用Quchers方法做蔬菜水果中的农残,称取15g样品,加15ml 1%的醋酸乙腈提取,加盐包净化处理,离心,取2ml上清液氮吹至近干,用1ml正己烷溶解过滤上机,结果六六六,狄氏剂,腐霉利,氯菊酯的回收率都在85-95之间,而氯氟氰菊酯和氯氰菊酯的回收率是272和275,这是为什么呢?造成回收率高的原因是什么?
用[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质[/color][/url]检测果蔬中菊酯类(氯氰菊酯、氟氯氰菊酯、氯氟氰菊酯,氰戊菊酯,溴氰菊酯)农药,刚开始进标准混合溶液时,它们都有出峰的,可是再进了十几个果蔬样品后,再重新进标准溶液后,质控的标准溶液就不出峰了,也就是说后面出峰的菊酯类走着走着就没有响应了,前面出现出峰的比如有机磷等出峰还是正常的,然后重新老化了VF-1701MS色谱柱,后面出峰的菊酯类标准溶液还是没有出峰?现在要如何解决?
三聚氰胺检测中,脂肪含量高用 三氯乙酸溶液饱和的正己烷 怎么配置和添加操作
已知原样品(平菇)中未检出氰戊菊酯,加标样品中氰戊菊酯含量为0.12ppm。测定含量为0.206,加标回收率高达170%,下面是我处理样品的过程,请教各位大神看是哪里出了问题:1.前处理:(1)称取平菇样品4个,每个各取25.00g,编号为1、2、3、4,1号为原样品,不加任何东西,2、3、4号为加标平行样品,每个样品加入一定量氰戊菊酯标准样品,氰戊菊酯含量为0.12ppm(2)4个样品均加入50ml乙腈,振荡30min(3)4个样品均经过抽滤,得抽滤液,将抽滤液加入到100ml比色管中(比色管中提前加入5-7g氯化钠),振荡,盐析,静置1h得上清液(4)取10ml上清液至三角瓶中,旋转蒸干,再加入10ml正己烷溶解(5)正己烷:丙酮=9:1的混合溶液活化弗罗里硅小柱,活化完成加入正己烷溶解溶液,开始接样,用正己烷:丙酮=9:1的混合溶液淋洗小柱,得样品溶液(6)将上述所得样品溶液旋转蒸发,再加入5ml正己烷,过滤膜,得上机样品。2.上机:(1)标准样品:用原样品配制基质标准样品0.1ppm(2)前处理得到3个加标平行样品3,数据处理:(1)基质标样浓度0.1ppm(2)3个加标平行样品浓度分别为0.203,0.210,0.205以上就是全过程,但是回收率特别高,不知道什么原因,求各位老师指点!!!
各位老师:氰戊菊酯和S-氰戊菊酯在出报告时,两个是不是不能分开出报告,只能出成氰戊菊酯和S-氰戊菊酯的总和?类似的问题还有氯氟氰菊酯和高效氯氟氰菊酯
大家好,用HP-5MS的色谱柱、GCMS来检测氯氰菊酯与氟氯氰菊酯,由于氯氰菊酯-4与氟氰戊菊酯-1无法分开,但是它们的检测的碎片不一样,氯氰菊酯的碎片为163、165、181;而氟氰戊菊酯的碎片为199、157、451,是否一定要将这两种物质分成两组分别进行检测,有人将它们两个物质放在同一组中进行检测的吗?如果放在一起检测,在定量上会有干扰吗?
高效氯氰菊酯水悬浮剂的定量分析方法孙文琴 闻柳 摘 要:采用高效液相色谱法,在正相硅胶柱上,以正己烷+乙晴,醋酸乙酯的混合物为流动相,于254nm波长下,对高效氯氰菊酯进行分离与测定,该法简便、快速,准确。
问题1.氰戊菊酯重复性差,一般如何解决? 2.氰戊菊酯加标回收率超过150%,为什么?前处理方法:正己烷:丙酮=1:1超声提取,浓缩,过佛罗里硅土,定容。仪器方法:进样口220,程序升温:150保持3分钟,20/min,升到210保持3分钟,10/min,升到260,1/min 升到270,保持5分钟
想请问各位大侠:5.7%的氟氯氰菊酯农药检测方法?用液相色谱可以做吗?具体检测条件是怎样?流动相用正己烷和乙腈吗?
二氯甲烷,丙酮,正己烷,四氢弗喃这些试剂都是实验室长期使用的,危害程度如何?操作需要注意哪些
要用液相测土壤中农残含量。有机磷类:辛硫磷(提取剂:丙酮;定容:正己烷)菊酯类:功夫(高效氯氟氰菊酯)(提取剂:石油醚+丙酮;定容:丙酮)流动相怎么选择?原因?波长怎么选择?根据?其他的条件都怎么确定?新手,不懂,望指教
最近用的方法检测菊酯类农药氟氰戊菊酯、溴氰菊酯及顺式氰戊菊酯回收率超低的,请问有什么好的方法做比较好
各位老师好,请问溴氰菊酯标液证书上说标液用正己烷稀释,但是国标书上做这个项目的流动相又是环己烷,我可以用环己烷稀释溴氰菊酯的标液吗?
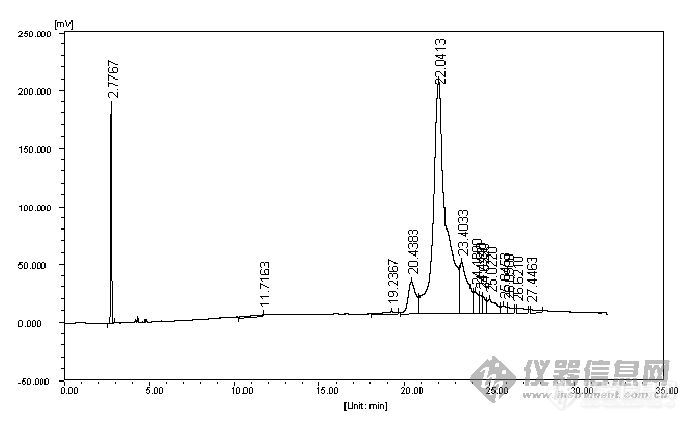
Fuli9790Ⅱ[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url]色谱条件如下:柱温200℃,检测器280℃,进样口280℃程序升温:200℃保持2min,5℃/min升到250℃,保持20min进样量:2μL氰戊菊酯用正己烷配制,浓度为0.64mg/L氰戊菊酯出峰不正常,请问专家和老师怎么解决啊???[img]http://ng1.17img.cn/bbsfiles/images/2008/01/200801231300_77704_1488514_3.jpg[/img]
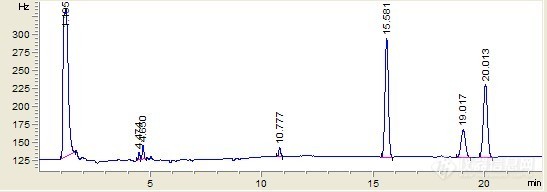
仪器是安捷伦7890A,色谱柱为HP-5,方法180 °C 用于 1 分钟 然后 7 °C/min 到 240 °C 用于 10 分钟 然后 10 °C/min 到 270 °C 用于 0 分钟做出来的几个菊酯单标都出了3个峰。。。图1:氰戊菊酯 0.1ppmhttp://ng1.17img.cn/bbsfiles/images/2010/11/201011111519_258721_2192483_3.jpg图2:溴氰菊酯 0.1ppmhttp://ng1.17img.cn/bbsfiles/images/2010/11/201011111520_258722_2192483_3.jpg图3:氟氰戊菊酯 0.1ppmhttp://ng1.17img.cn/bbsfiles/images/2010/11/201011111520_258724_2192483_3.jpg图4:顺式氰戊菊酯 0.1ppmhttp://ng1.17img.cn/bbsfiles/images/2010/11/201011111522_258726_2192483_3.jpg求达人解惑~~~另:氰戊菊酯与顺式氰戊菊酯出峰时间完全相同,可为何3个峰的峰形高低不一样,峰面积也差别很多?
问大家一个很菜鸟的问题,高效氟氯氰菊酯 高效氯氟氰菊酯 三氟氯氰菊酯 氟氯氰菊酯 是同一种东西吗?我自己搞糊涂了,有的说高效氟氯氰菊酯 三氟氯氰菊酯 氟氯氰菊酯是同一种东西,也叫功夫菊酯,那高效氟氯氰菊酯 高效氯氟氰菊酯是同一种东西吗?
求助:样品(茶叶)中含有氟氰戊菊酯和氯氰菊酯,怎样分离?平时这两种标样是分为两组的,但现在问题是样品中的这两种农药在GC条件下实现分离?不考虑MS。
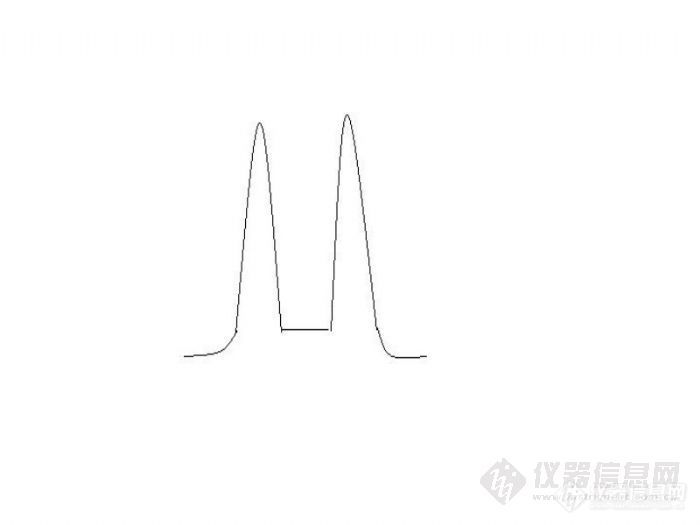
氯氰菊酯第四个峰和氟氰戊菊酯(氟氰菊酯)第一个峰无法分离,色谱柱CP-Sil 8 CB(0.25*0.25*30),使用zb-1701p和CP-Sil 19 CB(0.25*0.25*30),峰能分开,但各个异构体峰分离较差http://ng1.17img.cn/bbsfiles/images/2011/07/201107011433_302579_1830074_3.jpg
样品中加有氟氰戊菊酯,当天做与室温下放两天做,回收率高了,是前处理的问题,还是农药挥发的问题。
如题 因为没在实验室,请做过试验的人指点指点.:若样品中脂肪含量较高,可以用三氯乙酸溶液饱和的正己烷 液-液分配除脂后再用SPE柱净化
我用的是Agilent 7890A,ECD检测,HP-5,30m×0.32mm×0.25um的柱子,用了很多程序升温方法都无法分离氯氰菊酯和氟氰菊酯(即氟氰戊菊酯)。[em0702] 请问版上的各位兄弟有没有出现过该问题,即氯氰菊酯四个峰中的最后一个峰与氟氰菊酯的第一个峰重合(经单标进样后确认的),各位是如何通过程序升温或其他方法将此两种农药分开的?能否赐教,不胜感激[em0706]
请问氰戊菊酯、溴氰菊酯、三氟氯氰菊酯用液质检测应该选择哪些离子对?我的液质型号是安捷伦液相1200-AB质谱3200Q trap,选择上述菊酯的分子量为母离子,扫Q1的时候响应不高,打碎后MS2也找不到子离子,跪求高手指教!
第一组,氯氰菊酯和高效氯氰菊酯,GB2300.8种查到氯氰菊酯的离子为181,152,180但是没找到高效氯氰菊酯,找到了一个顺式-氯氰菊酯离子为163,181,165.这个顺式-氯氰菊酯是高效氯氰菊酯么,但是英文名对不上啊,氯氰菊酯的四个峰是不是包括了高效的,求高手解答。第二组,氟氯氰菊酯和高效氟氯氰菊酯,23200.8只查到了,高效的181,197,141氟氯氰菊酯没有找到,氟氯氰菊酯又名三氟氯氰菊酯,能不能用高效的离子呢?
求助,有做农残[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url]的老师吗?我们扩GB23200.121氟氰戊菊酯、氰戊菊酯、联苯菊酯、溴氰菊酯这四个菊酯,但是优化不出来。
氯氟氰菊酯跟高效氯氟氰菊酯标准品都有,计算的时候怎么计算,是把每一种的含量算出来再加和还是只算一种替代两种就可以?
请问大家,如果我想用氯氟氰菊酯的标准品(两个峰差不多大)去定量样品中的高效氯氟氰菊酯(一个峰大一个峰小)),该如何操作哇?是把两个峰加合在一起定量吗?
各位老师好,想问一下氟氰戊菊酯冷冻后容易降解吗?为什么我们第一次检测很明显,而且浓度很高,第二次复检就什么也没有,昨天又有样品检出今天复检又没了,求助一下老师们具体是什么原因?[img]https://ng1.17img.cn/bbsfiles/images/2022/05/202205261636465530_7381_5546384_3.png[/img][img]https://ng1.17img.cn/bbsfiles/images/2022/05/202205261636465478_5720_5546384_3.png[/img]
国抽中很多这种的,例如顺势氰戊菊酯+氰戊菊酯,氯氟氢菊酯+高效氯氟氢菊酯。例如氯氟氢菊酯出两个峰,后面的峰跟高效氯氟氢菊酯的峰是重合的,各位大佬,在做这种农残的时候配置标品需要分别配置两种物质的标品吗,在工作站中再分别计算总和?
[sub]?正己烷萃取血浆药物时,在药物的定量离子对那里刚好出现一个峰,和药物的峰完全重叠,这可能是什么原因,试过了正己烷空气吹干后,甲醇复溶,就是有峰,而且同样的步骤,血浆基质加标,发现峰的面积变成正己烷和药物的峰面积之和,正己烷是分析纯的,[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质联用[/color][/url],或许除了正己烷,还可以用什么进行萃取,提高响应值,定量限需要做到 0.03ng/ml 求助各位大神了[/sub]
茶叶氯氟氰菊酯和高效氯氟氰菊酯2763中要求按照SN/T1117检测,但是SN/T1117-2008已经废止,并且没有替代标准。请问茶叶氯氟氰菊酯和高效氯氟氰菊酯可以用什么标准检测?是否还可以用2763判定?







 我要推广仪器
我要推广仪器
 下载APP
下载APP