前言:米格列奈钙片(Mitiglinide Calcium Hydrate Tablets)主要由米格列奈钙组成,其化学名:双〔(2s)-2-苄基-3- (顺-六氢异吲哚-2-羰基)丙酸〕单钙二水合物。本品可以单独用于经饮食和运动疗法不能有效控制高血糖的Ⅱ型糖尿病病人。米格列奈是继瑞格列奈、那格列奈后第三个美格列脲类药物,是苯丙氨酸的衍生物。米格列奈钙片的原料药有本公司自己生产,其质控指标之一:有关物质的两个已知杂质,及(2S)-2-苯甲基丁二酸对照品和杂质C。此色谱柱(序列号:W10212097)在上篇文章中已经宣布退役,后来因色谱柱紧缺,摸索条件后启用了。以前的色谱条件:用十八烷基硅烷键合硅胶为填充剂;以0.05mol/L磷酸盐溶液(用0.05mol/L磷酸二氢钾溶液调节0.05mol/L磷酸氢二钠溶液pH值至7.0)-甲醇(40:60)为流动相;检测波长为290nm;柱温30℃。理论板数按雷贝拉唑钠峰计算不低于1000,雷贝拉唑钠峰与相邻杂质峰的分离度应符合要求。现在的色谱条件:用十八烷基硅烷键合硅胶为填充剂,以乙腈-0.01mol/L磷酸二氢钾溶液(用磷酸调节pH值至3.0)(45:55)为流动相;检测波长210 nm。理论板数按米格列奈钙峰计算应不低于2000。试验步骤: 取本品,加甲醇制成每1ml中含1.0mg的溶液,作为供试品溶液;另精密量取含量测定项下的对照品溶液适量,用甲醇稀释制成每1ml中约含2μg的溶液,作为对照溶液。取有关物质(2S)-2-苯甲基丁二酸对照品和杂质C对照品适量,用供试品溶液溶解并稀释制成每1ml中含米格列奈钙为1mg和有关物质对照品为5μg的溶液,作为系统适用性试验溶液。照含量测定项下的色谱条件,取对照溶液20μl,注入液相色谱仪,调节检测灵敏度,使主成分色谱峰的峰高约为满量程的10%~20%;取系统适用性试验溶液20μl,注人液相色谱仪,理论板数按米格列奈峰计箅不低于3000,米格列奈峰与有关物质对照品峰的分离度应符合要求。精密量取供试品溶液与对照品溶液各20μl,分别注人液相色谱仪,记录色谱图至主成分峰保留时间的2.5倍。其主要色谱图加下:系统适用性试验色谱图http://ng1.17img.cn/bbsfiles/images/2013/05/201305031602_438152_1621890_3.gifhttp://ng1.17img.cn/bbsfiles/images/2013/05/201305031603_438153_1621890_3.gifhttp://ng1.17img.cn/bbsfiles/images/2013/05/201305031626_438157_1621890_3.gif供试液样品色谱图:http://ng1.17img.cn/bbsfiles/images/2013/05/201305031639_438158_1621890_3.gif3.2.S.4.3.2.5检测限与定量限(色谱图见附件63~70)精密称取对照品10.49mg置10ml量瓶中,加甲醇适量使溶解并稀释至刻度,摇匀,滤过,取续滤液作为供试储备液,取供试储备液用甲醇逐级稀释,分别取20µl注入高效液相色谱仪,经测定,米格列奈钙主峰保留时间约为12.5分钟,在±1分钟的时间范围计算基线噪音约为361μV,当S/N≒3时,检测浓度为0.5245μg/ml,检测限为10.49ng,当S/N≒10时,定量限浓度为2.0980μg/ml,定量限为41.96ng,试验结果见下表。检测限的确定序号峰高(μV)Δε检测限(μV)13611083检测限验证浓度名称浓度(µg/ml)进样量(ng)峰高(µV)平均峰高(µV)S/N供试液110.4900209.8197991979954.8[/t
今天做的一药物的有关物质检测,对照溶液浓度标准要求配制成为1ug/ml,可我配成了0.4ug/ml,直接进样了,不知结果可不可信哦,我是新手,望指教!
基本信息通用名称:富马酸喹硫平商品名称:启维其它名称:舒思、思瑞康英文名称:Quetiapine Fumarate Tablets http://f.hiphotos.baidu.com/baike/s%3D220/sign=6a863f019d16fdfadc6cc1ec848f8cea/c8177f3e6709c93dbda6cadd9f3df8dcd1005440.jpg汉语拼音:Fumasuan Kuiliuping Pian中文别名:11--1-哌嗪基]二苯并[b,f][1,4]硫氮杂卓半富马酸盐[sup][1][/sup]英文名称:quetiapine fumarate英文别名:2-[2-(4-dibenzo[b,f][1,4]thiazepin-11-yl-1-piperazinyl)ethoxy]ethanol hemifumarate; Quetiapine hemifumarate; quetiapine fumarate main intermediates; 2-[2-(4-dibenzo[b,f][1,4]thiazepin-11-ylpiperazin-1-yl)ethoxy]ethanol (2E)-but-2-enedioate (salt)适应症喹硫平片用于治疗[url=http://baike.baidu.com/view/1580.htm]精神分裂症。质量标准详见附件截图:[img]http://ng1.17img.cn/bbsfiles/images/2013/08/201308151502_457839_1621890_3.gif[img]http://ng1.17img.cn/bbsfiles/images/2013/08/201308151502_457840_1621890_3.gif色谱柱信息:150MM PN:WEL518415 SN:W11212195 LN:W1811.02本次试验目的是进行上市品有关物质对比。1.自制样品有关物质情况:[img]http://ng1.17img.cn/bbsfiles/images/2013/08/201308081039_456793_1621890_3.gif2.上市样品(思瑞康)有关物质检测情况:[img]http://ng1.17img.cn/bbsfiles/images/2013/08/201308081039_456794_1621890_3.gif3.上市样品(舒思)有关物质检测情况:[img]http://ng1.17img.cn/bbsfiles/images/2013/08/201308081040_456795_1621890_3.gif4.自制样品和上市样品杂质谱图对比:[img=,707,519]http://ng1.17img.cn/bbsfiles/images/2013/08/201308081042_456797_1621890_3.gif结论:单纯从杂质谱来看,自制品和上市品(舒思)杂质谱基本一致,但量较上市品(舒思)低,较上市品(思瑞康)高,当自制品杂质总量是较低的,如再比上市品(思瑞康)低点就最好了,初步判断为原料药不同产生的。另外,本品有关物质检测如使用其他品牌的色谱柱会有较大的拖尾,以及主峰前杂质分离不好。同样色谱条件下,含量测定中,若进样针数较多,其他品牌的色谱柱会产生肩峰,若使用其他品牌的要在进针次数在20次的时候,要多走几针空白,估计是本品的辅料有影响,具体情况没有进行继续研究。
关于HPLC主成分自身对照法检查有关物质时检测波长确定的讨论审评二部张玉琥有关物质检查,包括对产品中残留合成原料、中间体、副产物及可能的降解产物的检查,是控制药品质量的重要指标,目的是检查药品中所含的上述杂质是否符合安全性的要求,同时也是药品稳定性评价中需重点考察的项目。有关物质检查常用的方法之一是HPLC主成分自身对照法(紫外检测器),即将HPLC色谱图中杂质峰面积与主成分自身对照液峰面积进行比较,以确定杂质限度是否合格。采用此方法时确定的检测波长是否合理直接影响到方法的可行性,因此检测波长的选择是方法学研究的重要内容。在审评中发现一些申报单位在采用HPLC主成分自身对照法检查有关物质时直接或间接地以主成分的最大吸收波长作为检测波长,由于有关物质检查的对象是杂质,若将主药的最大吸收波长确定为检测波长,则杂质在此波长下的吸收可能偏低,某些杂质甚至无吸收,这样会造成对杂质含量的低估甚至漏检,从而不能反映产品的真实质量,影响了对品种质量可控性及稳定性的评价。在有关物质检测波长确定方面,申报资料中比较常见的做法有:1.直接将主药的最大吸收波长选作检测波长。2.简单地套用含量测定的色谱条件。在HPLC法进行含量测定时,为提高方法的灵敏度,降低干扰,往往选用主成分的最大吸收波长作为检测波长。若套用含量测定的色谱条件,实际仍是以主药的最大吸收波长作为有关物质检测波长。3.以样品进行破坏性试验(酸、碱、热、光照、氧化等)后的溶液做紫外扫描,将扫描图谱中最大吸收波长确定为有关物质的检测波长。因破坏性试验后溶液中存在尚未破坏的主药、降解产物、辅料等,此溶液的紫外吸收为各成分紫外吸收的加和,并不能反映降解产物的紫外吸收特性。由于未破坏主药所占比例较大,故破坏性试验后溶液的最大吸收波长一般仍为主药的最大吸收波长。采用HPLC主成分自身对照法检查有关物质,其前提之一是需检查的杂质与主成分在确定的检测波长下应有相近的紫外吸收(响应值接近),选择检测波长时需对产品中可能存在的杂质(合成原料、中间体、副产物以及降解产物)的紫外吸收特性进行研究。已知杂质的紫外吸收特性可采用对其流动相溶液直接进行扫描的方法考察,未知杂质(如未知降解产物等)可通过二极管阵列检测器考察其紫外吸收情况,根据各主要杂质及主成分的紫外吸收特性,选取响应值基本一致的波长作为有关物质的检测波长。若对不同杂质难于找到均适宜的检测波长,可考虑选择在不同波长下分别测定,也可考虑采用加校正因子的主成分自身对照法。只有经试验研究确认主成分的最大吸收波长符合有关物质检查对测定波长的要求时,为方便操作,可选作有关物质的检测波长,以与含量测定的色谱条件一致。另外,HPLC主成分自身对照法检查有关物质比较适用于对微量杂质总量的控制,也可用于单个杂质的限度(一般不超过0.5%)控制。对于具有明确归属的已知杂质,建议采用杂质对照品法进行检查。对于有毒有害杂质,更应采用质对照品法单独测定,并制定严格的限度。
相关物质检测的时候通常会用到杂质对照品,关于这个杂质对照品你是如何管理的?对它的含量与纯度有木有做过分析检测?有效期是怎么规定的?
根据2020版药典中的丙二醇检测有关物质 对照一共有四个 但是只出了两个浓度大的峰 其中二甘醇和环氧丙烷都未出峰 用的是岛津的wax柱 色谱条件是根据药典设的 求帮助 [img=,690,765]https://ng1.17img.cn/bbsfiles/images/2023/12/202312211008204466_8776_5460811_3.png[/img]
请教下各位,一般做溶出曲线时,一个介质配一份该介质下的对照;但是遇到多个介质一起做的时候,是否可以共用一份对照?假如,同时配制的多介质对照品f值只相差2%以内,是否可以认为这几个介质的对照可以共用呢?就不再需要每个介质单独配对照?
继“宝刀未老,古木发新枝,米格列奈钙有关物质方法学部分”,进行的含量测定方法学研究。项目:含量测定(3.2.P.5.2.8)检查方法:照高效液相色谱法(中国药典2010年版二部附录Ⅴ D)测定试验条件:色谱柱(柱长:150mm,内径:4.6mm,填料:C18,填料粒径:5μm,序列号:W10212097)UV检测器(检测波长:210nm)流动相:乙腈-0.01mol/L磷酸二氢钾溶液(用磷酸调节pH值至3.0)(45:55)流速:1.0ml/min运行时间:约12min系统适用性:理论板数以米格列奈峰计算应不低于2000。具体试验操作:取本品20片,精密称定,研细,取细粉适量(约相当于米格列奈钙25mg),精密称定,置250ml的量瓶中,加甲醇溶解并稀释至刻度,滤过,取续滤液作为供试品溶液,取20ml注入液相色谱仪,记录色谱图;另取米格列奈钙对照品适量,同法测定。按外标法以峰面积计算,即得。计算公式:标示量百分含量(%)=××100%式中:Cs为对照品的浓度(mg/ml);At为供试液的主峰面积;Nt为供试液的稀释倍数;AS为对照品溶液的主峰面积;W为供试品取样量(mg)。3.2.P.5.3.6含量测定(色谱图见附件936~1079)http://ng1.17img.cn/bbsfiles/images/2013/06/201306201604_446667_1621890_3.gif3.2.P.5.3.6.1波长选择本品含量测定检查波长为210nm,同有关物质检查项。3.2.P.5.3.6.2流动相选择本品含量测定流动相为乙腈-0.01mol/L磷酸二氢钾溶液(用磷酸调节pH值至3.0)(45:55),同有关物质检查项。3.2.P.5.3.6.3进样精密度试验(色谱图见附件936~941)精密称取米格列奈钙对照品10.23mg,置100ml量瓶中,用甲醇适量振摇使溶解并稀释至刻度,摇匀,滤过,作为供试液,取供试液20μl注入液相色谱仪,记录色谱图。试验结果见下表。http://ng1.17img.cn/bbsfiles/images/2013/06/201306201735_446698_1621890_3.gifhttp://ng1.17img.cn/bbsfiles/images/2013/06/201306201735_446699_1621890_3.gif精密度试验结果进针次数123456RSD%峰面积(A)54718135472559546690954676315460382
自制杂质对照品,新药申报时杂质含量方面需要做哪些工作?按照CTD格式要求,对于自制对照品应该“简述含量和纯度标定的方法及结果”,具体做应该怎么做?请各位专家指导,谢谢!
现有一方法,能很好的检测出 自制片子中的有关物质,但确检测不出对照品中的有关物质,连主成分的峰也没出来,大家能提供些意见吗?
最近做了一个品种 :盐酸维拉帕米片,参照标准中国药典2010年版二部P779。发现一个问题,按照标准上的色谱条件,含量测定项下的对照品溶液和供试品溶液中主峰出峰时间都比较的一致,都在30.0分钟左右,但是在有关物质项下,出现了异常,对照溶液主峰在30.0分钟,供试品溶液主峰在28.0分钟,出峰时间明显有差异。在一台仪器上重复试验,在不同仪器上重复试验都做了,都出现了前述情况,请教专家如何解决?谢谢!
如题,请知道的朋友告诉我,先谢谢了! 我即将参与做的一个3.1类新药项目,其杂质定量研究正需要考虑这个响应因子的问题,以前做的都是补充申请,还没有考虑响应因子不一致的情况,现在考虑了一个方案,那就是液相色谱检测,液相色谱和紫外同步进行,如果我们这个新药项目杂质可得,并且有对照品,那么就可以绘制杂质和对照品的标准曲线,杂质标准曲线的斜率除以对照品标准曲线的斜率的比值就是响应因子吧?我想参照指导原则确定到底什么范围时可以用主成分的自身对照法计算含量,什么范围时,宜用杂质对照品法计算含量,也可用加校正因子的主成分自身对照法?您有好的建议吗?期待您的帮助。
做有关物质的时候,有关对照只需要得出主峰的峰面积,用来跟有关物质的杂志峰做对比,那有关对照也需要扣除空白溶剂吗?
有关物质检查,包括对产品中残留合成原料、中间体、副产物及可能的降解产物的检查,是控制药品质量的重要指标,目的是检查药品中所含的上述杂质是否符合安全性的要求,同时也是药品稳定性评价中需重点考察的项目。 有关物质检查常用的方法之一是HPLC主成分自身对照法(紫外检测器),即将HPLC色谱图中杂质峰面积与主成分自身对照液峰面积进行比较,以确定杂质限度是否合格。采用此方法时确定的检测波长是否合理直接影响到方法的可行性,因此检测波长的选择是方法学研究的重要内容。 在审评中发现一些申报单位在采用HPLC主成分自身对照法检查有关物质时直接或间接地以主成分的最大吸收波长作为检测波长,由于有关物质检查的对象是杂质,若将主药的最大吸收波长确定为检测波长,则杂质在此波长下的吸收可能偏低,某些杂质甚至无吸收,这样会造成对杂质含量的低估甚至漏检,从而不能反映产品的真实质量,影响了对品种质量可控性及稳定性的评价。1.直接将主药的最大吸收波长选作检测波长。2.简单地套用含量测定的色谱条件。在HPLC法进行含量测定时,为提高方法的灵敏度,降低干扰,往往选用主成分的最大吸收波长作为检测波长。若套用含量测定的色谱条件,实际仍是以主药的最大吸收波长作为有关物质检测波长。 3.以样品进行破坏性试验(酸、碱、热、光照、氧化等)后的溶液做紫外扫描,将扫描图谱中最大吸收波长确定为有关物质的检测波长。因破坏性试验后溶液中存在尚未破坏的主药、降解产物、辅料等,此溶液的紫外吸收为各成分紫外吸收的加和,并不能反映降解产物的紫外吸收特性。由于未破坏主药所占比例较大,故破坏性试验后溶液的最大吸收波长一般仍为主药的最大吸收波长。 采用HPLC主成分自身对照法检查有关物质,其前提之一是需检查的杂质与主成分在确定的检测波长下应有相近的紫外吸收(响应值接近),选择检测波长时需对产品中可能存在的杂质(合成原料、中间体、副产物以及降解产物)的紫外吸收特性进行研究。已知杂质的紫外吸收特性可采用对其流动相溶液直接进行扫描的方法考察,未知杂质(如未知降解产物等)可通过二极管阵列检测器考察其紫外吸收情况,根据各主要杂质及主成分的紫外吸收特性,选取响应值基本一致的波长作为有关物质的检测波长。若对不同杂质难于找到均适宜的检测波长,可考虑选择在不同波长下分别测定,也可考虑采用加校正因子的主成分自身对照法。只有经试验研究确认主成分的最大吸收波长符合有关物质检查对测定波长的要求时,为方便操作,可选作有关物质的检测波长,以与含量测定的色谱条件一致。 另外,HPLC主成分自身对照法检查有关物质比较适用于对微量杂质总量的控制,也可用于单个杂质的限度(一般不超过0.5%)控制。对于具有明确归属的已知杂质,建议采用杂质对照品法进行检查。对于有毒有害杂质,更应采用杂质对照品法单独测定,并制定严格的限度。 转自——中国植提论坛
初步看了下基础物质种类不多。看了不少相关资料。。。本人以前就是实验室的,可是有些东西还是蛮难买到的!希望大家交流下购买经验等。。。供相互学习!!!!!!!! 最近需要不少杂质对照品,相当难买。基本全进口。。。 基准物质具体有多少,有哪位高人可以列下神马的。。。感激 本人在南京某药厂。。。望同仁现身。。。。其他行业也欢迎。。毕竟采购东西很广泛!!! 主旨在于互相讨论各种采购问题。。。
[color=#DC143C]请问大家在用TLC测定有关物质的时候,有没有遇到过把供试品溶液稀释N倍后作为对照溶液的情况?我很疑惑:供试品溶液的浓度比对照溶液深,为什么杂质斑点不得比对照溶液深呢?这样的对照能说明什么问题呢?谢谢![/color]我是在中国药典上见到的这种方法:乙胺嘧啶的有关物质(05版CP第3页):取本品,加三氯甲烷-甲醇(9:1)制成每1ml中含20mg的溶液,作为供试品溶液;精密量取适量,加同一溶剂稀释成每1ml中含50ug的溶液,作为对照溶液。照薄层色谱法检验,……供试品溶液如显杂质斑点,与对照溶液的主斑点比较,不得更深。
高效液相色谱法测定有关物质的方法建立的过程 有关物质系药品中除主成分以外的杂质,有关物质来源的两种途径:1.合成过程中的中间体和副产物;2.储存过程中产生的降解产物。 由于有关物质的含量较少,所以选择专属性强、灵敏度高、重现性好的检测方法至关重要。目前常用的方法有HPLC法和TLC法,后法不讨论。现主要阐述HPLC法的建立过程。 HPLC法检测有关物质的方法有:(1)杂质对照品法(适用于已知杂质);(2)主成分自身稀释对照法(适用于一般杂质检查,杂质成分少且尚不能取得其对照品,简称“自身对照法”);(3)归一化法 (现已不多用)。前法为外标法、定量检测;后两法均为限量检测。一、色谱条件的确定 1、查阅文献,看看是否同行已经做过类似的工作,有的话可以参考。 2、没有文献,进行以下工作。 1)分析结构,看看有无紫外吸收,有就比较简单,选用紫外检测器就行了,没有的话那就麻烦,可以选用蒸发光散射检测器等。 2)分析结构,看看选用何种色谱方式进行分离(正相或者反相色谱)。 3)分析结构,看看流动相该如何选择,要不要选用一些离子对试剂。 4)与同类产品进行比较。 3、紫外扫描一下,找到最大吸收波长。将能得到的中间体、副产物、分解产物、样品配成相同浓度,在紫外扫描分光光度计上扫描一下,选择所有物质具有相同的最大吸收处的波长作为测定波长。要是有二极管阵列检测器就不需要这一步了。 4、还是得找一些资料,看看类似的样品的流动相,以它为起始流动相进行选择,如果没有,那就找专业书,看看它是怎么教你选择流动相的。 5、通过专属性验证所选色谱条件能否将各杂质与被测物分离检出 。应按0.5%(w/w)被测物浓度的各杂质量添加至被测物中,模拟被测物中可能存在杂质的状态,即有少量(约0.5%)杂质存在时能否与被测物达到完全分离(分离度大于2.0),以验证系统适用性。二、最低检测限 得出达到S/N=2或S/N=3时的样品药品浓度。三、供试品溶液浓度的确定 根据最低检出浓度,采用“上推法”来确定供试品溶液浓度:如一般设定杂质总量小于1.0%对照液,对照溶液的浓度至少应为最低检出浓度的20---50倍,供试品溶液浓度则应是最低检出浓度的2000~5000倍。同时还应考虑仪器、色谱柱等因素对最低检出浓度和最大进样浓度的影响(即耐用性因素),所以供试品溶液的浓度应在保证小于最大进样量的情况下,适当设定得高些,以保证该浓度在任何试验条件下,均有足够的检测灵敏度。四、线性及精密度试验 在稳定性考察中,如某杂质含量不断增加,则说明被测物降解的途径稳定、可循,则有必要对该杂质进行针对性地监控,即采用该杂质对照品 (经确证结构后,由人工合成获得)以外标法准确测定。此时,与含量测定相似,应进行线性试验。通常将杂质线性验证范围0.05%~1.5%)。且精密度试验也应符合要求,但RSD可根据实际情况,适当放宽至2.0~3.0。五、加样回收率试验 回收率试验采用在已知杂质含量的被测物中加入定量杂质的方法来评价。将各杂质以0.4%、0.5%、0.6% (w/w)的浓度加至被测物溶液中,以验证所采用的色谱条件是否可分离检测相应的各杂质以及与被测物中已存在的杂质是否累加,并观测累加量的准确性。六、强力破坏试验 强力破坏试验条件有强光、高温、高湿、强酸、强碱、氧化破坏等。破坏时不能过于剧烈,一般以产生20%~3O%杂质的条件为宜(《化学药物杂质研究技术指导原则》,国家食品药品监督管理局药品审评中心)。同时,还可采用二极管阵列检测器(DAD)或质谱(MS)监测器进一步验证主峰纯度,观察主峰中是否包含有被破坏产生的杂质峰。七、其他色谱参数的确定 流速的选择:主峰保留时间应在l0min以后,这样主峰不易出现拖尾、堆积的现象。柱温的选择:不应超过35℃,既可增强被测物与杂质的分离度,也可避免因温度过高使主峰降解,导致测定误差。溶剂的选择:由于有关物质测定的供试品溶液浓度高,应首先考虑被测物在溶剂中的稳定性 和溶解性。一般选流动相作溶剂为宜, 以排除溶剂峰的干扰。但由于有关物质的测定必须扣除溶剂峰,因此只要溶剂峰不干扰被测物质峰,即使不选用流动相作溶剂,也完全可以。在流动相溶解性不佳时,可采用甲醇或乙腈作溶剂以提高溶解性。 色谱图记录时间的设定:应能洗脱出有可能存在的全部杂质和经强力破坏试验产生的杂质,并规定至主成分保留时间的几倍为止。在测定制剂的有关物质时,个别辅料出峰滞后,此时应在质量标准中注明。八、杂质的限量。 必须根据《化学药物杂质研究技术指导原则》来定。
在做有关物质时,供试品峰面积为4000万,1%对照只有20万,这是正常的吗,如果不正常那是什么原因导致的?有什么办法解决?
最近看到欧洲药典有关物质时,有杂质对照品的,不太明白最后是怎么计算杂质含量的,哪位大侠给我解释一下,越详细越好,不胜感激!!1[em0808]
有关物质自身对照法,批次较多的时候各位大师们都是批批稀释自身对照还是这些批次用同一个自身对照? 说明一下这些批次是浓度很接近的, 我想做个提议给领导说明一下这些批次可以使用同一个对照,我应该从哪方面着手写? 或者这些溶液的浓度都在多少范围之内用同一个对照没有问题
比如有关物质定量时,没有杂质对照品,需要自制,那自制的对照品都要测试些什么项目呢?先把我知道的写出来,求各位补充1、结构鉴定相关内容(UV,IR,H-NMR,C-NMR或更多,MS(是否必须做高分辨呢?))2、自制对照品纯度或含量测定(色谱纯度(HPLC或GC等),水分残留,有机溶剂残留,重金属残留等,最后的含量=色谱纯度-水分残留-有机溶剂残留-重金属残留等)还有其它的要做吗?
我公司做某药品有关物质时,用外标法 , 需要进两针:一针有关物质样品,一针有关物质对照。在打图时我们一般都只积分相应保留时间(4.7min)有关物质的峰,而其他小峰都不积分,有天药检所的来检查说,其他小峰也最好积上,请问各位你们做有关物质时积分其他小峰吗?谢谢
想求一张用WAX(30m*3.2mm,0.5um)做的丙二醇有关物质对照品溶液的色谱图,希望能附上升温程序,急需
我公司做某药品有关物质时,用外标法,需要进两针:一针有关物质样品,一针有关物质对照。在打图时我们一般都只积分相应保留时间(4.7min)有关物质的峰,而其他小峰都不积分,有天药检所的来检查说,其他小峰也最好积上,请问各位你们做有关物质时积分其他小峰吗?谢谢

自身对照法,对照溶液制备:取供试品溶液5ml到100ML容量瓶中。请问这种类型的有关物质该如何计算?!感谢大家!另外还有个问题,就是杂质峰面积不得大于对照峰面积的5.0%,这种情况该如何计算!?http://ng1.17img.cn/bbsfiles/images/2014/10/201410111503_517810_2858060_3.jpg
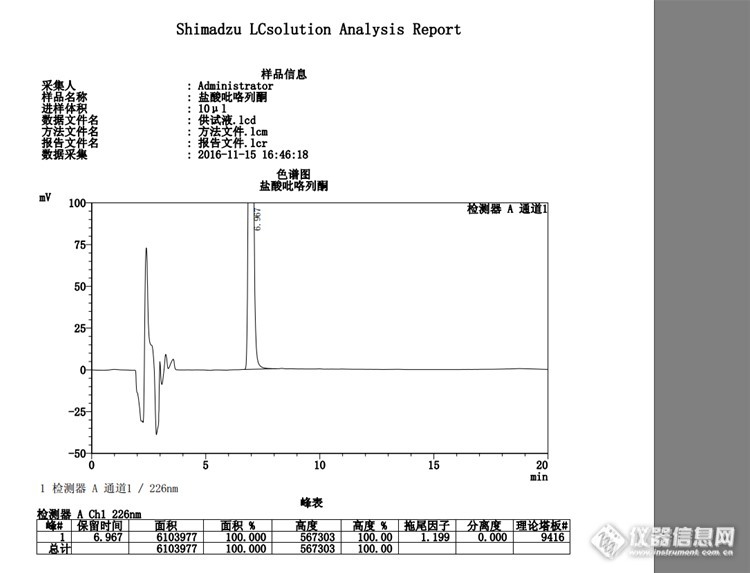
迎接2017年,原创1-Welchrom® C18测定盐酸比格列酮有关物质色谱柱sn:W13211841,pn:00310-02041(5μm,150mm×4.6mm)色谱条件自拟:流动相为乙腈-0.1M醋酸铵溶液(50:50),用醋酸调节pH值至5.0,检测波长为226nm,进样量为10μl,分析时间约为20分钟。操作:取本品适量置20ml容量瓶中,加流动相适量使溶解并稀释制成每1ml约含盐酸比格列酮0.5mg的溶液作为供试液,精密量取1ml,置100ml量瓶中,加流动相稀释至刻度,摇匀,作为对照溶液。本品空白溶剂为流动相。盐酸比格列酮简介药品名称: 盐酸吡格列酮别名:安可妥;盐酸匹格列酮;盐酸吡格列酮;盐酸皮格列酮;匹格列酮盐酸盐英文: Pioglitazone Hydrochloride;ACTOS中文化学名(±)5-苄基]-2,4-噻烷二酮盐酸盐剂型: 原料药性质:外观 白色晶体粉末熔程 193-194℃可溶性:在甲醇中溶解,在乙醇、氯仿中微溶分子式:C19H20N2O3S·HCl分子量:392.90CAS号:112529-15-4用途:新一代二型糖尿病治疗药,副作用小。本品主要成分是盐酸吡格列酮,其化学名称为(±)-5--苯甲基]噻唑烷-2,4-二酮单盐酸盐结构式:[url=http://p1.qhmsg.com/t01f3c03f21df4dc8b0.jpg][img]http://p1.qhmsg.com/dr/220__/t01f3c03f21df4dc8b0.jpg[/img]分子式:C19H20N203S·HCl分子量:392.90本品主要检测有关物质:其典型色谱图如下:空白溶剂:[img=,690,597]http://ng1.17img.cn/bbsfiles/images/2017/01/201701012253_01_1621890_3.png[/img]对照溶液:[img=,690,640]http://ng1.17img.cn/bbsfiles/images/2017/01/201701012254_01_1621890_3.png[/img]供试液:[img=,690,527]http://ng1.17img.cn/bbsfiles/images/2017/01/201701012256_01_1621890_3.png[/img]总结:现在该药品正在处于摸索阶段,结果显示本品较为“干净”,到底干净是否,要等组织长期的调查,我们相信群众的眼睛的,目前色谱数据较为理想,月旭的色谱柱的柱效较高均达到了9000,拖尾因子均小于1.3。一般我们在进行新的项目摸索方法的时候也在摸索色谱柱,初步确认该品种用月旭这款色谱柱较为理想。
一直很纠结,杂质对照品的稳定性需要做吗?我曾请教过别人说是不用作,可是如果不做,当作为对照品时,无论结构还是浓度一旦发生变化,岂不是杂质的检测就受影响吗?
我在做氯霉素滴眼液的有关物质时,在药典上有一句“另精密称取氯霉素二醇物对照品与对硝基苯甲醛对照品适量,按氯霉素二醇物每10mg加甲醇1ml使溶解,用流动相定量稀释成每1ml中含氯霉素二醇物8ug与对硝基苯甲醛2ug的混合溶液,作为杂质对照品溶液”其中“每1ml中含氯霉素二醇物8ug与对硝基苯甲醛2ug的混合溶液”这句话怎么理解啊?是先两种对照品各称,氯霉素二醇物8ug/ml,对硝基苯甲醛2ug/ml,然后各取1ml混合(这样混合之后混合液中含两种对照品的量就减半);还是说最终的混合溶液中含两种对照品的量是8ug/ml和2ug/ml啊? 我都头大了 谢谢赐教啊[em0812] [em0812] [em0812]
高效液相色谱法测定有关物质的方法建立的过程 有关物质系药品中除主成分以外的杂质,有关物质来源的两种途径:1.起始原料及合成过程中的中间体和副产物;2.储存过程中产生的降解产物。 由于有关物质的含量较少,所以选择专属性强、灵敏度高、重现性好的检测方法至关重要。目前常用的方法是高效液相色谱法。现主要阐述高效液相色谱法的建立过程。 高效液相色谱法检测有关物质的方法有:(1)杂质对照品法(适用于已知杂质);(2)主成分自身对照法(紫外检测器),采用HPLC主成分自身对照法检查有关物质,其前提之一是需检查的杂质与主成分在确定的检测波长下应有相近的紫外吸收(响应值接近)。一、色谱条件的确定 1、查阅文献,看看是相关的资料,有的话可以参考。 2、没有文献,进行以下工作。 1)分析结构,看看有无紫外吸收,有就选用紫外检测器,没有的话可以选用蒸发光散射检测器等。 2)分析结构,看看选用何种色谱方式进行分离(正相或者反相色谱)。 3)分析结构,看看流动相该如何选择,要不要选用一些离子对试剂。 4)与同类产品进行比较。 3、紫外扫描一下,找到最大吸收波长。将能得到的中间体、副产物、分解产物、样品配成相同浓度,在紫外扫描分光光度计上扫描一下,选择工艺中最容易带入或产生的杂质的最大吸收处的波长作为测定波长。要是有二极管阵列检测器就不需要这一步了。 4、查找资料,看看类似的样品的流动相,以它为起始流动相进行选择,如果没有,那就找专业书,看看它是怎么选择流动相的。 5、通过专属性验证所选色谱条件能否将各杂质与被测物分离检出 。应按0.5%(w/w)被测物浓度的各杂质量添加至被测物中,模拟被测物中可能存在杂质的状态,即有少量(约0.5%)杂质存在时能否与被测物达到完全分离(分离度大于2.0),以验证系统适用性。二、最低检测限 得出达到S/N=2或S/N=3时的样品药品浓度。三、供试品溶液浓度 一般在样品不过载的情况下,适当设定得高些,以保证该浓度在任何试验条件下,均有足够的检测灵敏度;也可以根据峰面积确定(经验),面积七位数时9开头,八位数时1开头。四、线性及精密度试验 按药典规定,即采用该杂质对照品 (外购或合成获得)以外标法准确测定。此时,与含量测定相似,应进行线性试验。通常将杂质线性验证范围0.05%~1.2%)。精密度试验是以杂质百分浓度为0.5%进样,相对标准偏差不大于2.0。五、回收率试验 杂质的定量试验可向原料药或制剂中加入已知量杂质进行测定。将各杂质以0.4%、0.5%、0.6% (w/w)的浓度加至被测物溶液中,以验证所采用的色谱条件是否可分离检测相应的各杂质以及与被测物中已存在的杂质是否累加,并观测累加量的准确性。六、强降解试验 强降解试验条件有酸、碱、氧化、强光、高温破坏等。破坏时不能过于剧烈,一般以产生10%~20%杂质的条件为宜(《化学药物杂质研究技术指导原则》,同时采用二极管阵列检测器(DAD)验证主峰峰纯度,验证物料平衡。七、其他色谱参数的确定 流速的选择:主峰保留时间约在l0min左右,这样主峰不易出现拖尾、堆积的现象,运行时间也不会太长。柱温的选择:不应超过35℃,既可增强被测物与杂质的分离度,也可避免因温度过高使主成分降解,导致测定误差。 溶剂的选择:首选流动相作溶剂为宜,以排除溶剂峰的干扰。也可以先用和流动相同比例有机溶剂溶解,再加入同比例水相。难溶物质,可采用甲醇或乙腈作溶剂以提高溶解性。 色谱图记录时间的设定:根据强力破坏试验和样品稳定性试验来规定色谱图记录时间,应能洗脱出有可能存在的全部杂质和经强力破坏试验产生的杂质,并规定运行时间主成分保留时间的几倍。八、杂质限度的确定 杂质限度的制订应考虑如下因素:杂质及含一定限量杂质的药品的毒理学研究结果;给药途径;每日剂量;给药人群;杂质药理学可能的研究结果;原料药的来源;治疗周期;在保证安全有效的前提下,药品生产企业对生产高质量药品所需成本和消费者对药品价格的承受力。严格按照《化学药物杂质研究技术指导原则》来制定。
标准物质/对照品很贵啊,一定要省着用!!!如果让你 称取 总量20%的 液体 标准物质(总重就10mg或ul),你如何称量?提示:本帖只对有建树者进行奖励!!!版友们,你们可以先取大约10mg或ul的水,放在我们购买的对照品或标准物质的瓶子中(或者HPLC的进样瓶),然后再去尝试如何去用!!







 我要推广仪器
我要推广仪器
 下载APP
下载APP