各位大侠好,有谁能提供苄基三甲基氯化铵分析方法吗?
聚丙烯酰胺和十六烷基三甲基氯化铵分析检测,用什么仪器?
查了一些网上的,十八烷基三甲基氯化铵(简称1831,OTMC)熔点80℃,十六烷基三甲基氯化铵(1631,CTMAB)熔点250-237℃、234℃。 有人提出碳链越长熔点越高,可以上数据刚好相反。 请教大家这两者熔点到底是多少?
有哪位大侠做过三(三甲基硅烷)硼酸酯或三(三甲基硅烷)磷酸酯的分析啊?用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]的话选用何种柱子合适啊?
分析二甲基丁醇,三甲基丁醇,样品浓度大约为99%。请教分析方法及使用何种色谱柱?谢谢了!![em58]
求助给位,我想问一下四丁基溴化铵、十六烷基三甲基溴化铵、苄基三甲基溴化铵怎么做红外,它们都很容易潮解的,吸水性很强,我问一下,做红外的时候样品怎么处理,或者这些物质的标准红外数据在哪可以查到啊
有谁做过三甲基丙酮酸钠液相含量分析,可以给予指导帮助吗?谢谢!
求助:原乙酸三甲酯分析方法或分析标准
各位老师,哪位知道十六烷基三甲基溴化铵或季铵盐类物质的液相色谱分析条件,选用什么分析柱?哪个检测器好?请指导下呀。多谢了!
求 JIS K9555-1992 三甲基氯硅烷标准
各位大侠:那位知道最新纯品三甲胺的国家标准?纯品三甲胺(99%以上)的分析测试方法?请给本人一份!不胜感谢!在线等!
急需要购买氯化甲基汞或者甲基汞(要分析纯不要标准品),请提供相关的信息及联系方式。购买时要不要去公安局办购买证运输证?
求《HG/T 4415-2012 2,3,5-三甲基氢醌》标准,谢谢!
最近买了一支甲基三氯化锡的原标,证书上没有提供质谱图,CAS在普库中也没有查到响应的标准谱图,在气质上DB-5ms柱我觉得没走出来,所以请教一下有没有人做过,用的什么柱子,有什么注意事项,能能不能帮忙提供个质谱图或提供几个参考碎片离子的,谢谢了
求助[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法分析三甲基乙酰氯及其合成液的研究Analysis and research of trimethyl acetyl chloride and its synthetic fluid 2005年 第19卷 第06期 作者: 薛勇江, 张玉亨, 祝安娜, 张进, 期刊 ISSN : 1002-1124(2005)06-0025-03
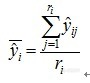
[align=center][b]基于[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱[/color][/url]分析技术的2,3,5-三甲基苯醌粗品萃取过程定量模型优化研究[/b][/align][b]中文摘要:目的[/b]实际工业生产工艺中,萃取是一项耗时耗力的过程,萃取终点的确定通常采用离线的HPLC, [url=https://insevent.instrument.com.cn/t/Mp]gc[/url]或由熟练工人根据经验判断,这些方法操作较复杂或是不够准确,在实际生产中缺乏一种快速有效的检测手段以判断萃取终点,节省操作时间,避免过分萃取浪费溶剂。利用[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱[/color][/url]技术可以明显改善萃取工艺。[b]方法[/b]本实验针对2,3,5-三甲基苯醌(TMBQ)粗品萃取环节,采用偏最小二乘法(PLS)建立模型,考察了不同预处理方法与变量选择方法对模型的影响以优化模型,采用[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱[/color][/url]结合PLS算法建立TMBQ萃取过程含量快速检测模型,并使用不同预处理方法与波段选择方法对模型进行优化,最终确定使用一阶导数+SG15点平滑预处理结合iPLS选择波段建立PLS模型。[b]结果[/b]建立模型的各项参数为:波普区间4385.33cm[sup]-1[/sup]-5152.86cm[sup]-1[/sup], 5928.11cm[sup]-1[/sup]-6309.94cm[sup]-1[/sup],模型决定系数R[sup]2[/sup]=0.996, RMSEP=0.1350。[b]结论[/b]建立的模型精密度与准确度良好,可以满足含量分析的需要,是TMBQ萃取过程含量快速检测的有效方法,可以快速准确的对三甲基苯醌粗品萃取过程进行在线监测,提供了一种用于该工艺环节的快速检测手段,如果应用于生产,可以节省操作时间,避免溶剂浪费。[b]关键词:[/b][url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱[/color][/url]分析;2,3,5-三甲基苯醌;萃取 2,3,5-三甲基苯醌是维生素E的主要中间体。2,3,5-三甲基苯醌在国外已有生产, 但国内尚未见文献报道。国内用2,3,5-三甲基苯醌主要依赖进口。因此,开展2,3,5-三甲基苯醌的合成研究对发展国内维生素 E 的生产具有重要意义。TMHQ的合成工艺国内外己有多种报道,较为先进的是TMP法与异佛尔酮法,TN[b]B[/b]Q粗品萃取过程是合成TMBQ的关键环节。在制药领域,NIRS作为一种重要的PAT工具,已成功用于药物的原辅料评价、关键过程的监测和控制、成品的快速放行和质量监测等各个环节,为保证产品质量、降低生产成本、革新生产过程发挥了重要的作用。[b]1实验材料与仪器1.1仪器[/b] Antaris Ⅱ傅里叶变换[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱仪[/color][/url](美国Thermo Fisher公司),7890A[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url]-氢焰离子化检测器(美国Agilent公司),HP-1毛细管色谱柱(美国Agilent公司)BT224S电子分析天平(德国Sartorius公司),容量瓶,100ml圆底烧瓶,分液漏斗,[url=https://insevent.instrument.com.cn/t/9p][color=#3333ff][url=https://insevent.instrument.com.cn/t/9p][color=#3333ff]移液枪[/color][/url][/color][/url](美国ThermoFisher公司)。RESULT[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱[/color][/url]采集软件,TQAnalyst[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱[/color][/url]分析软件,Matlab数据处理软件。[b]1.2试剂[/b] 2,3,6-三甲基苯醌(合成步骤见第二章),石油醚(天津富宇精细化工有限公司,沸程60℃-90℃)。[b]2方法2.1样品制备和处理[/b] 按照第二章步骤合成得TMBQ得其石油醚溶液,萃取水相合并有机相,旋蒸浓缩除去石油醚至橙黄色油状液体,称重,再用石油醚作为溶剂配置1ug/ml~50mg/ml一系列溶液。[b]2.2光谱采集[/b] 波长范围4000 cm[sup]-1[/sup]-10000cm[sup]-1[/sup];扫描次数32;分辨率8 cm[sup]-1[/sup],使用4mm光程的玻璃样品管乘装液体样品,采集样品前采集背景以消除背景干扰,每个样品重复采集三次光谱。光谱采集在恒定室温(24℃)与恒定湿度的条件下进行。[b]2.3样品集划分[/b] 使用K-S分类法将所有66个样品换分为48个校正集与18个验证集。[b]2.4模型建立与优化[/b] 采用导数、平滑等方法对原始光谱进行预处理,应用偏最小二乘法(PLS)建立模型,结合RMSEP等评价参数,通过变量选择方法选择特征波段优化模型。[b]2.5 重复性考察[/b] 选择3个验证集样品,每个样品连续采集10次光谱,使用建立好的模型预测每张光谱,并计算出每个样品十次预测值的均值和标准偏差。是第i个样品的第j张光谱,第i个样品共测定ri个光谱,第i个样品的预测平均值为:[align=center][img=,90,83]http://ng1.17img.cn/bbsfiles/images/2017/08/201708311044_01_1626619_3.png[/img][/align] 复测定的标准偏差为:[align=center][img=,164,102]http://ng1.17img.cn/bbsfiles/images/2017/08/201708311044_02_1626619_3.png[/img][/align] 用c[sup]2[/sup]检验来考察这些重复性标准偏差是否属于同一总体:[align=center][img=,271,245]http://ng1.17img.cn/bbsfiles/images/2017/08/201708311045_01_1626619_3.png[/img][/align] z为需要重复测定的样品数,将所得χ[sup]2[/sup]与自由度(z-1)临界值比较,若χ[sup]2[/sup]在临界值以下,则重复测定的所有方差属于同一总体,标准偏差均值σ可以作为近红外测定的标准偏差,近红外分析方法的重复性为z××σ[sub]max[/sub]。如果χ[sup]2[/sup]大于临界值,近红外分析方法的重复性随样品组分浓度不同而不同,这时,近红外分析方法的重复性不大于z××σ[sub]max[/sub](σ[sub]max[/sub]为σi中的最大值)。[b]2.6[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]检测[/b] 初始温度180℃恒温5min,以10℃/min的速率升温至240℃。进样口温度:300,检测器温度:300,载气:氮气,载气流速:3ml/min,进样量:0.5ul。[b]3结果3.1校正集与验证计划分[/b] 使用K-S分类法将所有66个样品换分为48个校正集与18个验证集。校正集与验证集的第一第二主成分分布图如图1,其中黑色符号代表校正集样品,红色符号代表验证集样品,验证集均匀分布于校正集中,可见使用该方法分类合理。[align=center][img=,553,217]http://ng1.17img.cn/bbsfiles/images/2017/08/201708311047_01_1626619_3.png[/img][/align][align=center]图1 所有样品主成分分布图[/align][b]3.2预处理方法的选择[/b] 考察无预处理、一阶导数+SG5点平滑、一阶导数加SG9点平滑、一阶导数+SG15点平滑、二阶导数加15点平滑这几种方式的建模结果,以RMSEC、RMSECV、RMSEP以及R[sup]2[/sup]作为评价指标,结果见表1。[align=center]表1 预处理方法评价参数[/align][align=center][img=,566,164]http://ng1.17img.cn/bbsfiles/images/2017/08/201708311104_01_1626619_3.png[/img][/align] 无预处理的模型结果最差,说明噪声对模型结果有较大影响,原始光谱如图2。SG15点平滑+一阶导数的预处理结果RMSEC、RMSECV以及RMSEP最小,R[sup]2[/sup]最高。因此选择SG15点平滑+一阶导数作为模型的预处理方法,预处理后光谱如图3。[align=center][img=,524,224]http://ng1.17img.cn/bbsfiles/images/2017/08/201708311048_01_1626619_3.png[/img][/align][align=center]图2 原始光谱图[/align][align=center][img=,532,210]http://ng1.17img.cn/bbsfiles/images/2017/08/201708311049_01_1626619_3.png[/img][/align][align=center]图3 一阶导数+SG15点平滑预处理光谱图[/align][b]3.2异常样本的剔除[/b] 图4为校正集样品在学生残差-杠杆值图中的分布。图中5号(红色方框标记)样品学生残差值与杠杆值都非常高,判定为异常样品,猜测为溶液配制错误或者在光谱采集过程中出现错误,因此在后期模型优化中剔除这一异常值。[align=center][img=,563,217]http://ng1.17img.cn/bbsfiles/images/2017/08/201708311050_01_1626619_3.png[/img][/align][align=center]图4 学生残差-杠杆值关系图[/align][b]3.3波段选择结果[/b] 以一阶导数+SG15点平滑为最优预处理方法进行波段选择,主要考察ForwardiPLS、SPA、相关系数法三种方法。[b]3.3.1iPLS波段选择结果[/b] 设定20为最大主成分数,分别考察以50、100、200个变量为波段基础的建模效果。红色虚线是全波段建模的RMSECV,红色与绿色条带的高度代表以此条带的变量建模所得RMSECV,从图5中可见,绿色条带的RMSECV值最小,因此绿色条带是被选择用于建模的波段,红色条带则表示不被选择的区域。表2为各变量基础的模型参数。[align=center][img=,558,268]http://ng1.17img.cn/bbsfiles/images/2017/08/201708311051_01_1626619_3.png[/img][/align][align=center]图5 以50个变量为基础的iPLS法波段选择效果图[/align][align=center][img=,572,266]http://ng1.17img.cn/bbsfiles/images/2017/08/201708311052_01_1626619_3.png[/img][/align][align=center]图6 以100个变量为基础的iPLS法波段选择效果图[/align][align=center][img=,618,262]http://ng1.17img.cn/bbsfiles/images/2017/08/201708311052_02_1626619_3.png[/img][/align][align=center]图7 以200个变量为基础的iPLS法波段选择效果图[/align][align=center]表2 不同变量基础的建模结果[/align][align=center][img=,646,111]http://ng1.17img.cn/bbsfiles/images/2017/08/201708311053_01_1626619_3.png[/img][/align][b]3.3.2 SPA法波段选择结果[/b] SPA算法首先通过完成n个波长分组各M个波长选择,然后通过多元定量校正模型完成m(1£m£M)个最优波长的选定。图8为SPA法选择变量的效果图。 运行SPA算法共选择3个变量,对应波数为4188.65cm[sup]-1[/sup],4885.50cm[sup]-1[/sup],7503.50cm[sup]-1[/sup],为图中红色方框标注,以此3个变量建立PLS模型,结果如表 所示,RMSECV与RMSEP均有所增加,R[sup]2[/sup]降低,表明模型预测能力与线性都有所降低。分析原因可能是此方法在选择波段过程中由1557个变量减少到3个,光谱变量删除过多,去除大量无关变量的同时导致许多有价值信息的丢失。[align=center][img=,501,246]http://ng1.17img.cn/bbsfiles/images/2017/08/201708311053_02_1626619_3.png[/img][/align][align=center]图8 SPA算法变量选择结果图[/align][b]3.3.3相关系数法波段选择结果[/b] 将相关系数阈值设定为0.6、0.7、0.8,使用相关系数法计算出TMBQ含量值与波数的相关系数图,如图9,图中虚线为设定的相关系数阈值,虚线以上及以及的部分代表相关系数大于阈值的波段,阈值越高,被选择的波段越少,当阈值设为0.8时,大于阈值的波段已经较少。以超过阈值的波段建立PLS模型。模型结果如表3,可见将阈值设为0.6时模型结果最好。[align=center] a[img=,402,175]http://ng1.17img.cn/bbsfiles/images/2017/08/201708311055_01_1626619_3.png[/img][/align][align=center] b[img=,409,187]http://ng1.17img.cn/bbsfiles/images/2017/08/201708311056_01_1626619_3.png[/img][/align][align=center] c[img=,409,176]http://ng1.17img.cn/bbsfiles/images/2017/08/201708311056_02_1626619_3.png[/img][/align][align=center]图9 不同阈值的波数相关图(a阈值设为0.6,b阈值设为0.7,c阈值设为0.8)[/align][align=center]表3 相关系数法建模参数[/align][align=center][img=,496,105]http://ng1.17img.cn/bbsfiles/images/2017/08/201708311058_01_1626619_3.png[/img][/align][b]3.4 小结[/b] 综合比较全波段建模与三种波段选择方法建模结果,参数如表。其中使用iPLS法选取600个变量,波段区间为4385.33cm[sup]-1[/sup]-5152.86cm[sup]-1[/sup],5928.11cm[sup]-1[/sup]-6309.94 cm[sup]-1[/sup],分别对应双键上C-H第一组合频与一级倍频吸收,建模后具有最高的决定系数和最低的各项方差值,这些参数表明使用该方法建立的模型预测能力最好,与真实值最接近。因此本实验主要选择iPLS方法选择变量,结合一阶导数+SG15点平滑建立模型,应用于TMBQ萃取过程含量的快速检测。[align=center]表4 各变量选择方法比较[/align][align=center] [img=,374,136]http://ng1.17img.cn/bbsfiles/images/2017/08/201708311059_01_1626619_3.png[/img][/align][align=center][img=,524,214]http://ng1.17img.cn/bbsfiles/images/2017/08/201708311059_02_1626619_3.png[/img][/align][align=center]图10 优化后模型预测线性图[/align][b]3.5重复性试验考察[/b] 采集验证集8号、25号、36号样品,对TMBQ含量模型进行重复性测试,每样品采集10次光谱。预测结果见表5。[align=center]表5 重复性考察结果[/align][align=center][img=,578,337]http://ng1.17img.cn/bbsfiles/images/2017/08/201708311100_01_1626619_3.png[/img][/align] 自由度为2时,χ[sup]2[/sup]临界值为5.99。实际χ[sup]2[/sup]小于临界值,[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱[/color][/url]分析方法重复性为0.154,可以满足分析应用。[b]3.6NIR预测考察[/b] 第一次使用20ml石油醚萃取,之后每次使用等体积10ml石油醚萃取,共萃取8次,使用[url=https://insevent.instrument.com.cn/t/Mp]gc[/url]测定TMBQ峰面积,并使用NIR采集8次萃取液[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱[/color][/url],使用优化好的定量模型对其含量进行预测。[align=center][img=,490,255]http://ng1.17img.cn/bbsfiles/images/2017/08/201708311102_01_1626619_3.png[/img][/align][align=center]图11 NIR预测值[/align] 图11为NIR对萃取过程的预测结果,第一次萃取即将大部分产品萃取出,随后的每次萃取量呈逐渐下降的趋势,在第五次萃取后,萃取液中产品含量几乎为0,并且随后没有变化,表明已达到萃取终点。使用[url=https://insevent.instrument.com.cn/t/Mp]gc[/url]检测第4~8次萃取液,记录TMBQ峰面积,结果如表6。[align=center]表6 [url=https://insevent.instrument.com.cn/t/Mp]gc[/url]检测结果表[/align][align=center][img=,529,66]http://ng1.17img.cn/bbsfiles/images/2017/08/201708311103_01_1626619_3.png[/img][/align] 第五次萃取后,TMBQ峰面积已经很小,并且基本没有变化,因此在4次萃取完全可以将水相中的TMBQ萃取完全,继续萃取已经没有意义,[url=https://insevent.instrument.com.cn/t/Mp]gc[/url]检测与NIR预测结果相符,表明此模型预测能力良好,对萃取工艺具有一定指导意义。[b]4讨论[/b] 本实验采用[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱[/color][/url]结合PLS算法建立TMBQ萃取过程含量快速检测模型,并使用不同预处理方法与波段选择方法对模型进行优化,最终确定使用一阶导数+SG15点平滑预处理结合iPLS选择波段建立PLS模型,建模所用波段区间为4385.33 cm[sup]-1[/sup]-5152.86cm[sup]-1[/sup],5928.11 cm[sup]-1[/sup]-6309.94cm[sup]-1[/sup],模型决定系数R[sup]2[/sup]=0.996,RMSEP=0.1350。使用[url=https://insevent.instrument.com.cn/t/Mp]gc[/url]验证了NIR模型对萃取过程与终点的预测能力。以上结果表明模型精密度与准确度良好,可以满足含量分析的需要,是TMBQ萃取过程含量快速检测的有效方法。[b]5参考文献[/b]孙月婷. 维生素E 的合成与分析研究现状. 广州化工, 2011, 39(6): 34-35.O.A.Kholdeava Synthesis of Vitamia E J.Mol.Cotal,1992,88(5):235~ 244孔黎明, 周涛, 菅盘铭. 2, 3, 5- 三甲基苯醌和2, 3, 5- 三甲基氢醌的一种合成方法: 中国, 102219665. 2011-10-19.A BShishmakov, Yu V Mikushina, O V Koryakova. Oxidation of 2,3,6-Trimethylphenolon Titanium Dioxide Xerogel by Hydrogen Peroxide in the Absence of an OrganicSolvent. RUSSIAN JOURNAL OF APPLIED CHEMISTRY, 2011, 84(9):1555-1559. O V Zalomaeva, N N Trukhan,I D Ivanchikova, et al. EPR study on the mechanism of H[b][sub]2[/sub][/b]O[b][sub]2[/sub][/b]-basedoxidation of alkylphenols over titanium single-site catalysts. J. Mol.Catal. A: Chem., 2007, 277(1-2), 185~192.褚小立. 化学计量学方法与分子光谱分析技术.北京 化学工业出版社. 2011.董学锋,戴连奎,黄承伟等.结合PLS-DA与SVM的[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱[/color][/url]软测量方法

气相色谱法对维生素E的原料三甲基氢醌的检测摘要 三甲基氢醌即2,3,5-三甲基氢醌,又名2,3,5-三甲基对苯二酚,是生产维生素E的中间体,其主要用途是用作生产维生素E的主要原料。目前,维生素E已成为国际市场上用途广泛、产销量极大的主要维生素品种,国内外市场前景广阔。目前全国生产能力不能满足国内市场供应不足,部分依赖进口。因此对三甲基氢醌的需求日益增加。而对于三甲基氢醌检测目前国家和行业都没有一个统一的检测标准。为此南京科捷分析仪器应用研究所根据客户的要求应用GC5890C气相色谱仪对2,3,5-三甲基氢醌进行方法研究。实验结果表明:本方法简便,分析速度快。能满足生产质量控制的要求,从而降价低生产成本。关键词 2.3.5- 三甲基氢醌 2,3,5-三甲基对苯二酚 维生素E中间体 气相色谱法一.2.3.5三甲基氢醌气相色谱图 http://ng1.17img.cn/bbsfiles/images/2011/06/201106171053_300271_2242538_3.jpg三、仪器配置 检测项目2,3,5-三甲基氢醌及其杂质色谱仪器型号GC5890C型色谱仪 配有FID检测器毛细管色谱柱0.32*30*0.25专用柱色谱工作站N2000(电脑1台自备)氮氢空发生器 HGT300E 1台或高纯氮、氢气、空气钢瓶各一瓶
GC测定三甲基丙酮酸时,进第三针就出现鬼峰,保留时间固定。清洗衬管,更换石英棉,鬼峰变小了20倍。每天反复清洗衬管比较烦。各位同仁分析一下到底是什么原因,有何解决办法!
十六烷基三甲基溴化铵含量及游离胺的测定方法.
药物杂质中有三甲胺、溴代十六烷和溴代十八烷(沸点超过300度)。其中三甲胺标准品的溶剂为甲醇,浓度为100mg/L。做药物杂质的含量分析时,如用DB-5ms柱,进样口温度为310度,则柱温25度甲醇峰掩盖三甲胺的峰;溴代十六烷和溴代十八烷在220度保持20分钟会出峰。如用DB-wax柱,进样口温度为240度,溶剂为乙醚。如何证明溴代十六烷和溴代十八烷在进样口中完全气化。
需要购买氯化甲基汞、氯化乙基汞的纯品标准,请提供相关的信息及联系方式。
十六烷基三甲基溴化铵?请教大虾:十六烷基三甲基溴化铵 溶液需要在什么条件下保存?我配置8g/l 的溶液放置半个月左右后性质就不稳定了,溶液中主要成分有氯化钡,含量100g/l,我是在80摄氏度左右的热水中将十六烷基三甲基溴化铵和氯化钡溶解后定容常温保存。
求助各位老师,有没使用过 氯化甲基汞 汞的同位素 标准品,或是做过水产品中甲基汞检测的,麻烦告知一下,谢谢?
本人在做铁矿石全铁分析时,之前用的是氯化亚锡-三氯化钛方法测定全铁含量,用盐酸溶样180℃,10分钟左右,然后用氯化亚锡还原大部分三价铁,呈淡黄色后加钨酸钠,再用三氯化钛还原至乌兰。。。但是我发现在加钨酸钠的时候,会产生白色不溶沉淀,这种情况有时又不会产生,不知是什么原因,求指出问题!在加钨酸钠产生沉淀后我就用甲基橙作指示剂了,然后加氯化亚锡还原至甲基橙无色,冷却后,加硫磷混酸和二苯胺磺酸钠开始滴定,这样就省略了三氯化钛还原的步骤,不知用甲基橙作指示剂指示氯化亚锡还原三价铁终点可不可行,看到有文献这样做,而且比国标的方法简单,不知精确度怎么样,需要注意些什么?
十六烷基三甲基溴化铵是否溶于二氯甲烷,氯仿或者四氯化碳阿?在其他溶剂中的溶解度如何?谢谢了
甲基,乙基和氯化汞形态分析 原子荧光与液相色谱联用,做混合标准溶液时,每天测的都不一样,差别极大,怀疑是溶液不稳定,或者污染,重新配制标准溶液,又有变化,十分不解?ps但是标准溶液都是参照方法用水直接溶解的,器皿都浸泡过。http://ng1.17img.cn/bbsfiles/images/2012/01/201201171633_346172_1619679_3.jpg
我用气相色谱分析脂肪醇,用吡啶溶解,再加三甲基硅烷时,出沉淀,不分层!继续加六甲基二硅胺烷时,出现分层,下面是沉淀,请问沉淀是什么物质?请问有知道的吗?急!
连、偏、均三甲苯的标准品,要有标准品证书的在哪里可以购买?
对于三氯化镓的分析 ,不知各位高手怎么进行的?我没有查到国标, 恳请哪位能传一份 行标 企标 或国际标准?在下邮箱haoyang8003@yahoo.cn
[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法测定碳九加氢产品中三甲苯含量的测定方伟民宁波广昌达新材料有限公司摘要:建立了用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url]、DB-FFAP色谱柱分析碳九加氢产品中各三甲苯含量的方法。该方法采用单色谱柱、单氢火焰离子化检测器(FID),分流进样分析,采用校正面积归一化法对分析结果进行定量。该方法的特点是仪器配置简单,样品用量少,分离效果显著,结果准确。关键词:[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]DB-FFAP色谱柱碳九加氢三甲苯前言随着国内乙烯工程的大量投入使用,裂解制乙烯的副产品乙烯胶油大量产出,这些副产品因含有大量的九个碳原子的芳烃馏分,统称碳九芳烃。主要组分有异丙苯、正丙苯、乙基甲苯、均三甲苯、偏三甲苯、邻三甲苯、茚等。碳九芳烃馏分组分复杂,沸点相近,难以一一分离,目前主要分离出偏三甲苯和均三甲苯用于制偏苯三酸酐和均苯四甲酸二酐等,用于涂料,合成树脂等。对这些副产品进行加氢再利用,加氢后的产品中三甲苯的含量的高低直接影响产品销售对象。因此建立分析加氢产品中三甲苯含量的方法尤其重要。本方法仪器配置简单,样品用量少,分离效果显著,结果准确,可用于工厂常规分析。1 实验部分1.1 试剂与仪器设备1.1.1 试剂试剂名称 规格 生产厂家高纯氮气 纯度≥99.999% 宁波亚大气体有限公司高纯氢气 纯度≥99.999% 宁波亚大气体有限公司压缩空气 —— 现场装置直供邻二甲苯 纯度:99.9% 阿拉丁试剂1,3,5-三甲苯 纯度:89.0% 阿拉丁试剂1,2,4-三甲苯 纯度:99.5% 阿拉丁试剂1,2,3-三甲苯 纯度:99.9% 阿拉丁试剂1,2,4,5-四甲苯 纯度:90.0% 阿拉丁试剂1,2,3,5-四甲苯 纯度:98.0% 阿拉丁试剂1.1.2仪器和设备1.1.2.1 电子天平:赛多利斯BSA124S电子天平,精确到0.0001g1.1.2.2 [url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url]:常州盘诺仪器有限公司的A90[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url],配置有氢火焰离子化检测器(FID),B75型16位自动进样器,10μL进样针,1.1.2.3 色谱柱:DB-FFAP,60×0.25×0.50,J&W1.2 色谱操作条件选择及探讨1.2.1载气的选择由于反应体系中异构体较多,选择载气主要从柱效考虑用氢火焰离子化检测器一般多用氦气和氮气做载气。用氦气做载气,有点可以缩短分析时间,但分析成本较高,选用氮气做载气,柱效高,分离效果好,但分析时间较长,综合考虑选用氮气做载气。1.2.2色谱柱的选择三甲苯与四甲苯的同分异构体比较多,各同分异构体的沸点如表1:表1三甲苯和四甲苯各同分异构体的沸点组分 1,3,5-三甲苯 1,2,4-三甲苯 1,2,3-三甲苯 1,2,4,5-四甲苯 1,2,3,5-四甲苯 1,2,3,4-四甲苯沸点/℃ 164.7 169.4 176.1 196.8 199.0 205.0从表1中我们可以看出三甲苯和四甲苯的同分异构体的沸点非常接近,特别是三甲苯的三种同分异构体。要使组分完全分离需选用强级性的分离柱,如FFAP毛细管柱、OV101毛细管柱、SE-30毛细管柱、聚乙二醇-20等色谱柱。1.2.3色谱柱及色谱条件从分析要求、分析效果和分析方法的经济消耗等方面进行考虑选用如下的色谱柱及操作条件。如表2:表2色谱柱及色谱条件色谱柱固定相 聚乙二醇TPA,极性固定相柱长/m 60柱内径/mm 0.25固定液膜厚度/μm 0.50载气 氮气检测器 检测器类型 氢火焰离子化检测器 温度/℃ 250 空气流量/(mL/min) 400 氢气流量/(mL/min) 30汽化室 温度/℃ 230 分流比 100:1柱箱 初温/℃ 80 初温保持时间/min 2 一阶升温速率/(℃/min) 4 一阶终止温度/℃ 136 一阶温度保持时间/min 0 二阶升温速率/(℃/min) 2 二阶终止温度/℃ 148 二阶温度保持时间/min 0 三阶升温速率/(℃/min) 2 三阶终止温度/℃ 166 三阶温度保持时间/min 0 四阶升温速率/(℃/min) 5 四阶终止温度/℃ 230 四阶温度保持时间/min 3柱流量/(mL/min) 1.5进样量/μL 0.42 结果与讨论2.1 色谱图中组分的定性在相同实验条件下对标准物质对各组分进行分析,采用保留时间定性,用标准物质配制混合标样进行分析结果如图1。各组分都能在色谱图上出峰,且分离效果明显。 图1 混合标准物质在色谱上的色谱图1)1,3,5-三甲苯 2) 1,2,4-三甲苯 3) 1,2,3-三甲苯4) 1,2,4,5-四甲苯 5)1,2,3,5-四甲苯2.2 定量分析样品中的各物质都能在色谱图上出峰,所以采用校正面积归一化法进行定量。用称量法配制含1,3,5-三甲苯、1,2,4-三甲苯、1,2,3-三甲苯、1,2,4,5-四甲苯、1,2,3,5-四甲苯和邻二甲苯,用邻二甲苯做标准物质计算其它组分的相对质量校正因子。各组分称量应精确至0.0001g,含量计算应精确至0.0001%(质量百分数)。并按下式进行计算: fi—标样中i相对邻二甲苯的质量校正因子A— 标样中邻二甲苯的峰面积Ai—标样中组分i的峰面积m—标样中邻二甲苯的质量,gmi—标样中组分i的质量,g2.2.1 校正因子的测定用邻二甲苯做标准物质测定各组分的质量相对校正因子,其结果如下:表3 各组分重复质量校正因子的测定标样 组分 称重(g) 含量(%) 次数 峰面积 校正因子 1,3,5-三甲苯 2.2643 9.0897 1 7114.08 0.99 2 6190.29 0.99 3 6313.75 1.00 1,2,4-三甲苯 9.0575 36.5057 1 28312.47 1.00 2 2463.46 1.01 3 2511.18 1.00 1,2,3-三甲苯 1.1245 4.5094 1 3676.47 0.95 2 3199.86 0.96 3 3263.33 0.96 1,2,4,5-四甲苯 1.2738 5.1083 1 4123.67 0.96 2 3578.28 0.97 3 3681.98 0.96 1,2,3,5-四甲苯 0.2218 0.8894 1 675.38 1.02 2 586.63 1.03 3 603.26 1.02 邻二甲苯 10.7673 43.1785 1 33396.17 1.00 2 29238.58 1.00 3 29511.96 1.00表4 校正因子测定结果分析组分 实验1 实验2 实验3 平均值 相对偏差(%)1,3,5-三甲苯 0.99 0.99 1.00 0.99 1.011,2,4=三甲苯 1.00 1.01 1.00 1.00 1.001,2,3-三甲苯 0.95 0.96 0.96 0.96 1.041,2,3,5-四甲苯 0.96 0.97 0.96 0.96 1.041,2,4,5-四甲苯 1.02 1.03 1.02 1.02 0.98在相同的测试条件下,连续三次测量标准混合液,计算各组分的质量校正因子的偏差小于5%,符合分要求。2.2.2检测限为了确定各组分的检测限,通过对混合标样的分析,以色谱峰高度基线噪声的3倍做为标准计算检出限。用邻二甲苯做基液,配制样品浓度0.0005%左右,连续分析样品6次,取平均用于计算各组分的检测限,其结果如下:表5 各组分检测限计算组分 平均峰高 配制浓度(% m/m) 信号噪声 检测限(% m/m)1,3,5-三甲苯 0.32 0.0005 0.05 0.000231,2,4-三甲苯 0.24 0.0004 0.05 0.000251,2,3-三甲苯 0.30 0.0005 0.05 0.000251,2,4,5-四甲苯 0.29 0.0005 0.05 0.000261,2,3,5-四甲苯 0.19 0.0003 0.05 0.000242.2.3方法验证和精密度分析用已知纯度的标准样品配制2组已知浓度的标准试样,在规定的实验方法条件下,用标样进行6次测试分析,6次的分析结果的平均值与该组分的理论值进行比对,确定方法的回收率和精密度,实验结果如表6:表6 标样回收和精密度计算结果组分 标样1# 配制浓度%(m/m) 平均值%(m/m) RSD,% 回收率,%1,3,5-三甲苯 4.1829 4.1746 2.82% 99.81,2,4=三甲苯 30.8511 30.5734 1.35% 99.11,2,3-三甲苯 2.3698 2.3390 2.52% 98.71,2,3,5-四甲苯 3.0255 2.9317 1.89% 96.91,2,4,5-四甲苯 1.1277 1.1085 1.05% 98.3组分 标样2# 配制浓度%(m/m) 平均值%(m/m) RSD,% 回收率,%1,3,5-三甲苯 6.5346 6.4889 2.68% 99.31,2,4=三甲苯 20.1885 20.0674 1.16% 99.41,2,3-三甲苯 4.3328 4.2158 2.15 % 97.31,2,3,5-四甲苯 8.1271 7.8589 2.57% 96.71,2,4,5-四甲苯 3.0554 3.0310 2.16% 99.2通过对标准混合样的分析,结果表明该方法相对偏差小于5%,满足分析要求。2.3实际样品分析在规定的色谱条件下,取罐样产品分析,其分析色谱图如下:图 2 G5209产品罐样色谱分析图表5 G5209产品罐样测定结果组分 结果%(m/m)1,3,5-三甲苯 5.71,2,4=三甲苯 32.51,2,3-三甲苯 3.21,2,3,5-四甲苯 2.91,2,4,5-四甲苯 0.82.4结论采用DB-FFAP色谱柱,进行分流进样,用校正面积归一化法分析碳九加氢产品中的三甲苯和四甲苯各同分异构体的含量,操作简单,分离效果好,分析结果稳定,准确度高。为产品进一步处理和销售提供了准确的可靠的分析数据。参考文献陶克毅.石油学报,1989,5(1):33~38SH/T 1773-2012 1,2,4-三甲苯纯度及烃类杂质的测定[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法傅若农,[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]概论第二版







 我要推广仪器
我要推广仪器
 下载APP
下载APP