使用[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质联用[/color][/url]技术测硝基咪唑及其代谢物 使用内标法 在做液相优化的时候标准品和内标物的峰没有办法分开 内标物和标准品的需要完全分开吗
大家有知道这些代谢物哪里能提供?CL 288511 /CL 182704是什么编号呀? 咪唑乙烟酸咪唑乙烟酸 的代谢物 CL 288511 咪唑乙烟酸 的代谢物 CL 182704
有做硝基咪唑,及其代谢物的吗?好做吗?
5,7-二氯-4-(2,4,5-三氯苯氧基)-2-(三氟甲基)-1H-苯并咪唑大家如何测试?求大神带。
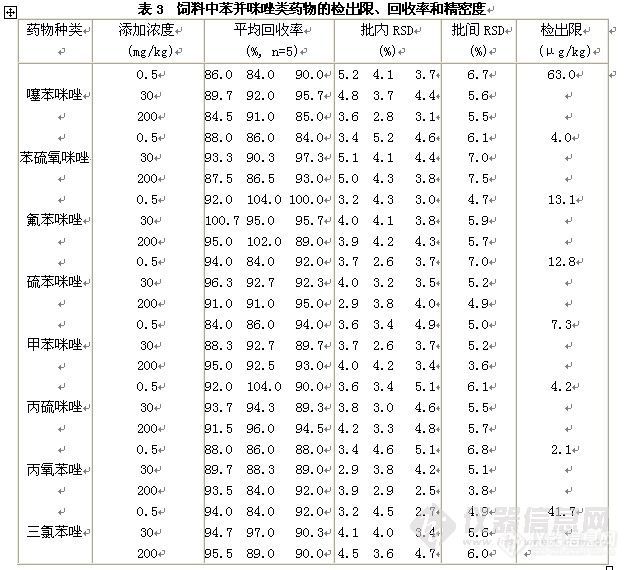
液相色谱串联质谱法测定饲料中8种苯并咪唑类药物摘 要 建立了同时测定饲料中8种苯并咪唑类药物(噻苯咪唑、丙硫咪唑、硫苯咪唑、苯硫氧咪唑、氟苯咪唑、甲苯咪唑、丙氧苯唑和三氯苯唑)的液相色谱串联质谱分析方法。饲料样品直接用酸化乙腈提取,提取液用甲酸溶液稀释后直接进行分析。分析时采用XBridgeTM C18色谱柱,以甲酸溶液-乙腈体系进行梯度洗脱,MRM方式测定,基质外标法定量。苯并咪唑类药物在0.02~10 mg L-1浓度范围内呈良好的线性,线性相关系数均大于0.990,苯并咪唑类药物在饲料样品中最低检测限为2.1~63.0μg/kg。饲料中苯并咪唑类药物在0.50~200 mg/L范围内的回收率为84.0%~104%之间,相对标准偏差(RSD)均小于10%。 关键词 苯并咪唑类药物;液相色谱串联质谱法;饲料 苯并咪唑类药物(benzimidazoles, BMZs)属于广谱、高效、低毒抗蠕虫药,由于对胃肠线虫具有很强的驱杀作用,至今仍在广泛使用。但由于BMZs在实验动物和靶动物显示致畸和致突变作用,目前使用的BMZs多数是食品残留中重要的监控对象,且BMZs在体内转化的代谢产物仍具有毒理作用,所以我国以及联合国粮农组织、欧盟、美国、日本等国家和组织都将苯并咪唑类药物列入限制使用的兽药药物,并制订出各种苯并咪唑类药物在不同动物体内(肌肉、组织、奶等)的最高残留限量。饲料安全直接关系到动物性食品的安全,考虑到苯并咪唑类药物经常被添加到饲料中使用,故很有必要进行饲料中苯并咪唑类药物的分析研究。 目前对于动物组织中苯并咪唑类药物的分析方法较多,而饲料中苯并咪唑类药物分析方法国内未见发表,国外也较少,涉及的种类也较少,最多的仅有5种药物。动物组织和饲料中BMZs分析涉及的主要分析手段有:酶联免疫吸附法( ELISA) 、气相色谱-质谱法(GC-MS)、高效液相色谱法(HPLC)及高效液相色谱串联质谱法(HPLC-MS/MS),高效毛细管电泳法(HPCE)。考虑到苯并咪唑类药物在我国使用情况,本研究选择了8种常用苯并咪唑类药物,考虑到LC-MS/MS法灵敏度高的特点,样品酸化乙腈提取后直接稀释后进行液相色谱串联质谱分析。1 材料与方法1.1 仪器与试剂 Waters 2695 Quattro MicroTM API高效液相色谱串联质谱仪(美国Waters公司),配置电喷雾离子源;固相萃取仪(美国Supelco 公司);Sigma离心机。噻苯咪唑和丙硫咪唑标准品(Accustandard 公司);硫苯咪唑、苯硫氧咪唑、氟苯咪唑、甲苯咪唑、丙氧苯唑和三氯苯唑标准品(Dr. Ehrenstorfer)。乙腈、二甲亚砜和甲酸为色谱纯(Fisher公司)。1.2 仪器条件 XBridgeTM C18色谱柱(150 mm×2.1 mm,内径3.5 μm);流动相A为0.1%甲酸溶液,B相为乙腈,梯度洗脱条件:B相在1.0 min内从15%线性增加到25%,再在2.5 min内线性增加到95%,保持3.5 min,然后在0.1 min内降至15%,保持4.9 min;流速:0.3 mL/min;进样量:10 µL;柱温:30℃。 质谱条件:ESI源正离子模式电离;多反应监测(MRM);毛细管电压:3.0 KV;萃取锥孔电压:20 V;RF透镜电压:0.5 V;离子源温度:110 ℃;脱溶剂气温度:350 ℃;锥孔气流速:50 L/h;脱溶剂气流速:600 L/h;倍增器电压:650 V;二级碰撞气:氩气;其它条件详见表1。http://ng1.17img.cn/bbsfiles/images/2010/11/201011301506_262957_1759541_3.jpg1.3 样品处理 称取2g试样(精确到0.01g)于50 mL离心管中,加入20 mL0.5 %甲酸乙腈,涡旋1 min,然后超声提取10 min,以5000 r/min的速度离心5 min后吸取1.0 mL上清液于5 mL刻度试管中,加入3 mL0.1 %甲酸溶液于试管中,混匀后过0.22 μm滤膜,进行液相色谱串联质谱分析。1.4 线性实验 准确称取各10.0 mg BMZs标准品于相应的10mL容量瓶中,噻苯咪唑、甲苯咪唑、丙氧苯唑和丙硫咪唑用二甲亚砜溶解并定容至刻度,其余4种BMZs用甲醇:二甲亚砜(2:3 v/v)溶解并定容至刻度,即得均为1000 mg/L标准储备液。分别吸取1.0 mL各标准储备液于同一10mL容量瓶中,用甲醇稀释至刻度,即得100 mg/L的混合标准工作液。分别准确移取苯并咪唑类药物混合标准中间液适量,配制浓度为0.2.、0.8、2.0、10.0、40.0和100.0 mg/L的系列标准溶液,吸取0.1 mL于5 mL刻度试管中,再吸取空白试料提取液0.9 mL于该5 mL刻度试管中,加入3 mL0.1%甲酸溶液后混匀过膜,进行上机测定,以定量离子对峰面积为纵坐标,标准溶液浓度为横坐标,绘制基质校准标准曲线。2 结果与分析2.1 液相色谱质谱分析 苯并咪唑类药物色谱分析时,通常采用反相分离体系,主要有三类流动相体系:离子增强体系,pH2~3,一般使用乙腈-磷酸或磷酸盐体系;离子抑制流动相体系,pH5~7;离子对流动相体系,离子增强流动相中加入阴离子对试剂。对于多组分苯并咪唑类药物液相色谱质谱分析时,通常采用离子增强体系进行梯度洗脱,如0.1%甲酸溶液-乙腈体系,因为该体系和纯水-乙腈体系相比色谱峰的拖尾现象得到了明显改善。 苯并咪唑类药物属弱碱性物质,中等极性,在酸性条件下很容易质子化,于是本方法选择ESI+进行分析。以乙腈/0.1%甲酸溶液(3:7,v/v)为溶解液,用蠕动泵(20μL/min)对苯并咪唑类药物的质谱条件进行优化。经过优化的条件为:毛细管电压:3.0KV;离子源温度:110℃;脱溶剂气温度:350℃;锥孔气流速:50L/h;脱溶剂气流速:600L/h。其它条件详见表1。2.2 提取净化方法的选择和优化 [font=宋体
GB 29687-2013 食品安全国家标准 水产品中阿苯达唑及其代谢物多残留的测定 高效液相色谱法
大家都用的是什么标准的方法?我用的是: 氯霉素:NY5070--2002,无公害食品 水产品中渔药残留限量 附录A 己烯雌酚 SC/T3020-2004水产品中己烯雌酚残留量的测定 酶联免疫法 呋喃唑酮代谢物 DB32/T 1038-2007 水产品中呋喃唑酮代谢物残留量的测定有不同的吗?我是江苏的,呋喃唑酮代谢物测定时用的地标。
新手,第一次做食品中硝基呋喃类药物的检测。请做过的老师们指点一下。用硝基呋喃类代谢物标准品优化质谱参数还需要衍生么,样品前处理衍生的目的是什么? 衍生的话,它们的母离子跟子离子质量数还跟国标上一致么?国标21311里面衍生,不同浓度的基质标曲加入的衍生剂的量相同。这样会不会影响衍生的效果?
我门最近刚刚开展肉中硝基呋喃代谢物的检测。仪器是agilent 6410的质谱。但是突然发现在优化质谱条件,寻找母离子和子离子时,需要将标准品进行衍生,请教各位大侠,有没有具体的衍生化条件?越具体越好!不胜感激!
刚开始学习液质,检测呋喃代谢物代谢物的方法也是从别处学着做的,遇到了一个问题,请大家指教!测鸡肉中呋喃代谢物时,标准品混标加在空白试剂中衍生后,再配制成不同浓度,上机跑标准曲线。加标回收是加在阴性样品鸡肉中再加试剂衍生化的。发现其他三种外标内标都很好(时间稳定,响应也高),就是标准品中amoz是3.76分钟出峰(内外标),而样品加标回收的amoz都是在3.54分钟上出一个响应很好的峰(内外标)而3.76分钟时几乎没有峰。请问可以判定加标回收中,3.54分的峰是amoz么?
最近在做国标21169-2007蜂蜜中双甲脒及其代谢物的扩项(液相色谱法),按照方法做出来,双甲脒回收率在百分之七八十左右,2,4-二甲基苯胺在百分之三四十左右,死活都达不到标准方法里说的双甲脒回收率99.8%以上,2,4-二甲基苯胺71.7%以上。不知道有没有老师做过这个方法,怎么才能提高双甲脒和2,4-二甲基苯胺的回收率?
单位开展这个项目,但是目前没有呋喃代谢物的标准对照品,问了几家供应商都没有,不知道大家的都在什么地方买的,有没有电话可以联系
GB 29704-2013 食品安全国家标准 动物性食品中环丙氨嗪及代谢物三聚氰胺多残留的测定 超高效液相色谱-串联质谱法
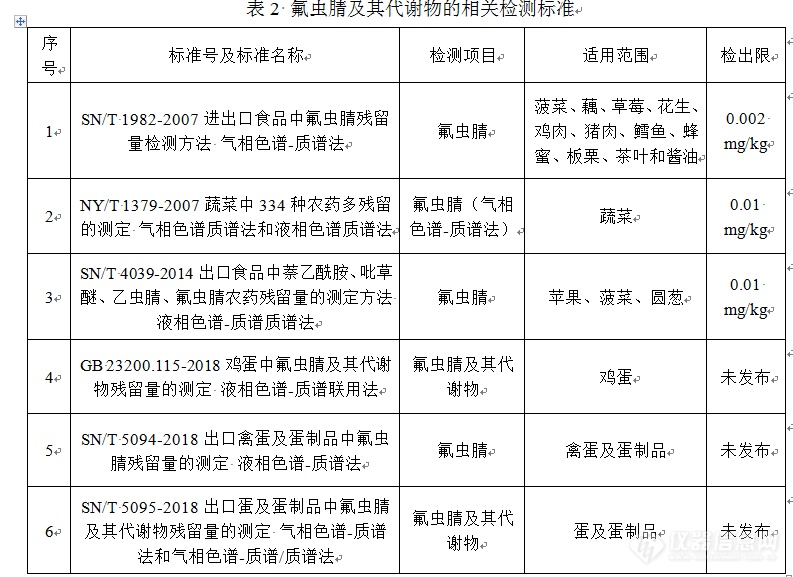
食品安全地方标准 :食品中氟虫腈及其代谢物残留量测定 高效液相色谱-串联质谱法编制说明[b]一、工作简况(一)任务来源与项目编号、起草单位、主要起草人[/b]本检测方法标准的制订工作,是受广西壮族自治区卫生和计划生育委员会委托,由广西出入境检验检疫局检验检疫技术中心负责研制,项目编号为2018004。标准主要起草单位为广西出入境检验检疫局检验检疫技术中心。主要起草人为:****。(二)简要起草过程广西出入境检验检疫局检验检疫技术中心接受制订任务后,成立由实验室专业技术人员组成的研制小组,全面负责标准的研制计划和工作安排。成员由正高1人,副高5人,博士、工程师4人共10人组成。制订本标准主要依据《中华人民共和国标准化法》,GB/T 1.1-2009《标准化工作导则 第1部分:标准的结构和编写》,GB/T 20001.4-2015《标准编写规则 第4部分:试验方法标准》和GB/T 27404-2008《实验室质量控制规范 食品理化检测》等标准或规范。制标小组为做好标准的制定工作,广泛收集国内外有关的检测技术资料、检测标准、限量指标,以及相关的法律、法规,召开了有关座谈会,听取各方建议。在研制过程中,遵循科学性、实用性和前瞻性等原则,尊重客观规律,融合科学理论和标准化方法。考虑到我国食品卫生标准分析方法应逐步与国际接轨,以及分析方法应有较强的可操作性的要求,确定了标准制定的关键环节,对本项目作了以下主要制订:(1)确定了检测项目为氟虫腈及其代谢物氟虫腈砜、氟虫腈亚砜和氟甲腈(2)限定了检测方法为高效液相色谱-串联质谱法;(3)根据食品安全法和标准制订的要求,规定检测对象和标准验证范围为卫生部发布的国家实施市场准入管理的28大类食品中畜禽肉及副产品、水产品、水果、蔬菜、鲜蛋、粮谷类及豆类等食品。二、与我国、我区有关法律法规和其他标准情况的关系新制地方标准与相关法律、法规及相关标准协调一致,没有冲突。氟虫腈及其代谢物的相关检测方法标准见表2。第一,新制地方标准的检测项目更全面。新制地方标准不仅对食品中氟虫腈原药残留量进行测定,还对食品中氟虫腈代谢物氟虫腈亚砜、氟虫腈砜和氟甲腈的残留量进行测定,符合GB 2763-2016对氟虫腈的定义(定义中包含氟虫腈的代谢物)。而相关的其他检测标准SN/T 1982-2007、NY/T 1379-2007和SN/T 4039-2014均只检测食品中的氟虫腈原药残留量。第二,新制地方标准的检测范围更宽。新制地方标准不仅适用于蔬菜、水果、粮谷类食品,还适用于畜禽肉、水产品、内脏、鲜蛋以及茶叶等食品。而即将实施的国家标准GB 23200.115-2018及行业标准SN/T 5094-2018和SN/T 5095-2018均仅适用于禽蛋及蛋制品。第三,新制地方标准的灵敏度、准确度和精密度更优。新制地方标准的灵敏度、准确度和精密度等相关指标不仅满足国家标准GB/T 27404-2008《实验室质量控制规范 食品理化检测》中的规定,同时也满足欧盟指令SANTE 11945 -2015《Guidance document on analytical quality control and method validation procedures for pesticides residues analysis in food and feed》中的相关要求。第四,新制地方标准采用优化的QuEChERS前处理净化方法,相对于其他国家标准或行业标准采用的固相萃取或液液萃取的前处理净化方法,QuEChERS方法具有快速、方便、高效、经济和安全等特点。[img=,690,502]https://ng1.17img.cn/bbsfiles/images/2019/02/201902061027187646_1901_2166779_3.png!w690x502.jpg[/img]三、与国外、国内有关法律、法规和标准情况的说明国家标准“GB 2763-2016食品中农药最大残留限量”将氟虫腈残留定义为氟虫腈、氟甲腈(MB46513)、氟虫腈砜(MB46136)和氟虫腈亚砜(MB45950)的残留之和,以氟虫腈表示。该标准规定了谷物、油料和油脂、蔬菜、水果、糖料和食用菌6类食品的最大残留限量为0.02 mg/kg (玉米为0.1 mg/kg除外),并规定了相应的检测方法:谷物、油料和油脂、蔬菜参照SN/T 1982-2007,水果、糖料和食用菌参照NY/T 1379-2007。其中SN/T 1982-2007采用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]-质谱法,检测范围为菠菜、藕、草莓、花生、鸡肉、猪肉、鳕鱼、蜂蜜、板栗、茶叶和酱油,检出限为0.002 mg/kg;NY/T 1379-2007 采用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]-质谱法,检测范围为蔬菜,检出限为0.01 mg/kg。此外,检验检疫行业标准SN/T 4039-2014采用液相色谱-质谱/质谱法,检测范围为苹果、菠菜、圆葱,检出限为0.01 mg/kg。这3个检测标准均没有涉及到检测食品中氟虫腈的3种代谢物。由此可见:我国目前仅对谷物、油料和油脂、蔬菜、水果、糖料和食用菌6类食品中的氟虫腈残留制定了最高限量,对畜禽产品和水产品均没有相关要求;目前只有检测食品中氟虫腈原药残留量的标准方法,而没有针对食品中氟虫腈代谢物氟甲腈、氟虫腈亚砜和氟虫腈砜的检测标准;现有的3个检测方法标准的适用范围较局限。而即将实施的国家标准GB 23200.115-2018及行业标准SN/T 5094-2018和SN/T 5095-2018虽然均对食品中氟虫腈原药及其3种代谢物残留量进行了测定,但其检测范围均局限于禽蛋及其制品。欧盟法规“动植物源性食品和饲料中农药残留最高限量(EC No3965/2005)”中对氟虫腈的定义同样包含氟虫腈的砜化代谢物即氟甲腈(MB46513)、氟虫腈砜(MB46136)和氟虫腈亚砜(MB45950)。2014年10月欧盟出台法规(EU) No1127-2014,在EC No3965/ 2005规定的基础上对水果、蔬菜、畜禽产品和水产品等食品和饲料中氟虫腈的最高残留量进行了修订,如下:Fruit vegetables, Cereals, Nuts, Seeds, Rhizome, Tea, Coffee, Herbal, Infusions and Cocoa均为0.005 mg/kg;Potatoes, Flowering brassica, Leek or Brussels sprouts 均为0.01 mg/kg Onions or Shallots为0.02 mg/kg;Tropical root and Tuber vegetables为0.05 mg/kg。相对于国家限量标准GB 276-2016,欧盟对氟虫腈的限量要求更加全面和细化,而且相应的检测标准均涉及到氟虫腈及其代谢物的检测。
毕业在即,毕业论文中涉及2-丙基苯并咪唑的红外标准图谱图,望各位朋友帮帮忙了!本人将万分感谢!如果哪位朋友能找到,请发到我的邮箱,piao23luo◎163.com,谢谢拉!
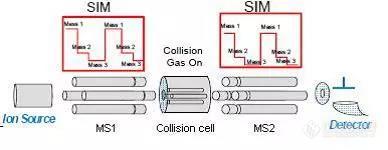
标准解读:国标GB 23200.113:208种农药及其代谢物残留量的测定[color=#333333]2018年6月21日,经食品安全国家标准审评委员会审查通过,国标GB23200.113-2018《食品安全国家标准 植物源性食品中208种农药及其代谢物残留量的测定 [url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]-质谱联用法》正式发布,并于2018年12月21日正式实施。[/color][color=#333333]新国标几乎囊括了所有的植物源性食品,并首次将 QuEChERS 样品前处理方法和[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]-质谱联用检测方法纳入多农药残留检测的国家标准方法中,因此吸引不少业内人士的关注。[/color][color=#333333][color=#333333]总结了新国标GB 23200.113-2018三大特点:[/color][/color][color=#333333][color=#333333][b]1[/b][color=#ffa900][b]基质涵盖范围广、涉及农药种类多[/b][/color][/color][/color]新国标几乎囊括了所有的植物源性食品,包括蔬菜、水果、食用菌,谷物、豆类、油料作物,茶叶、香辛料,植物油等[color=#ffa900][b]9大类23种样品基质[/b][/color]。检测目标针对[color=#ffa900][b]208种农药及其代谢物[/b][/color],包括有机磷类、有机氯类、拟除虫菊酯类、三唑类、酰胺类、三嗪类、苯氧羧酸类、氨基甲酸酯类等。[b]2[/b][color=#ffa900][b]首次将QuEChERS前处理技术引入国家标准[/b][/color][b][color=#0052ff][/color][/b]近些年,QuEChERS方法以其快速(Quick)、简便(Easy)、廉价(Cheap)、有效(Effective)、可靠(Rugged)、安全(Safe)的特点成为国际上最新发展起来的一种用于农产品检测的快速样品前处理技术。QuEChERS方法是美国农业部Anastassiades教授等人于2003年开发的[sup][/sup],该技术的原理是将均质后的样品经乙腈(或酸化乙腈)提取后,采用萃取盐盐析分层,利用基质分散萃取机理,采用PSA(primary secondary amine,N-丙基乙二胺)或其它吸附剂与基质中绝大部分干扰物(有机酸、脂肪酸、碳水化合物等)结合,通过离心方式去除,从而达到净化的目的[sup][/sup]。众所周知,前处理是食品检测中最费时费力的环节, QuEChERS技术的全面引入,大大简化了前处理过程。[b]3[/b][color=#ffa900][b]首个使用GC-MS/MS检测植物源性食品中农药残留的国家标准[/b][/color][url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]-三重串联四级杆质谱GC-MS/MS独有的多反应监测模式(MRM)能为目标物分析提供更专属的选择性。该模式下MS1选择母离子,在碰撞池中产生碎片离子,MS2监测该母离子的特征碎片离子(见图1),因此能有效消除基质噪音且灵敏度高,是在复杂基质中进行痕量目标物分析的最佳选择。同时,采用多反应监测(MRM) 模式可以简化样品的净化程序,节省实验成本,提高工作效率[sup][/sup]。[align=center][img=,388,150]https://ng1.17img.cn/bbsfiles/images/2018/09/201809241931374931_8053_2166779_3.jpg!w388x150.jpg[/img][/align][align=center] MRM原理图[/align][align=center][/align]新国标是我国首个将QuEChERS前处理法和GC-MS/MS分析法这两种高效方法结合起来针对植物源性食品中多农残分析的国家标准。它能应用于复杂基质背景下目标化合物的快速、准确鉴定,为广大的实验分析工作者减轻了负担。SMQ食品检测所现已拥有成熟的QuEChERS前处理技术,以及丰富的[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]-三重串联四级杆质谱仪使用经验,欢迎广大企业咨询送检(0755-27528676)。[color=#ffffff]参考文献[/color]Anastassiades M, Lehotay S J, Š tajnbaher D, Schenck F J. Fast andeasy multiresidue method employing acetonitrile extraction/partitioning and“dispersive solid-phase extraction” for the determination of pesticide residuesin produce. Journal of AOAC international, 2003, 86(2): 412-431.孙亚真, 巩卫东, 张淑霞, 宁水平. QuEChERS—色质联用技术在食品农药多残留检测中的应用与前景. 农产品加工学刊, 2012, 2: 89-92.马智玲, 赵文, 李凌云, 郑姝宁, 林桓, 张延国, 高青珍, 刘肃. [url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]-三重四极杆串联质谱法快速测定蔬菜水果中129种农药的残留量. 色谱, 2013, 31(3): 228-239.
跪求EN 14333.1-2004无脂食品.苯并咪唑杀菌剂carbendazim、噻唑苯并咪唑和苯菌灵的测定方法,中文英文的都行,E-mail:grant_su@sina.com
本人急需测定喹乙醇代谢物的农业部标准,不知哪位大虾有?可否给我提供一下,十分感谢。
10号走呋喃代谢物样品的时候,发现了一个很奇怪的现象,标准品保留时间正常,走空白和样品时AMOZ、SEM、AHD、AOZ四项内标保留时间全部偏离0.2min左右,最后一针标准品也没能幸免。可以肯定不是流动相的问题,柱子平衡时间也足够长。为了确认,又重新进了一遍,仍然重复上述情况。最后,把空白舍去,按照标准品、样品、标准品顺序运行,结果正常,空白的影响力这么大?一针空白可以导致后面序列全部异常(总共6针)?我真是第一次碰到这种奇怪的现象,问题的原因还没有找到,等看一下今天的样品结果再做判断吧。仔细看了一下,发现空白没有内标,测了一下空白的PH,大约在5-7,标准和样品都在3左右,难道真的是弄错了空白?查看了当天的样品,同时检测的还有克伦特罗、孔雀石绿、氯霉素和盐霉素,前三个都有内标监控,所以肯定没错,最有可能的是和盐霉素空白混了,想重新检测比较一下,结果进样小瓶已经处理掉了,哎,只能等有时间再验证了。17和18号进行了相关验证,发现使用盐霉素空白并不能导致呋喃代谢物保留时间漂移,看来当时的呋喃空白应该是预处理操作的时候出现了错误,因无法重现,可能要成为不解之谜了。。。。。。以下是当天保留时间偏离的三个样品和标准的TIC图谱,其中第一、三、五、七通道分别是AMOZ、SEM、AHD、AOZ的内标,每一通道前三个是样品,最后一个是标准品。大家看一下有没有遇到这样偏离的情况,讨论一下。http://ng1.17img.cn/bbsfiles/images/2010/11/201011202027_260858_1855403_3.gif
大家好: 我在做食品中呋喃妥因代谢物时,不知道什么原因,所有的样品都检出,标准品也不成线性,回收率很高有时能达到好几百,请问呋喃妥因代谢物检测过程中有什么因素可以影响?谢谢指点。(我用的是Thermo的TSQ Quantum [url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url])
硝基呋喃类药物(nitrofurans)广泛用于家禽、家畜、水产、蜂等动物传染病的预防与治疗,当含有硝基呋喃类抗生素残留的食品被食用,将会对人类健康造成危害。所以,在食品安全的检测中均要检测硝基呋喃代谢物。 农业部 783 号公告 -1-2006 规定了水产品中硝基呋喃类代谢物残留量的液相色谱 - 串联质谱测定法。 本标准适用于水产品中呋喃唑酮的代谢物3- 氨基-2- 噁唑烷基酮(AOZ)、呋喃它酮的代谢物5- 甲基吗啉-3- 氨基-2- 噁唑烷基酮(AMOZ)、呋喃西林的代谢物氨基脲(SEM)和呋喃妥因的代谢物1- 氨基-2- 内酰脲(AHD)残留量的测定。[align=center][img]http://img1.17img.cn/17img/images/201807/insimg/11425eb2-15a1-4463-8539-d09f50458dd9.jpg[/img][/align][align=center][img]http://img1.17img.cn/17img/images/201807/insimg/1750d0a1-3cfd-41a4-8db8-b7956f51938c.jpg[/img][/align][align=center][img]http://img1.17img.cn/17img/images/201807/insimg/81dd15e4-f3f0-4ded-b990-5cd4134d8d0e.jpg[/img][/align][align=center][img]http://img1.17img.cn/17img/images/201807/insimg/8d8ea9c4-74ab-4a44-81e3-6fcc7463b274.jpg[/img][/align]
本人新手,准备做呋喃唑酮代谢物AOZ的检测,用LC-MS做SRM。现在对于优化质谱条件有些不解,优化的时候优化的是AOZ的条件,还是AOZ衍生之后的条件?如果是衍生之后的话,我手里只有AOZ的标准品,应该怎样操作呢?第一次接触衍生,希望大家帮帮我。
原理:样品经盐酸水解,用2-硝基苯甲醛过夜衍生,调pH值7.1-7.5后,用乙酸乙酯提取,液相色谱-串联质谱检测,内标法定量。具体分析步骤如下:[b]1[/b] 试样的制备,水解和衍生化称取2.5g(粉体样品减半,精确至0.01g)制备的样品(鱼虾等取可食部分按标准要求取样),置于50mL塑料具塞离心管中,加入10mL 0.2mol/L盐酸溶液,用匀质机高速匀质1min,再依次加入200µ L 10ng/mL 内标标准液, 0.05mol/L的2-硝基苯甲醛衍生剂甲醇溶液400µ L,振摇1min,置于37°C,120rpm的恒温摇床中保持16小时。[b]1.2[/b]提取,净化和溶解[color=black]将上述衍生溶液取出放置至室温,加入5mL 0.2mol/L磷酸氢二钾溶液,振摇2min。用1mol/L氢氧化钠溶液,0.2mol/L盐酸溶液调节pH约7.1-7.5, (pH计调节,用水清洗电极于离心管中,控制体积不超过20mL)。5000转/min离心10min,取上清液于另一50m[/color]L[color=black]带盖塑料离心管中。加20mL乙酸乙酯, 振摇10分钟, 5000r/min离心5min,吸取上层乙酸乙酯溶液于另一带盖塑料离心管中;残渣中加20mL乙酸乙酯重新提取一次,合并乙酸乙酯层,提取液于 38°C用氮吹仪吹干,加入1.0mL20%甲醇水溶液,涡旋1min,过0.22µ m滤膜待进样。[/color][b]1.3 [/b]方法空白除不加样品外,其余步骤同上。[b]1.4 [/b]标准曲线和QC[b]1.4.1[/b]标准曲线:取5个50mL塑料具塞离心管,分别加入10μL,20μL,40μL,100μL,200μL硝基呋喃代谢物的标准溶液(50ng/mL),除不加样品外,其余步骤同1.1衍生,1.2项净化及溶解的操作。使最终定容浓度为0.5ng/mL, 1.0ng/mL, 2.0ng/mL, 5.0ng/mL, 10.0ng/mL。内标浓度为2.0ng/mL。[b]1.4.2 [/b]QC点:取1个50mL塑料具塞离心管,加入100μL 20 ng/mL的QC混合标准溶液,内标浓度为2.0ng/mL。除不加样品外,其余步骤同1.1衍生,1.2项净化及溶解的操作。使最终定容浓度为2.0ng/mL。[b]1.5 [/b]加标试验称取2.5g(粉体样品减半,精确至0.01g)样品,置于50 mL塑料具塞离心管中,加入40μL 50ng/mL标准溶液,其余步骤同1.1衍生,1.2项净化及溶解的操作。使最终定容浓度为2.0ng/mL。[b]1.6 [/b]仪器参数条件可参考GB/T 21311-2007。[b]1.7 [/b]定量计算 结果计算公式为 : [i]X[/i] = c • V / m式中:X---试样中被测组分残留量,单位为(µ g/kg);C---从标准工作曲线上得到的被测组分溶液浓度,机读数,单位为(µ g /L) V---样品溶液定容体积,单位为(mL);M---试样的质量,单位为(g);注:计算结果需要将空白值扣除。[b]1.8[/b] 方法检测限水产品及肉类基质:呋喃唑酮的代谢物3-氨基-2-唑烷基酮(AOZ): 0.2µ g/kg;呋喃它酮的代谢物5-甲基吗啉-3-氨基-2-唑烷基酮(AMOZ): 0.2µ g/kg;呋喃妥因的代谢物1-氨基-2-内酰脲(AHD) : 0.2µ g/kg;呋喃西林的代谢物氨基脲(SEM):0.5µ g/kg。基质干扰较大的粉类基质,方法检测限为: 呋喃唑酮的代谢物3-氨基-2-唑烷基酮(AOZ): 0.5µ g/kg;呋喃它酮的代谢物5-甲基吗啉-3-氨基-2-唑烷基酮(AMOZ): 0.5µ g/kg;呋喃妥因的代谢物1-氨基-2-内酰脲(AHD) : 0.5µ g/kg;呋喃西林的代谢物氨基脲(SEM):1.0µ g/kg。[b]1.9 [/b]质量控制[b]1.9.1[/b]每次试验做一个方法空白,方法空白0.1ng/mL, 仪器最多间隔20针进1针方法空白.[b]1.9.2 [/b]标准曲线的回归系数大于等于0.995.[b]1.9.3[/b] QC点的浓度为2.0 ng/mL, 回收率应在80%-120%,仪器最多间隔20针进1针QC.[b]1.9.4 [/b]样品加标试验的回收率在70%-130%。[b]1.9.5 [/b]超过MDL的样品,进行双样定量分析, 相对百分比差值小于20%。[b]1.9.6 [/b]每次实验的同类基质至少做一个平行样.[b]1.10 [/b]注意事项[b]1.10.1[/b]对于含油较多的样品,在净化氮气吹干后,用2mL乙腈溶解,加2mL用乙腈饱和的正己烷去油,弃去上层正己烷,氮气38℃吹干, 加入1.0mL [color=black]20%[/color]甲醇溶液,涡旋1min,过0.22µ m滤膜待进样。[b]1.10.2 [u]对于动物肌肉、内脏、鱼、虾和肠衣样品GB/T 21311-2007要求称取样品后加入10 mL 50%甲醇水溶液,振荡10 min后,以4000r/min离心5min,弃去液体,残留物加入10[/u][/b][u] [/u][b][u]mL 0.2mol/L盐酸溶液,之后步骤同1.1衍生,1.2项净化及溶解的操作。个人认为若样品测定值为阳性,经此步骤处理后结果会减小甚至变为阴性,建议可参考FDA的方法,省去(加入10 mL 50%甲醇水溶液)水洗步骤,更能客观反映样品真实含量。 温馨提示:夏天大家都喜欢吃小龙虾配冰镇啤酒,经检测发现虾类中硝基呋喃代谢物检出率非常高,希望大家最好不要经常吃,偶尔吃一次还是可以滴![/u][/b]
现在正在做农药代谢物的检测,不知道从那家公司能够买到农药代谢物的标准品。谢谢大家
请问各位大虾,硝基呋喃类代谢物的衍生物标准品为什么不可以直接用来做外标曲线的标准品?这样不就少了外标标准品也要衍生这步吗?
各位大侠,GB 29704-2013 食品安全国家标准 动物性食品中环丙氨嗪及代谢物三聚氰胺多残留的测定 超高效液相色谱-串联质谱法,有验证过的吗?SPE选用哪个牌子比较好?能达到1ppb的检测限呢?谢谢
请教各位,在哪里可以查到关于农药代谢物的限量标准?看过NY1500和GB2763,在前者里看到有关于克百威等少数几种农药,其残留限量中提到了它们的代谢物,其他农药则没有以前看到一个帖子说有些农药在检测时也要求检它们的代谢物,请问有没有相关的标准规定了那些农药要检代谢物?另外,国外很多农药都有代谢物的限量标准,这个标准制定的依据是什么?哪里可以得到比较全的这样的标准?先谢谢了~
请问各位老师,在GB/T 20769-2008中,有氟虫腈及其代谢物吗?GB23200.8里只有氟虫腈没有代谢物,虽说可以按这个方法或者NY/T761用ECD做,标准里没有心里没底。

最近实验室在准备扩项,扩的121这个标准,刚好有咪鲜胺这个项目,但是我看到论坛里有老师发了咪鲜胺不能用121去检测,因为少了一个三氯苯酚,而1456因为前处理太麻烦不考虑扩这个,我在网上有一个新的[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url]标准:SN/T 5444-2022 出口植物源食品中咪鲜胺及其代谢产物的测定 [url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]-质谱/质谱法,这个标准用的是Q法,并且所有的代谢物都有做到,但是就是不知道明年的国抽会不会用这个方法,还是说会继续用121或者1456,想问问各位老师有什么看法?还有个问题就是国抽和2763都提到了咪鲜胺锰盐,但是现今的标准却没有咪鲜胺锰盐的离子参数,我们也买到了咪鲜胺锰盐的标物,所以想问下各位老师有没有它的离子对和碰撞电压?[img=,690,101]https://ng1.17img.cn/bbsfiles/images/2022/11/202211291022467341_8503_4026362_3.png!w690x101.jpg[/img]
咪鲜胺是英国Boots公司现在的德国艾格福公司于1974年合成,1977年推向市场的一种咪唑类光谱杀菌剂,它通过抑制甾醇的生物合成而发挥作用,对子囊菌和半知菌特别有效,主要作用于水果,蔬菜采收后的保鲜剂、防腐剂、以及作为植物叶面喷施用于放置水稻恶苗病、胡麻叶斑病等病害。在GB2763-2016中4.323咪鲜胺和咪鲜胺锰盐 残留物:咪鲜胺及其2,4,6-三氯苯酚。规定的检验方法NY/T1456-2007 水果中咪鲜残留的测定气象色谱法,用GC-ECD检测。这个方法中是在210-240度条件下用吡啶盐酸盐将咪鲜胺及其代谢产物水解成2,4,6-三氯苯酚,需水解一小时,再萃取提取,上机检验,提取过程,水解过程,十分繁琐,而且吡啶盐酸盐有一定毒性,在大批量做样的时候,该方法占用的人力物力太多。下面研究了一下咪鲜胺在喷洒在农产品上一段时间后,会产生2种初级代谢物N-丙基-N-脲(BTS44595)和N-醛基-N-丙基-N-脲(BTS44596),再经过一段时间这两种初级代谢物最终转化为次级代谢物2,4,6-三氯苯酚(BTS45486),所以说要检农产品的咪鲜胺,在农作物身上可能存在咪鲜胺、BTS44595、BTS44596,和BTS45486这四种形态,所以按在NY/T1456-2007中的检验方法来做的话是十分合适和全面的,就是麻烦点,转化的过程不好控制。那么还有其他方法检测么,有[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质[/color][/url]法检测咪鲜胺主体23200.8,[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url]法检测20769咪鲜胺主体的方法,几种代谢物的检验方法暂时没有查询到,但是有用液相做咪鲜胺主体和代谢物的文献,也有用[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url]做咪鲜胺主体和2,4,6-三氯苯酚的,提取方法有很大不同,大概是代谢物要在酸性条件下提取效果才好,我觉得随着检验技术的进步,咪鲜胺也会出现克百威三羟基克百威、涕灭威、涕灭威砜,涕灭威亚砜这些代谢物一起检测的方法。大家对咪鲜胺检测有好的方法可以补充分享一下。







 我要推广仪器
我要推广仪器
 下载APP
下载APP