优化 提取溶液的选择:PMG和APMA属于强极性物质,易溶于水,难溶于有机溶剂,故一般采用极性溶剂提取,如纯水及KOH、NaHCO[sub]3[/sub]溶液等[sup][/sup]。实验发现,用碱性溶液提取后,大豆中脂肪、蛋白等物质会与碱性物质发生反应,导致离心后的提取液异常浑浊,不利于后期净化和衍生,因此本实验采用纯水作为提取试剂,再经二氯甲烷液液萃取去除脂溶性杂质。 净化柱的选择:研究发现,对提取后的溶液不经SPE净化直接进行衍生, PMG和APMA的回收率均不足30%,且精密度很差,这可能是由于大豆中富含脂肪、蛋白质等物质干扰衍生过程,故本实验比较了对脂肪、蛋白质有很好去除效果的C[sub]18[/sub]、中性Al[sub]2[/sub]O[sub]3[/sub]、HLB固相萃取SPE柱的净化效果,结果发现提取液经中性Al[sub]2[/sub]O[sub]3[/sub]净化后,PMG和APMA几乎检测不到;而C[sub]18[/sub]净化后目标物回收率为92.7%、90.8%,HLB为83.6%、80.5%。故本实验选取了净化效果更好,成本相对低廉的C[sub]18[/sub]固相萃取小柱。 衍生条件的优化:FMOC-Cl的衍生机制是在碱性环境下(pH=9.0)通过FMOC-Cl基团取代目标化合物氮原子上的氢,从而生成较稳定的化合物FMOC-Cl。参照行业标准[sup][/sup]及文献报道[sup][/sup]所选用的缓冲液浓度,本实验采用50g/L的硼酸钠水溶液缓冲液体系,设置的衍生试剂质量浓度为1、2、5、10、20 g/L FMOC-Cl丙酮溶液,按照本文1.4步骤对PMG和APMA质量浓度为0.5 mg/L的纯水溶液和大豆空白基质溶液分别进行衍生,结果见图1。结果表明,在纯水溶液中,FMOC-Cl浓度为2 g/L时,PMG和APMA的峰面积已达到最大,随着衍生化试剂浓度的升高,其峰面积无明显变化;而在大豆空白基质溶液中,FMOC-Cl低浓度(1、2g/L)时,PMG和APMA几乎检测不到,其峰面积随衍生化试剂浓度增加而加大,浓度到达一定程度(10 g/L)时,峰面积不再变化。产生这种现象的原因,可能是由于尽管大豆提取液经过了二氯甲烷和C[sub]18[/sub]小柱的净化,但还是会有少量水溶性蛋白、脂肪等杂质残留在净化液中,这些杂质可能会与衍生试剂反应,影响目标物的衍生效果。研究还发现,当FMOC-Cl浓度为20 g/L时,得到的PMG和APMA色谱峰产生拖尾现象,可能是由于衍生试剂化学性质较活泼,其用量大时,过量的FMOC-Cl会迅速转化成FMOC-OH,干扰目标物峰形。在50g/L硼酸钠水溶液、10 g/L FMOC-Cl丙酮溶液条件下,考察不同时间(0.5h、1h、2h、4h、8h和16h)对衍生效果的影响,结果发现,2 h后PMG和APMA的测定值无明显增加。因此,本实验最终选定的衍生条件为50g/L硼酸钠水溶液、10 g/L FMOC-Cl丙酮溶液,室温下衍生2 h。[img=,596,378]http://ng1.17img.cn/bbsfiles/images/2017/07/201707020904_01_2984502_3.png[/img][img=,690,530]http://ng1.17img.cn/bbsfiles/images/2017/07/201707020904_02_2984502_3.png[/img]2.3 基质效应的考察 基质效应(主要是抑制)是LC/MS/MS仪器检测时经常遇到的现象。由于本实验采用极性很强的水作为提取剂,大豆中的色素、脂肪酸等极性较强的物质也有少部分进入到最后的上机液中,在离子化带电过程中会与目标物产生竞争,抑制目标物的离子化效率。实验考察了用PMG和APMA的纯水标样去标定经过本文1.4步骤处理后的大豆空白基质溶液配置的同浓度标样,其色谱图见图2。结果发现,PMG在纯水和大豆空白基质中峰面积基本一致,而APMA在大豆空白基质中的峰面积仅为纯水中的55.7%,产生了明显的基质抑制效应。为了消除基质干扰,本实验选用大豆样品空白基质配置不同浓度的标准溶液来绘制标准曲线进行校准。2.4 线性范围和定量限 用大豆空白基质溶液分别配置0.001、0.005、0.01、0.05、0.1、0.2、0.5 mg/L的PMG和APMA混合标准溶液,按本文1.4步骤衍生后测定,以各自定量离子的峰面积为Y对应质量浓度X(mg/L)做标准曲线,得到的线性方程和相关系数见表2。结果表明,这两种物质在0.001~0.5 mg/L浓度范围内线性良好,相关系数R分别为0.9996和0.9993。以10倍信噪比(S/N)计算,该方法PMG和APMA的定量限(LOQ)均为0.01 mg/kg。[align=center]表2 PMG和APMA大豆基质标准溶液的线性方程、相关系数和定量限(LOQ)[/align][align=center]Table 2 Linear equations,correlation and LOQ of PMG and APMA in the soybean matrix standard solutions[/align][table][tr][td][align=center]Analyte[/align][/td][td][align=center]Linear range/(mg/L)[/align][/td][td][align=center]Linear equation[/align][/td][td][align=center]R[/align][/td][td][align=center]LOQ/(mg/ kg )[/align][/td][/tr][tr][td][align=center]PMG[/align][align=center]AMPA[/align][/td][td][align=center][sup]0.001~0.5[/sup][/align][align=center][sup]0.001~0.5[/sup][/align][/td][td][align=center]Y=889809x+1671.3[/align][align=center]Y=476982x+1161.9[/align][/td][td][align=center]0.9996[/align][align=center]0.9993[/align][/td][td][align=center]0.01[/align][align=center]0.01[/align][/td][/tr][/table]2.5 回收率和精密度 称取大豆空白试样1.0 g,分别添加0.02、0.2、2 mg/kg水平的PMG和APMA混合标样,每个水平重复6次,按照本文1.4步骤前处理方法处理后上机检测,实验结果见表3。从表3可以看出,PMG的平均回收率为80.2%~91.5%,相对标准偏差(RSD,n=6)为3.37%~6.96%;APMA的平均回收率和RSD分别为77.7%~89.3%和4.11%~8.27%。[align=center]表3 大豆中PMG和APMA的加标回收率和相对标准偏差(n=6)[/align][align=center]Table 3 Recoveries and relative standard deviations(RSD)of PMG and APMA spiked in the soybean(n=6) [/align][table][tr][td][align=center]Analyte[/align][/td][td][align=center]Spiked level(mg/kg)[/align][/td][td][align=center]Recovery/%[/align][/td][td][align=center]RSD/%[/align][/td][/tr][tr][td]PMGAMPA[/td][td][align=center]0.02[/align][align=center]0.2[/align][align=center]2[/align][align=center]0.02[/align][align=center]0.2[/align][align=center]2[/align][/td][td][align=center]80.2[/align][align=center]91.5[/align][align=center]86.8[/align][align=center]77.7[/align][align=center]89.3[/align][align=center]85.9[/align][/td][td][align=center]6.96[/align][align=center]3.37[/align][align=center]3.95[/align][align=center]8.27[/align][align=center]4.25[/align][align=center]4.11[/align][/td][/tr][/table][b]3 结语[/b] 本文建立了超高效液相色谱-串联质谱法(UPLC/MS/MS)测定大豆中草甘膦及其代谢物氨甲基膦酸残留的分析方法。该方法灵敏度高,PMG和APMA定量限(LOQ)达到0.01 mg/kg,能满足大豆产品相关限量标准要求。同时该方法具有较高的准确度和精密度,前处理步骤简单快速,特别适合大批量大豆样品的检测。
[size=16px][color=#ff0000][b][url=https://www.instrument.com.cn/job/position-93062.html]立即投递该职位[/url][/b][/color][/size][b]职位名称:[/b]销售工程师[b]职位描述/要求:[/b]1.专科及以上学历,综合素质好;2.市场营销、化学分析、生物制约或自动化等专业;3.对分析仪器的原理和结构有一定了解,或者使用过分析仪器;4.性格开朗、热爱挑战、富有激情、勤奋、吃苦耐劳;5.善于跟别人沟通,能很好地维护客户;6.热爱销售工作,很希望长期从事销售工作;7.具有市场开拓能力和团队精神,为人正直有信用;8.积极推动负责产品的销售工作,完成销售任务;9.有分析仪器相关工作经验者优先。[b]公司介绍:[/b] -吉林省安舍科技有限公司,位于长春市,是安捷伦消耗品,安捷伦服务产品,普兰德,ZETA,近岸等品牌的一级代理商,2013年成立以来,长期稳定的从事化学分析和生命科学领域的相关服务、仪器、消耗品、试剂的销售业务。...[url=https://www.instrument.com.cn/job/position-93062.html]查看全部[/url][align=center][img=,178,176]https://ng1.17img.cn/bbsfiles/images/2021/08/202108160948175602_3528_5026484_3.png!w178x176.jpg[/img][/align][align=center]扫描二维码,关注[b][color=#ff0000]“仪职派”[/color][/b]公众号[/align][align=center][b]即可获取高薪职位[/b][/align]
1.药物鉴别:具有旋光性的药物,在“性状”项下,一般都收载有“比旋度”的检验项目。测定比旋度值可用来鉴别药物或判断药物的纯杂程度。《中国药典》要求测定比旋度的药物很多,如肾上腺素、硫酸奎宁、葡萄糖、丁溴东莨菪碱、头孢噻吩钠等。 例如,肾上腺素比旋度的测定方法为:取本品,精密称定,加盐酸溶液(9→200)溶解并定量稀释制成每1ml中含20mg的溶液,照“旋光度测定法”测定,比旋度应为。 2.杂质检查:具有光学异构体的药物,一般具有相同的理化性质,但其旋光性能不同,一般有左旋体、右旋体和消旋体之分,通过测定药物中杂质的旋光度,可以对药物的纯度进行检查。 例如,硫酸阿托品中杂质莨菪碱的检查,硫酸阿托品为莨菪碱的外消旋体,无旋光性,而莨菪碱为左旋体,莨菪碱虽然作用较强,但毒性也大,常将其作为杂质加以控制。 检查方法为:取本品,按干燥品计算,加水制成每1ml中含50mg的溶液,依法测定,旋光度不得超过。已知莨菪碱的比旋度为,控制莨菪碱的限量为24.6%. 考试大-全国最大教育类网站(www.Examda。com) 3.含量测定:具有旋光性的药物,特别是在无其他更好的方法测定其含量时,可采用旋光度法测定。《中国药典》采用旋光度法测定含量的药物有葡萄糖注射液、葡萄糖氯化钠注射液、右旋糖酐氯化钠注射液、右旋糖酐葡萄糖注射液等。
老师让我测土壤中的氯氰菊酯、莠去津和利谷隆残留量,我想用固相萃取法来净化?(选用的是[B][color=#DC143C]C18[/color][/B]固相萃取小柱),但我不知道选什么淋洗液和洗脱液,只知道要根据极性选,我也看过一些常用溶剂的极性排列表,但还是不知道选什么,因为莠去津和利谷隆是极性较强的,都可以用同一种物质来淋洗和洗脱,但氯氰菊酯是弱极性的,要将这三者同时进行淋洗和洗脱,究竟用什么呢?请大家赶快帮忙出出主意啊,非常感谢!!!
建立了水产品肌肉组织中螺旋霉素、替米考星、泰乐菌素、北里霉素同时测定的超高效液相色谱一紫外检测(UPLC—TUV)方法。样品经乙腈提取后,浓缩至近干,用4% NaC1溶解残渣,正己烷除脂,经固相萃取小柱净化,乙腈洗脱;以乙腈一25 mmo~L磷酸二氢铵(pH 2.5,含10% 乙腈)为流动相,以ACQUITYUPLC BEH Cl8为分离柱,柱温为45℃,流速为0.3 mL/min,紫外检测。方法在0.100~20.0 mg/L范围内呈线性相关,螺旋霉素、替米考星、泰乐菌素和北里霉素的相关系数分别为0.998 7、0.999 3、0.999 4和0.998 0。平均回收率为70%~102%,相对标准偏差为2.9%~11.2% ,螺旋霉素、替米考星、泰乐菌素和北里霉素的检出限分别为25、25、50、75 kg。方法满足水产品肌肉组织中螺旋霉素、替米考星、泰乐菌素和北里霉素的残留量测定。







 我要推广仪器
我要推广仪器
 下载APP
下载APP