如题,在2.9min时乙酸乙酯出峰,在17min左右N,N-二甲基乙酰胺出峰,出峰时间为10min,峰形特别不好,四分之一圆形,我用的是岛津-2010,进样口和检测器温度均为200度,柱温为70度,分流比为10,急求!!!谢谢各位前辈!!
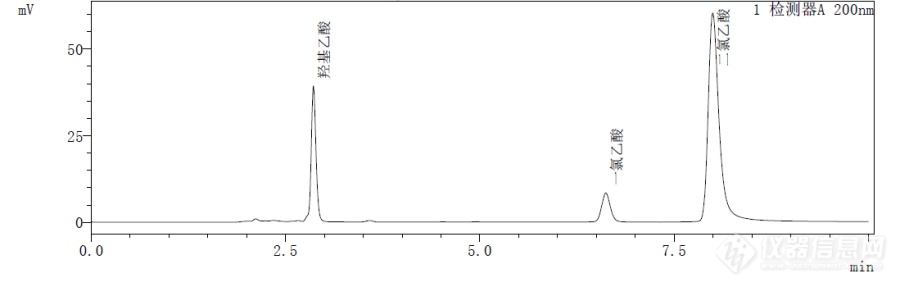
[align=center][b]椰油酰胺丙基甜菜碱中一氯乙酸、二氯乙酸和羟基乙酸的测定[/b][/align] 椰油酰胺丙基甜菜碱(CAB)是一种两性表面活性剂,因其对眼睛和皮肤刺激性低,对头发和皮肤有护理效果并产生大量稳定泡沫,在肥皂和硬水中有出色的起泡性和洗涤性,故广泛用于香波和泡沫浴液等洗涤用品中。 在工业生产中,常使用一氯乙酸(MCA)作为原料生产CAB。而工业MCA中含有少量的二氯乙酸(DCA),DCA是生物学证实具有潜在致癌风险的物质,同时在生产过程中残留的MCA对皮肤、黏膜有很强的腐蚀性,通常采用水解法将MCA转化为刺激性更小的羟基乙酸(GCA)。椰油酰胺丙基甜菜碱产品的指标含量分析中,一般要求一氯乙酸<20ppm,二氯乙酸<300ppm,羟基乙酸<0.5%。[b]色谱条件:[/b]色谱柱:[b]Kromasil C8(4.6*250mm,5μm)[/b]柱 温:24℃检测器:紫外检测器波 长:200nm流动相:乙腈:水=10:90(每1000mL中加入2.0mL磷酸)流 速:1ml/min进样体积:20μL采集时间:10min[img=,690,219]https://ng1.17img.cn/bbsfiles/images/2018/10/201810291003374445_9066_2428063_3.png!w690x219.jpg[/img] 图1 :一氯乙酸、二氯乙酸和羟基乙酸混标色谱图[img=,690,328]https://ng1.17img.cn/bbsfiles/images/2018/10/201810291003547039_780_2428063_3.png!w690x328.jpg[/img] 图2 :椰油酰胺丙基甜菜碱样品色谱图[b]总结[/b]参考国标GB/T 28193-2011表面活性剂中氯乙酸(盐)残留量的测定方法,建立高效液相色谱法,一次性测定样品中一氯乙酸、二氯乙酸和羟基乙酸的含量。其优点是以高比例水相作为流动相,样品不需要进行萃取、酯化等前处理,操作方便,快速高效。使用Kromasil C8色谱柱分离样品中一氯乙酸与其余组分,效率高,分离度好,结果可靠,可为椰油酰胺丙基甜菜碱生产厂家提高产品质量提供参考。[b]注:由深圳爱湾医学检验实验室验证 [/b]
溴乙酸乙酯样品,用DMSO溶解,进样Agilent 7890 + DB624柱 + FID样品不出峰,何解?bp 溴乙酸乙酯159°,DMSO189°难道溶剂与样品发生了什么反应?
分离活性炭管中苯、丙酮、乙酸乙酯,FID检测器,FFAP柱(30*0.32*0.50),用二硫化碳做溶剂,请问能用用同一个条件:进样口温度150℃,检测器温度180℃,柱温40℃,以5℃/min升至80℃。把物质分离出来吗?谢谢
如果同时想做苯,甲苯,乙酸乙酯,乙酸丁酯,丙酮,丁酮,三氯乙烯,二氯乙烷,环己酮的话应该悬着什么样的色谱柱,什么条件.......如果实在做不到一起的话,最合适的搭配,方法,条件是什么???.
各位兄弟姐妹,请问羟丙基-β-环糊精在乙酸乙酯的溶解度是多少?我查了很多文献都找不到答案,打算自己去测定了!但是具体该怎么做?希望各位能指点一下小弟!
测定乙酸乙酯残留,溶剂为二甲基亚砜,测定精密度,发现有一些溶剂峰没有出现,而有的溶剂峰却出现了。请教大家是怎么回事,应该主溶剂的峰都是应该出现的啊,而且很大。溶剂峰和乙酸乙酯峰分离度很好。乙酸乙酯出峰在10分钟,二甲基亚砜出峰在17分钟。
昨天做实验发现 往浓硫酸加入乙酸乙酯时 没有分层而是乙酸乙酯全部溶到浓硫酸里面。这时什么原因呀?浓硫酸能氧化乙酸乙酯吗?请高手指教?谢谢!
在对黄连进行薄层色谱的时候,用环己烷:乙酸乙酯:异丙醇:甲醇:水:三乙胺(3:3.5;1:1.5:0.5:1)进行展开,但是没有展开完,就感觉有二次展开的情况,想问问出现这种情况的原因,和解决方法
超高效液相色谱测甲苯,乙酸乙酯,丙酮的条件
有一个物料测试里面的4个溶剂,其中乙酸,乙酸乙酯,dmf用一种色谱条件,采用直接进样。其中丙酮采用顶空进样。是因为这两类溶剂的沸点有差异,才分别测试的吗
工作场所空气有毒物质测定 饱和脂肪族酯类化合物 [url=http://www.stdbuy.cn/Standard/StdInfo.aspx?ca=99VLGLHxWzU=]GBZ/T 160.63-2007[/url] 是作废了吧?它被 工作场所空气有毒物质测定 第122部分:甲酸甲酯和甲酸乙酯和工作场所空气有毒物质测定 第126部分:硫酸二甲酯和三甲苯磷酸酯 部分代替。那么 原先 [url=http://www.stdbuy.cn/Standard/StdInfo.aspx?ca=99VLGLHxWzU=]GBZ/T 160.63-2007[/url] 中的乙酸乙酯 乙酸丙酯 现行的标准是什么?
请问你们做的胶黏剂溶于乙酸乙酯吗?我做胶黏剂中的苯含量,标准上是用用乙酸乙酯溶解,可我做的样品不溶于乙酸乙酯?不知道怎么回事···请各位帮忙想想怎么回事,难道主含量不溶,只要苯溶就能测?不懂,请高手帮忙解答下,O(∩_∩)O谢谢
柱子是依利特Hypersil ODS柱 25um 4.6*250mm 流动相是乙腈:乙酸乙酯:0.1%磷酸,其中乙酸乙酯占到15%,峰形也还理想。因为样品极性中等,不溶于正己烷、甲醇,易溶于丙酮、氯仿、乙酸乙酯;流动相中没有乙酸乙酯或氯仿这些溶剂就不能从柱上洗脱出来。是不是键合上去的C18会被乙酸乙酯洗脱下来吗?望各位老师帮忙解释下 谢谢!
我现在准备做 丙酮(分析纯试剂)、乙酸乙酯(分析纯试剂)的含量测定和水分测定,我现有的色谱柱不适用,根据国标:GB/T 686 2008 (化学试剂 丙酮):GDX-104[0.180mm—0.154mm,(80目—100目)]或选用Porapak Q[ 0.180mm—0.154mm,(80目—100目)]GB/T 12589 2007 (化学试剂 乙酸乙酯): 10%聚乙二醇己二酸酯涂于石油醚经浸泡、丙酮洗涤过的401有机担体[0.18mm—0.28mm,(60目—80目)]PS:都是填充柱大家做过同时测丙酮和水分、乙酸乙酯和水分的测定吗,什么样的柱子同时适合这两种测试呢?岔下话题:TCD检测器只能用填充柱么?
[color=#444444]请教一下谁用过GC-FID检测溴乙酸乙酯呀?是用的什么色谱柱呀?色谱条件是什么呀?有没有相关的文献介绍呀?谢谢各位大神!!![/color]
[color=#444444]正在做一个物质的溶剂残留,测定目标为乙酸乙酯,样品已知易溶于碱性水溶液,微溶于常用有机溶剂(已经做过甲醇、DMF、DMSO的溶解度试验)。但是问题是乙酸乙酯不溶于水,在配置对照溶液的过程中乙酸乙酯会挥发,重复性惨不忍睹。并且碱用的是氨,挥发时也会带走乙酸乙酯,溶液稳定性相当差。[/color][color=#444444]尝试过向碱溶液加过甲醇等未果,依然难以使溶液稳定。[/color][color=#444444]求教:如何选择溶剂或者改善溶剂来使乙酸乙酯稳定于溶液中?是否可以通过萃取来得到乙酸乙酯?具体如何操作[/color]
用安捷伦6890N [url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质联用仪[/color][/url]器,测二硫化碳中乙酸乙酯,3000微克每毫升的标液只能测出来溶剂峰,乙酸乙酯不出峰。更换了好多种条件,初始柱温60,保持5分钟,5℃每分钟升到80,保持2分钟,进样口温度200,250,280,290都设置过,还是不出峰,后来把溶剂延迟设置成了0还是没有,怎么办啊
小弟我的样品用乙酸乙酯溶解,定容。想进100甲醇的流动相,不知可否。乙酸乙酯是否对C18有损害?溶剂峰会很大吗?
我现有一产品需要做有机溶剂残留量,可能的残留有机溶剂主要为二氯甲烷,乙酸乙酯,甲醇,乙醇.请问应该用什么样的色谱条件?
[color=#444444]我合成了一系列苄基叠氮化合物,然后与丙烯酸甲酯的双键试反应,为了检测是否打开双键发生反应,要用[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质联用仪[/color][/url]打图。请问我可不可以用乙酸乙酯做溶剂稀释一下,再进样。谢谢,急![/color]
求助各位老师,关于乙酸乙酯的测定,样品百分比含量大约在90-98%左右,要求是使用内标标准曲线法,在建立曲线的时候浓度应该怎么选择?溶剂是乙醇标液和内标物需要自己配内标物是使用乙酸正丙酯。
[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url]法检测瘦肉精,方法要求配置5%氨化乙酸乙酯做洗脱液,现配现用。配的过程中发现取5ml氨水用乙酸乙酯定容至100ml时,两者不能完全互溶,超声也不行,该怎么办?
哪位知道乙酸乙酯在[C4mim]Cl中的溶解度,谢谢

帮我看下乙酸乙酯的标线做的怎么样?1分钟多的是二硫化碳,我用的乙酸乙酯是分析纯的,我推算是1.6分钟那个是乙酸乙酯,其他是杂质峰,老师们觉得呢[img=,690,949]https://ng1.17img.cn/bbsfiles/images/2019/07/201907051351244896_5205_1791505_3.jpg!w690x949.jpg[/img]
请问做压溶剂峰时刻以压下乙酸乙酯这样同时有两处化学位移的峰吗?谢谢
正己烷,乙酸乙酯对苯的溶解效果哪个好?有这方面的资料吗?苯和邻苯可以同时测吗?有什么要注意的
合成人员的样品经常是利用乙酸乙酯萃取后直接拿来进样的感觉乙酸乙酯似乎会吸附在C18色谱柱上不知道是否能将这种乙酸乙酯作为溶解的样品进入HPLC色谱柱呢或者有没有什么好的建议来每天冲洗色谱柱呢谢谢赐教!
我在GDX-102气象色谱柱(热导池法)上测混合物(含乙酸。乙醇。正丙醇。异丁醇。异戊醇。乙酸乙酯。乙酸正丙酯。乙酸异丁酯。乙酸异戊酯),请问专家:怎样控制气象色谱的操作条件能把这些混合物各自的含量测出来,而且峰形较好
以前很少做气相,现在要做溶剂残留,主要是测乙酸乙酯,仪器是岛津2014,顶空进样。有以下几个问题,求助各位,先谢谢啦1、溶剂应该选什么,我们目前是用二甲亚砜做溶剂,是否可以,二甲亚砜的沸点比较高,应该比乙酸乙酯出峰要晚吧2、大家有没有用内标,是不是自动进样就可以不用内标呢?3、用什么色谱柱合适,我用的是HP-5的柱子4、气相做样品时,每做完一针是否需要在初始温度平衡一段时间







 我要推广仪器
我要推广仪器
 下载APP
下载APP