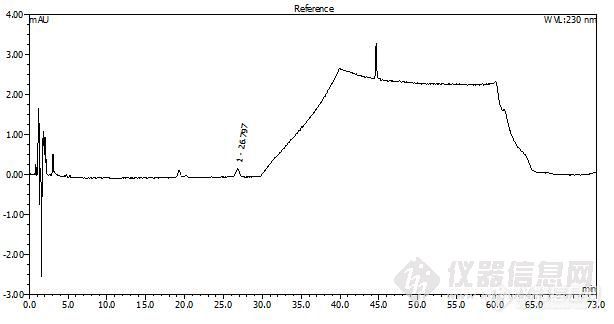
[size=3][font=宋体]各位有做这个项目的么? 按照药典相关规定,摸索梯度洗脱条件得到下图。但是按照药典规定,主成分色谱峰的峰高为满量程的20%(见下文红色标示)。可是在本色谱条件下,梯度系统峰的变化比苯甲酸雌二醇的主峰峰高大多了,如果按照药典规定出图,30min以后就看不到基线了。“【[/font][font=黑体]检查[/font][font=宋体]】[/font][font=Times New Roman] [/font][font=黑体]有关物质[/font][font=Times New Roman] [/font][font=宋体]用内容量移液管精密量取本品适量(约相当于苯甲酸雌二醇[/font][font=Times New Roman]2mg[/font][font=宋体]),置[/font][font=Times New Roman]100ml[/font][font=宋体]量瓶中,加无水乙醇适量,充分振摇,待溶液澄清后,用无水乙醇稀释至刻度,摇匀,作为供试品溶液。精密量取[/font][font=Times New Roman]1ml[/font][font=宋体],置[/font][font=Times New Roman]100ml[/font][font=宋体]量瓶中,用无水乙醇稀释至刻度,摇匀,作为对照溶液。照苯甲酸雌二醇有关物质项下的色谱条件,量取对照溶液[/font][font=Times New Roman]20μl[/font][font=宋体]注入液相色谱仪,调节检测灵敏度,[color=#fe2419]使主成分色谱峰的峰高约为满量程的[/color][/font][color=#fe2419][font=Times New Roman]20%[/font][/color][font=宋体][color=#fe2419]。[/color]再精密量取供试品溶液与对照溶液各[/font][font=Times New Roman]20µ l[/font][font=宋体],分别注入液相色谱仪,记录色谱图。供试品溶液的色谱图中如有杂质峰,单一杂质峰面积不得大于对照溶液主峰面积([/font][font=Times New Roman]1.0%[/font][font=宋体]),各杂质峰面积的和不得大于对照溶液主峰面积的[/font][font=Times New Roman]1.5[/font][font=宋体]倍([/font][font=Times New Roman]1.5%[/font][font=宋体])(供试品溶液中任何小于对照溶液主峰面积[/font][font=Times New Roman]0.01[/font][font=宋体]倍的色谱峰可忽略不计)。”[/font][/size][img=middle]http://ng1.17img.cn/bbsfiles/images/2010/02/201002071025_200720_1632027_3.jpg[/img]
同位素内标UP[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url]/MS法测定婴儿配方奶粉中性激素苯甲酸雌二醇残留 摘要:建立测定婴儿配方奶粉中性激素苯甲酸雌二醇的UP[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url]/MS法。样品前处理采用酶解、甲醇提取,固相萃取柱净化。本方法回收率在76.5-86.3%范围内,相对标准偏差9.7-15.2%。原文在资料中心。[url]http://www.instrument.com.cn/download/[/url]
[table=100%][tr][td=2,1,650][font=宋体][/font][align=center][b][size=5]产品概要[/size][/b][/align][size=5] 雌二醇[b](β-Estradiol)[/b]是应用广泛的甾类同化激素,被大量用于促进反刍动物的生长。但由于雌二醇存在明显的致癌性,欧美等国家已相继禁止或严格禁止使用。我国农业部第235号文件中规定,动物源性食品中雌二醇的残留限量为不得检出。由于[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url](GC)分析法样本前处理及测定操作繁琐且费用高,使推广应用受到限制。[/size][align=center][b][size=5]产品用途[/size][/b][/align][size=5] 可定性、定量检测动物组织(肌肉、肝脏、虾、鱼等)、尿液、饲料、奶粉等样本中雌二醇的残留量。[/size][align=center][b][size=5]产品性能参数[/size][/b][/align][size=5] [/size][table][tr][td][size=5]产品规格[/size][/td][td][size=5]96T[/size][/td][/tr][tr][td][size=5]适用范围[/size][/td][td][size=5]动物组织(肌肉、肝脏、虾、鱼等)、尿液和饲料,奶粉等[/size][/td][/tr][tr][td][size=5]检测时间[/size][/td][td][size=5]1.5个小时[/size][/td][/tr][tr][td][size=5]检测下限[/size][/td][td][size=5]鸡肉/肝、猪肉/肝、虾、鱼 :5ppb饲料 :80ppb尿液 :2.5ppb[/size][/td][/tr][tr][td][size=5]灵敏度[/size][/td][td][size=5]0.1ppb[/size][/td][/tr][tr][td][size=5]前处理流程[/size][/td][td][size=5]组织:均质、振荡离心、吹干、溶解、反萃取、振荡离心、吹干、复溶[/size][/td][/tr][tr][td][size=5]交叉反应率[/size][/td][td][size=5]雌 二 醇 :100%雌二醇-3甲醚:30%炔 雌 醇 :7%雌酚酮 :小于0.1%苯甲酸雌二醇 :小于0.1%雌 三 醇 :小于0.1%[/size][/td][/tr][tr][td][size=5]结果判定[/size][/td][td][size=5]定性:根据样本孔颜色与标准品孔颜色进行比对定量:运用酶标仪进行计算[/size][/td][/tr][/table][/td][/tr][tr][td=2,1][size=5][i][b]无锡康纳生物科技有限公司 0510-85191271,13771068995,杨小姐[/b][/i][/size][/td][/tr][/table]
做液相苯甲酸时,样品中杂质太多怎么处理?
测定精对苯二甲酸(PTA)中的主要杂质对羧基苯甲醛(4-CBA)和对甲基苯甲酸(p-TOL)高效毛细管电泳仪法和高效液相色谱法,在测定精对苯二甲酸(PTA)中的主要杂质对羧基苯甲醛(4-CBA)和对甲基苯甲酸(p-TOL)时,哪种方法更好?听客户提到国标有用极普仪测试的,请问有没有对这个比较了解的。谢谢
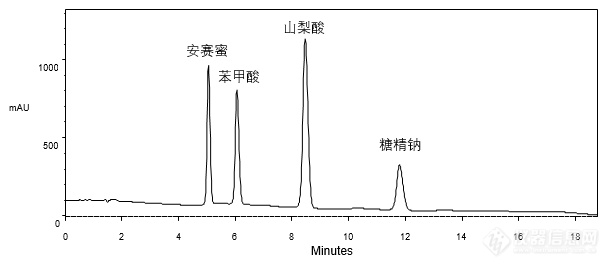
[align=center][b]GB 5009.28-2016食品安全国家标准 食品中苯甲酸、山梨酸和糖精钠的测定[/b][/align][align=center][b] ——标准品与乳品实际样品的分析[/b][/align][align=center][/align][align=left]本实验按照《GB5009.28-2016 食品安全国家标准食品中苯甲酸、山梨酸和糖精钠的测定》方法,分别对安赛蜜、苯甲酸、山梨酸、糖精钠的标准品混合溶液及加标乳品样品进行了分析。首先,使用CAPCELL PAK C[sub]18[/sub] MG S5 4.6 mm i.d. × 150mm色谱柱,对标准品混合溶液进行分析,如图1,安赛蜜、苯甲酸、山梨酸、糖精钠标准品均得到了良好的分析结果。[/align][align=left][/align][align=center][img=,611,268]http://ng1.17img.cn/bbsfiles/images/2018/03/201803221532276656_9890_2222981_3.png!w611x268.jpg[/img][/align][align=center]图1 标准品混合溶液分析色谱图[/align][img=,400,200]http://ng1.17img.cn/bbsfiles/images/2018/03/201803221532280132_6863_2222981_3.png!w400x200.jpg[/img][align=left][/align][align=left]其次,对乳品加标样品进行分析,如图2,糖精钠(Rt 12 min)与其后杂质峰之间未能取得基线分离,分离度仅为1.02。[/align][align=left][/align][align=center][img=,668,335]http://ng1.17img.cn/bbsfiles/images/2018/03/201803221533054905_2223_2222981_3.png!w668x335.jpg[/img][/align][align=center]图2 加标乳品样品分析色谱图[/align][align=left][img=,406,203]http://ng1.17img.cn/bbsfiles/images/2018/03/201803221533317202_2333_2222981_3.png!w406x203.jpg[/img][/align][align=left][/align][align=left]为改善糖精钠与杂质间的分离,在国标方法基础上,将流动相由[b]乙酸铵 / 甲醇 = 95 / 5[/b]调整为[b][b]乙酸铵 / 甲醇[/b][color=red]([/color][color=red]2 mmol/L [/color][color=red]甲酸)[/color]= 92 / 8[/b],再次对混合标准溶液和加标样品进行分析,结果如图3所示。[/align][align=left][/align][align=center][img=,690,545]http://ng1.17img.cn/bbsfiles/images/2018/03/201803221534141056_4073_2222981_3.png!w690x545.jpg[/img][/align][align=center]图3 混标与加标乳品样品分析色谱图[/align][align=left][img=,464,171]http://ng1.17img.cn/bbsfiles/images/2018/03/201803221535548985_7176_2222981_3.png!w464x171.jpg[/img][/align][align=left][/align][align=left]如图3,在酸性条件下,出峰顺序发生了变化,安赛蜜保留时间略有缩短,糖精钠保留时间明显缩短,由12 min缩短至8 min,苯甲酸和山梨酸保留时间分别延长至2 min和6 min;在分离度方面,糖精钠与苯甲酸之间分离度为2.79,苯甲酸与峰后杂质间分离度为2.04,所有色谱峰之间都达到了基线分离。[/align][align=left][/align][align=left]为使客户有更多选择,实验室又在国标原方法条件下继续筛选色谱柱,最终使用SUPERIOREX ODS S5 4.6 mm i.d. × 250 mm色谱柱时,仅微调有机相比例即可实现加标乳品样品的良好分析结果。如图4,杂质峰与糖精钠之间分离度达到2.48,达到基线分离要求。[/align][align=left][/align][align=center][img=,580,332]http://ng1.17img.cn/bbsfiles/images/2018/03/201803221537130173_1058_2222981_3.png!w580x332.jpg[/img][/align][align=center]图4 加标乳品样品分析色谱图[/align][align=left]*注:峰上标所示数字由下至上依次为分离度与不对称因子。[/align][align=left][img=,326,177]http://ng1.17img.cn/bbsfiles/images/2018/03/201803221537540634_9437_2222981_3.png!w326x177.jpg[/img][/align][align=left][/align][align=left]综上所述,按照国标《GB 5009.28-2016 食品安全国家标准食品中苯甲酸、山梨酸和糖精钠的测定》方法进行分析,使用CAPCELL PAK C[sub]18 [/sub]MG色谱柱对标准品混合溶液能得到良好分析结果,但在对加标乳品样品进行分析时,糖精钠与样品中的杂质未能实现基线分离,通过在流动相中添加甲酸可实现安赛蜜、糖精钠、苯甲酸、山梨酸及杂质的基线分离;另一方面,使用SUPERIOREX ODS色谱柱,在原条件基础上微调即可实现乳品中安赛蜜、苯甲酸、山梨酸、糖精钠及杂质间的良好分离。[/align]
[align=center][b]苯甲酸山梨酸标准溶液的配置方法探讨[/b][/align]苯甲酸、山梨酸是常用的食品添加剂,也是液相色谱经典的检测项目。但是苯甲酸山梨酸不溶于水,溶于甲醇,所以苯甲酸、山梨酸的标准溶液如何配置也是目前的难点之一!根据 GB5009.28—2016苯甲酸、山梨酸标准溶液的配置方法有以下两种:1、 确称取苯甲酸钠、山梨酸钾,用水溶解并分别定容。2、 当使用苯甲酸和山梨酸标准品时,需要用甲醇溶解并定容。根据GB/T5009.29—2003苯甲酸、山梨酸标准溶液的配置方法为:3、 称取苯甲酸、山梨酸,加入碳酸氢钠溶液5毫升,加热溶解,随后定容。 总结这几种方法,因为标准品的溶解介质不一样,与实际样品的基质不一样,样品的检测结果还是有较大区别的。因为标准品的问题,有的能力验证甚至难以通过。我想和大家请教一下,究竟哪种标准品溶液配制更加合理。
[color=#454545]液相色谱测定苯甲酸时,配制苯甲酸标液,标准品是苯甲酸固体,可是苯甲酸在水中几乎不溶解,要怎么配制苯甲酸标液,又能不产生误差?[/color]
对羟基苯甲酸甲乙丙酯的标准溶液有很多杂峰正常吗?我感觉理论上应该是只有三个大的色谱峰,结果出了十多个[img]https://ng1.17img.cn/bbsfiles/images/2023/10/202310191022364344_1417_5979722_3.png[/img]
我们是用水杨酸捕捉羟基自由基,以前的做法是用C18的色谱柱,5%的乙醇做流动相,流速是0.3,把标准样用35%乙醇稀释400倍进样,以前的标准样是可以检测出两个峰的(水杨酸和2,5-二羟基苯甲酸),但后来柱子坏了换了一根色谱柱,也是C18的,但不同型号的,用回原来的方法就无法出峰,只有一个溶剂峰,但两种主要物质出不来,后来把流动相换成10%的乙醇、30mMol醋酸钠醋酸缓冲液(ph4.9)都试过还是无法出峰。后来老师让我们用样品原液不稀释进样,峰是出来了,但峰高度很低而且面积也不大,而且很多杂质峰,峰型也很奇怪,重点是水杨酸以及2.5二羟基苯甲酸的保留时间几乎一样,无法分离。以前是标准样稀释400倍就能出峰的,现在为什么用原液峰面积也这么小?还有保留时间一样应该怎么处理?很急啊,谢谢。
有高手用液谱做过食品中的对羟基苯甲酸酯的测定吗?我们现在做标准曲线,发现4个标准品的峰出峰不是很好,1,4个峰分得很开,各有各的时间段。2,每个峰分得不是太清晰,旁边都有杂峰3,要出第三个峰或者第四峰前,基线下降有高手做过的请指教!国标法是用气相的,但山东省有个标准是用液相,二极管检测器(DB37/T1100-2008),因为有甲乙丙丁四酯,这个标准上图谱是有的,但没指明各个是哪个峰。我怀疑,DB这个标准上标准溶液没指明有什么溶剂配制,我们就用水配制,但样品是用甲醇定容取上清液的,有做过的高手请说一下经验!
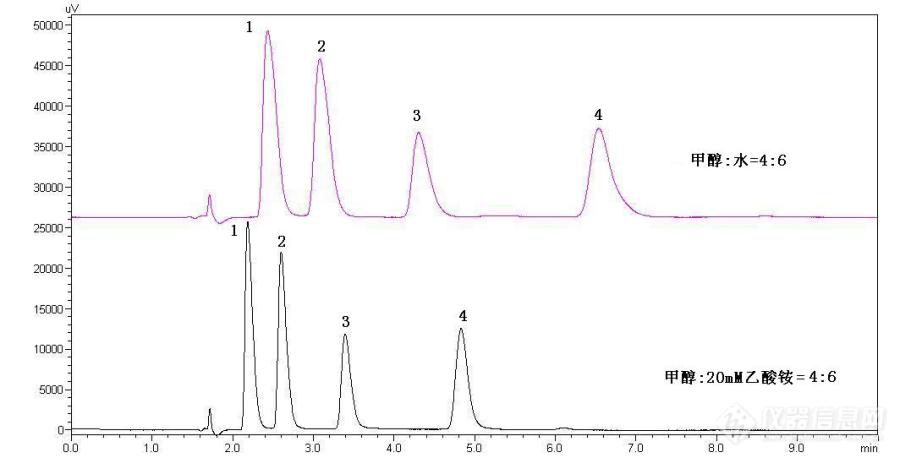
食品中对羟基苯甲酸酯类液相与[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法的测定方法比较摘要:本文详细讲述了液相色谱与[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法测定食品中对羟基苯甲酸酯类各自的优缺点,GB 5009.31-2016只收录了[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法测定标准的测定方法。关键词:对羟基苯甲酸酯类;液相色谱;[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url];优缺点;GB 5009.31-2016引言[font=黑体][size=18px][b] [/b][/size][/font][font=宋体][size=12px]食品中常见的对羟基苯甲酸酯类又称为对羟基安息香酸酯或尼泊尔金酯,包括对羟基苯甲酸甲酯、乙酯、丙酯、丁酯,是一类新一代高效低毒消毒杀菌防腐剂,它的抗菌能力、pH应用范围及用量比苯甲酸和山梨酸及其盐类广(见表1),且使用安全,经济方便,对人体刺激较小。在国外,已被广泛用于食品、饮料、化妆品和医药等方面。作为食品防腐剂,它可用于饮料、果蔬加工品、海产加工品、禽畜加工品、调味品、啤酒、米酒等加工品中,还可用于水果、蔬菜和海产品的防腐保鲜。它不但可完全替代苯甲酸钠和山梨酸钾,其使用范围比苯甲酸钠和山梨酸钾更广。在国外,已被广泛用于食品、饮料、化妆品和医药等方面。在日本,对羟基苯甲酸酯和山梨酸是主要的防腐剂产品。而我国,对羟基苯甲酸酯类防腐剂的用量也在逐年增加,成为防腐剂的第二个主要产品。[/size][/font][align=center]表1 GB2760-2014对羟基苯甲酸酯类限量要求[/align][img]https://ng1.17img.cn/bbsfiles/images/2020/08/202008192125459013_3455_2166779_3.png[/img]实验部分讨论液相色谱1.前处理及色谱分析条件称取5g(精确至0.01g)试样于50ml比色管中,加入15mL95%乙醇,混匀,超声波清洗器提取10min,冷却至室温后用95%乙醇定容至50mL刻度线,摇匀。静置分层,取上清液经0.22um微孔滤膜过滤后待测。色谱柱:C18柱,150×4.6 mm(i.d),5 μm,或性能相当者;流动相:甲醇(B)+20 mmol/L乙酸铵溶液(A) = 40+60(体积比);洗脱梯度见表2:[font=黑体]表2. 对羟基苯甲酸酯类的洗脱程序[/font][table][tr][td]时间/min[/td][td]2.00[/td][td]4.00[/td][td]12.00[/td][td]12.01[/td][td]15.00[/td][/tr][tr][td]B%[/td][td]40[/td][td]60[/td][td]60[/td][td]40[/td][td]stop[/td][/tr][/table]流速:1 mL/min;柱温:35 ℃;进样体积:10 μL;检测波长扫描范围:210 nm—390 nm,定量波长256 nm。由于对羟基苯甲酸酯在[font=times new roman]pH4~8的[/font]范围内稳定存在且有很好的抗菌效果,但水溶性较低,易溶于乙醇,而乙醇的毒性较甲醇低,所以采用乙醇做为标准使用液和样品提取液。大部分添加对羟基苯甲酸酯类的食品都具有含水性高、不易长期保存的特点,因此在取样时选择被测物稳定保存的状态,采用浸泡过夜、超声及振荡提取的方式,能达到较好的提取效果。2、色谱条件的选择及优化2.1 检测波长的选择以浓度为[font=times new roman]0.02 mg/ml的标准使用液依次在高效液相色谱—二极管阵列检测器190 nm-410 nm波长[/font]范围进行扫描,以确定被[font=times new roman]测物质的最大吸收波长,对羟基苯甲酸酯类的最大吸收波长均十分相似,在256nm处有紫外最大吸收峰,这是与自身具有苯环和羰基结构所决定,实验选择256nm为检测波长,能有效满足四种物质的分析检测。[/font] [size=12px] 2.2 分析条件的选择[/size]分别使用水和20mM乙酸铵做流动相在相同色谱条件下进行分析(见图2),UPLC(超高速液相)在5分钟内完全分离且重现性好,出峰顺序依次为对羟基苯甲酸甲酯、乙酯、丙酯、丁酯。由图可以看出在峰形和分析时间上利用缓冲盐做流动相要优于纯水,特别是面对食品样品的复杂性,选择20mM乙酸铵和甲醇作为流动相能有效排除基质干扰。[img]https://ng1.17img.cn/bbsfiles/images/2020/08/202008192125462304_3941_2166779_3.jpg[/img]图2 混合标准样品色谱图1.对羟基苯甲酸甲酯[font=arial][color=black];[/color][/font][font=times new roman]2.[/font]对羟基苯甲酸乙酯[font=arial][color=black];[/color][/font][font=times new roman]3.[/font]对羟基苯甲酸丙酯[font=arial][color=black];[/color][/font][font=times new roman]4.[/font]对羟基苯甲酸丁酯实验选择C18色谱柱进行分离,是根据尼泊尔金酯类的物理化学性质,在C18柱上具有良好的保留,当提高有机相甲醇的比例时,能快速得到洗脱。选择20m M乙酸铵作为流动相,能较好的平衡食品复杂基质的p H值,有效避免杂质干扰。实验尝试使用了日本岛津HPLC-20A,美国Grance Alltima 4.6mm×150mm C18色谱柱与日本岛津UPLC,shim-pack XR-ODS 3.0mm×75mm C18色谱柱对市售果蔬汁饮料、酱腌菜及酱油制品和糕点等所含的对羟基苯甲酸酯类进行比对分析,发现均能得到良好分离,且结果一致。在实验中发现,相同条件下,采用HPLC-20A,美国Grance Alltima 4.6mm×150mm C18色谱柱对酱腌菜类食品进行分析,在对羟基苯甲酸乙酯处会有干扰(见图3),通常情况下,改变流动相的比例能避免此类问题的发生,但若直接选用岛津UPLC,shim-pack XR-ODS 3.0 mm×75 mm C18色谱柱进行分析,则能有效避免(见图4)。考虑到两种C18柱的价格及维护费用,选择使用Grance Alltima 4.6mm×150mm C18进行大批量的实验分析,在保证实验数据的准确可靠前提下,能有效降低成本。[img]https://ng1.17img.cn/bbsfiles/images/2020/08/202008192125466015_1144_2166779_3.jpg[/img] [img]https://ng1.17img.cn/bbsfiles/images/2020/08/202008192125467275_3703_2166779_3.jpg[/img]图3 酱腌菜中对羟基苯甲酸酯类色谱图(HPLC-20A)峰序同图2 图4 酱腌菜中对羟基苯甲酸酯类色谱图(UPLC)2.3 基质的干扰与条件优化 [size=16px] [/size][size=12px]实验选择糕点、果蔬汁饮料及酱油及酱腌菜类为基质,采用上述方法进行前处理,岛津HPLC-20A,Grance Alltima 4.6mm×150mm C18进行分析检测,发现果蔬汁及饮料等,基质简单,峰形对称(见图5);在进行酿造酱油基质的加标回收分析时发现,甲酯由于出峰时间较早,容易被杂质峰包埋,致使检测的检出限降低(见图6);酱腌菜类食品由于基质复杂,特别是在腌制过程中产生的不明物质较多且各有差异,在实验中稍有不慎,极容易与对羟基苯甲酸乙酯、丙酯产生干扰(见图7),因此在进行分析过程中应注意色谱条件的选择,改变流动相中甲醇的比例能有效避免杂质的干扰。[/size][img=,690,331]https://ng1.17img.cn/bbsfiles/images/2020/08/202008192131109762_6420_2166779_3.png!w690x331.jpg[/img][img=,690,308]https://ng1.17img.cn/bbsfiles/images/2020/08/202008192131386200_7631_2166779_3.png!w690x308.jpg[/img][img=,690,325]https://ng1.17img.cn/bbsfiles/images/2020/08/202008192131527267_1909_2166779_3.png!w690x325.jpg[/img][url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]国标GB5009.31-2016《食品安全国家标准 对羟基苯甲酸酯类的测定》检测范围为酱油、醋、饮料及果酱,采用GC酸化提取后进行分析检测,实验原理与本方法可互为验证和补充,但操作步骤复杂,耗时长,乙醚试剂消耗大,对检测人员伤害较大,且效率低,但基质处理的干净,不存在干扰而产生假阳性的情况。仪器条件:Agilent 7890 检测器:FID 进样量:1 μL 色谱柱:[color=black]HP-1(30m*320μm*0.25μm);HP-5(30m*320μm*0.25μm) ;DB-17 30m*320μm*0.25μm ; [/color][color=red]HP-5MS(30m*320μm*0.25μm)[/color][color=red] [/color]检测器温度:240[font=宋体]℃[/font] 进样温度:250[font=宋体]℃[/font]程序升温:100[font=宋体]℃[/font](1min) 20 [font=宋体]℃[/font]/min 160[font=宋体]℃[/font](3min) 15 [font=宋体]℃[/font]/min 250[font=宋体]℃[/font](3min)样品处理:取样5.0 g(±0.01 g)于50 mL塑料离心管中,加入1 mL盐酸(1:1)溶液酸化,再加入10 mL饱和氯化钠水溶液摇匀,分别每次30 mL乙醚提取三次,涡旋振荡4000 r/min离心,取上清液乙醚合并入250 mL分液漏斗中,先加入10 mL饱和氯化钠水溶液洗涤一次弃去水层,分别每次30 mL 1g/100mL的碳酸氢钠溶液洗涤洗三次 ,静置,弃去水层。过装有10 g无水硫酸钠的漏斗至鸡心瓶中,35[font=宋体]℃[/font]浓缩至干,用无水乙醇定容至2 mL上机测试。注释:[color=red](1)使用HP-5MS色谱柱主要是为了增加分离度,使得目标峰与分析纯乙醚中的干扰峰分离开。[/color](2)对羟基苯甲酸乙酯处有试剂干扰,来自乙醚。(3) 标准中使用125 mL的分液漏斗,实验室直接使用50mL的塑料离心管更易于提取。(4) 标准中用75 mL、50 mL、50 mL乙醚提取三次,实验室用离心管离心取上层乙醚层更方便,由于离心管容积只有50 mL,所以减少乙醚量,分别每次用20 mL乙醚提取三次。(5) 标准中用分液漏斗萃取,静置弃去水层,实验室用离心的方法分层,取上清液乙醚层。(6) 标准中在分液漏斗中加10 g无水硫酸钠于室温放置30 min脱水,实验室过装有10 g无水硫酸钠的漏斗脱水。色谱图分析比较:通过图8、图9([url=https://insevent.instrument.com.cn/t/Mp]气相[/url]法前处理)与图5~图7(液相法前处理)比较,[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]的图谱明显干净多了,而且不存在一点的基质干扰的现象。 [img]https://ng1.17img.cn/bbsfiles/images/2020/08/202008192125470058_269_2166779_3.png[/img] [img]https://ng1.17img.cn/bbsfiles/images/2020/08/202008192125470897_3417_2166779_3.png[/img]果酱食品基质加标(2ppm) b. 果酱类食品空白基质图8 果酱食品中对羟基苯甲酸酯类空白基质及基质加标(2ppm) [img]https://ng1.17img.cn/bbsfiles/images/2020/08/202008192125471786_7777_2166779_3.png[/img] [img]https://ng1.17img.cn/bbsfiles/images/2020/08/202008192125472549_3883_2166779_3.png[/img] a. [size=12px]果酱食品基质加标(2ppm) b. 果酱食品空白基质 [/size] [size=12px]图9 果酱食品中对羟基苯甲酸酯类空白基质及基质加标(2ppm) [/size]按国标GB5009.31-2016检测方法线性范围和测定低限:在1~500 μg/mL浓度范围内,以峰面积(y)与目标化合物浓度(x,μg/mL)绘制标准工作曲线。结果表明,在1.0~500 μg/mL质量浓度范围内,目标化合物良好线性关系,线性方程式、线性关系和测定低限(LOQ,S/N=10)见表2。表2 线性方程式、线性相关系数和定量限[table][tr][td][align=center]化合物[/align][/td][td][align=center]线性方程式[/align][/td][td][align=center]线性相关系数[/align][/td][td][align=center]定量限/(mg/kg)[/align][/td][/tr][tr][td][align=center]对羟基苯甲酸甲酯[/align][/td][td][align=center]Y=0.532847x-1.26878[/align][/td][td][align=center]0.99915[/align][/td][td][align=center]2.0[/align][/td][/tr][tr][td][align=center]对羟基苯甲酸乙酯[/align][/td][td][align=center]Y=0.563352x-1.13532[/align][/td][td][align=center]0.99972[/align][/td][td][align=center]2.0[/align][/td][/tr][tr][td][align=center]对羟基苯甲酸丙酯[/align][/td][td][align=center]Y=0.554134x-2.50160[/align][/td][td][align=center]0.99915[/align][/td][td][align=center]2.0[/align][/td][/tr][tr][td][align=center]对羟基苯甲酸丁酯[/align][/td][td][align=center]Y=0.530932x-4.29406[/align][/td][td][align=center]0.99971[/align][/td][td][align=center]2.0[/align][/td][/tr][/table] 回收率和精密度分别向空白蚝油中添加目标物,做空白添加回收试验,添加水平为2.0、5.0、50.0 mg/kg,各添加水平分别做6次平行试验。加标回收率为88.7%~108%,相对标准偏差(RSD)为1.3~5.2%,方法的精密度及回收率均满足定量测定的要求,试验结果见表3。[size=12px][font=times new roman] [img=,690,371]https://ng1.17img.cn/bbsfiles/images/2020/08/202008192133361979_6058_2166779_3.png!w690x371.jpg[/img][/font][/size] 总结: [font=宋体][size=12px]国标方法是用盐酸酸化样品,乙醚提取,浓缩后,用具有氢火焰离子化检测器的[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url]进行分离检测,外标法定量;耗时长、成本高、消耗大、毒性强、工作效率低等弱点,极大的限制了在食品检测过程中的广泛运用,优点是谱图干净,几乎没有基质干扰的现象。针对食品检测工作中样品量大、基质干扰多、成分复杂且易变质腐败等特点,在检测过程中也需要建立一种准确高效的检测方法运用于实际工作;液相法是一种快捷、准确、适用范围广的方法,以满足检测工作的需要,达到提高工作效率的目的。遇到有基质干扰,不合格的样品时改用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法进行准确定量检测。[/size][/font] [size=12px] [/size]
[b][font=宋体]问题描述:[/font][font=宋体]用安捷伦[/font]1260[font=宋体]检测苯甲酸山梨酸,标准溶液的保留时间与样品的保留时间不一致,样品的保留时间较标准品延后,该如何处理?[/font][font=宋体]解答:[/font][/b][font=宋体]理论上,色谱条件不变的前提下,样液中化合物峰保留时间应与标准溶液中化合物峰保留时间一致,这是液相色谱定性的前提与要求。受实际条件的制约,两者间的保留时间相对允许差为[/font]5%[font=宋体]。[/font][font=宋体]导致苯甲酸山梨酸标液的保留时间与样品的保留时间不一致的原因可能有:[/font][font=宋体]([/font]1[font=宋体])样液中杂质的干扰[/font][font=宋体]由于苯甲酸、山梨酸极性的原因,在国标[/font]GB 5009.28-2016[font=宋体]《食品安全国家标准[/font] [font=宋体]食品中苯甲酸、山梨酸和糖精钠的测定》中,使用[/font]C[sub]18[/sub][font=宋体]色谱柱对苯甲酸、山梨酸进行色谱分离时,采用了低有机相比例的流动相进行等度洗脱,即甲醇:乙酸铵([/font][i]V/V[/i][font=宋体])[/font]=5[font=宋体]:[/font]95[font=宋体]。低有机相比例的流动相使得样品中部分非极性化合物不能及时洗脱出色谱柱,进而影响苯甲酸、山梨酸在[/font]C[sub]18[/sub][font=宋体]色谱柱的保留时间。在实际的食品苯甲酸、山梨酸检测中,样液中杂质干扰是引起保留时间不一致的主要原因。解决的办法是:在色谱分离的后期增加一步强溶剂洗脱步骤,或延长色谱洗脱时间。[/font][font=宋体]([/font]2[font=宋体])流动相的不稳定[/font][font=宋体]在现代液相色谱中,二元及其以上的液相色谱仪是主流。二元或四元液相色谱仪可以通过比例阀将不同溶剂按不同比例在线混合而成所需的流动相。对低比例流动相而言,比例阀的稍微波动,就会影响目标化合物保留时间的较大变化。解决的办法是:提前按比例混合好流动相。[/font][font=宋体]([/font]3[font=宋体])柱温不稳定[/font][font=宋体]柱温是影响化合物在色谱上的保留行为的因素之一。通常,在反相色谱体系中,柱温的降低会引起化合物保留时间的延长。在缺乏柱温控制装置条件下,实验室的日夜温差会引起化合物在色谱保留时间的漂移。解决的办法是:安装柱温控制装置,将柱温控制在设定温度[/font]1[font=宋体]℃[/font][font=宋体]范围内。[/font][font='微软雅黑','sans-serif'][color=black][back=white]领取更多《实战宝典》请进:[url]http://instrument-vip.mikecrm.com/2bbmrpI[/url][/back][/color][/font][font='微软雅黑','sans-serif'][color=black][back=white] [/back][/color][/font]
GB 29698-2013 食品安全国家标准 奶及奶制品中17β-雌二醇、雌三醇、炔雌醇多残留的测定 气相色谱-质谱法
朋友们谁有苯甲酸甲酯、对甲苯磺酸、TEBAC、甲醇钠、4-二甲胺基吡啶、苯甲酰氯、乙硫醇、巴豆醛、乙酰乙酸甲酯、丙酰氯、DCP的国标、行标或企业标准啊?帮忙找一下啊,谢谢!
食品添加剂苯甲酸检测方法研究进展 王俊红 (浙江大学动物科学学院, 浙江杭州 310029) 摘 要:苯甲酸作为食品添加剂,其添加量多少将直接影响到人体健康。该文综述高效液相色谱法、毛细管电泳法、气相色谱法、紫外分光光度法及薄层层析法等检测方法基本原理及其近几年在苯甲酸检测技术方面研究进展,最后展望这些检测方法在苯甲酸检测方面发展前景。 关键词:苯甲酸;食品添加剂;食品安全 中图分类号:TS202.3文献标识码:A文章编号:1008―9578(2010)12―0037―02 苯甲酸外观为白色结晶体,有安息香或苯甲气味,其蒸气具有很强刺激性,主要用作食品添加剂,具有抑制食品中微生物繁殖或杀灭、防止食品腐败变质、保持食品鲜度作用。但若过量添加,不仅能破坏维生素B1,还能使钙形成不溶性物质,影响人体对钙的吸收,同时对胃肠道有刺激作用;过量食用可诱发癌症〔1〕,长期使用可诱发哮喘、荨麻疹及血管性水肿等变态反应,对人体健康造成不利影响。 近年来,食品安全已成为全社会共同关注热点。苯甲酸作为食品添加剂,我国GB2760–1996《食品添加剂使用卫生标准》规定其使用限量应<0.1g/kg,故有关苯甲酸含量检测研究也备受关注。本文就苯甲酸检测方法研究进展作一综述,为我国开展食品添加剂苯甲酸检测提供参考。 1·高效液相色谱法 高效液相色谱是从20世纪60年代后期开始发展起来的,具有填料颗粒小、且均匀,小颗粒具有高柱效特点,该法是目前应用最多一种色谱分析方法。与经典液相色谱相比,其优点是分辨率高、灵敏度高、样品量少、易回收和色谱柱可重复使用等〔2〕。 中国国家质量监督局发布乳与乳制品中苯甲酸和山梨酸测定方法为高效液相色谱法,标准规定奶制品中苯甲酸检测制样和高效液相色谱测定方法,适于乳与乳制品中苯甲酸和山梨酸含量测定。祝伟霞〔1〕、Pingqi〔3〕分别以醋酸锌和亚铁氰化钾溶液作为沉淀剂,对奶粉、酸奶和其它发酵乳制品进行处理,然后采用反相色谱法测定其中苯甲酸含量。证明该法具有前处理简单、方便、灵敏度高、重现性好等优点。 液相色谱法除可检测乳制品中苯甲酸含量,还能测定其在其它食品中含量。Tfouni等〔4〕采用高效液相色谱法测定巴西食品中苯甲酸含量。分别将饮料、果汁、黄油及奶酪等食品进行粉碎处理,然后与蒸馏水混合,再用氢氧化钠溶液将pH值调为碱性,最后离心处理,取上层清液进行反相色谱法测定,其检测精密度和准确度均能满足分析要求。Liu等〔5〕采用液相色谱仪测定面粉和油炸食品中苯甲酸含量,样品经乙醇超声破碎后提取苯甲酸,然后用C18柱进行梯度洗脱。经实验测得苯甲酸线性检出范围为0.50~15.06mg/L,最低检出限为0.22 mg。 2·毛细管电泳法 毛细管电泳(简称CE),是上世纪80年代初发展起来一种新型高效分离技术〔6〕。以毛细管为分离通道,以高压支流电场为驱动力,通常使用内径为25,000~100,000 nm弹性涂层熔融石英管。该毛细管特点是容积小、侧截比大,可用自由溶液或凝胶等为支持介质,在溶液介质下能产生平面状电流场〔7〕。该法具有高效、快速、样品量少、测定成本低等优点。 Han等〔8〕以对羟基苯甲酸为内标,采用毛细管电泳系统对食品中苯甲酸含量进行检测,经实验证实,检出限范围为10~20 ng/mL。胡美珍等〔9〕对样品进行超声、除二氧化碳处理,然后用乙醚进行抽提、净化,再将提取物用高效毛细管电泳仪进行检测。通过对实验条件优化选择,最终测得苯甲酸在浓度5 mg/L~50 mg/L时线性关系良好,样品回收率95%以上:实验过程快速、回收率高,在食品分析领域具有良好应用前景。李利军等〔10〕采用ACS2000高效毛细管电泳仪对苯甲酸进行测定,通过对缓冲体系、检测波长、分离电压等条件优化,测得苯甲酸缓冲体系为含有15 mmol/L十六烷基三甲基溴化铵体积分数1%乙酸,检测波长285 nm,分离电压20 KV,线性范围为5~40μg/mL。该法具有良好重现性和准确度,可用于苯甲酸生产过程质量控制和检测。 3·气相色谱法 气相色谱法出现于1952年,已成为分离科学中较为成熟、使用最普遍、运行最容易一种分离分析方法〔11〕。该法以气体作为流动相,除应用于分析气体试样,还可分析易挥发或可转化为易挥发液体和固体;不仅可分析有机物,也可分析部分无机物。随着检测技术发展,还出现与气相色谱仪联用气相色谱―质谱检测技术。 刘敏红等通过样品酸化,用乙醚提取苯甲酸,然后用带氢火焰离子检测器气相色谱仪进行分离测定。通过对柱温箱、载气流量等条件进行优化,并通过精密度试验、准确度实验、灵敏度实验,最终证明该检测方法可行性,为苯甲酸检测研究提供另一种思路。Farahani等〔12〕运用气相色谱―质谱仪(GC/MS)分析饮料和生活用水中苯甲酸含量。首先将样品进行前处理,然后采用GC/MS对样品进行分析,最终测得苯甲酸线性检出范围为0.5~500μg/mL,相关性≥0.99,重复性良好(RSID<10.3%,n=8),回收率为90%~113%,从而证明该法准确、有效。 4·紫外分光光度法 该法检测原理是苯甲酸为共轭型有机化合物,在近紫外光区具有较强吸收〔13〕,经实测证实,苯甲酸在230 nm处有最大吸收峰。苯甲酸在10°时溶解度为0.21 g/100 mL水,20°时为0.28 g/100 mL水〔14〕。因此可将标样和样品用水溶解后采用紫外分光光度计进行含量检测。 曾启华〔15〕运用7520型紫外分光光度计测定酸性食品中苯甲酸含量,经试验证实,该法操作简单、易于掌握,在操作和结果准确性上具有一定优势,与气相色谱法比较,其结果极为接近,不会影响到对结果判断。杜向东〔16〕用751G紫外分光光度计检测面粉中添加苯甲酸,其检测原理是将苯甲酸与乙醚混合后,在260~280 nm之间有典型吸光特性,最终测得苯甲酸最低检出量为1 mg/kg,回收范围84.4%~95.3%,从而证明紫外分光光度法稳定性、抗干扰性及准确性。 5·薄层层析法 该法原理是将试样酸化后,用乙醚提取苯甲酸,将试样提取液浓缩,点于聚酰胺薄层板上展开,显色后,根据薄层板上苯甲酸比移值与标准比较定性,并可进行概略定量,〔GB/T2009.29–2003〕。张秀尧〔17〕采用聚酰胺薄膜层析检测食品中苯甲酸,经实验测得苯甲酸检出下限为1μg。 6·展望 随着苯甲酸在食品中添加量检测方法相继建立,理想分析方法应是简单、快速、准确、有效、灵敏、专一、经济等特点。薄层层析法对样品前处理繁琐、复杂、耗时,易受时间、杂质等因素干扰,准确性相对较低。高效液相色谱法、气相色谱法、毛细管电泳法是近年发展起来检测方法,此类检测方法快速、准确、稳定;但所需仪器设备投资大,对操作技术要求高,尚未能得到广泛应用。而紫外分光光度法具有灵敏度高、分析时间短、成本低等优点,适于批量检测。随着待测食品种类扩大和食品基质成分复杂化,来自食品中非测定成分干扰将日益增多。因此对检测样品纯化、检测手段要求将越来越高,发展廉价、灵敏、专一、快速净化手段和检测方法是今后研究方向。 〔参考文献〕略
苯甲酸标准品是用水定容还是甲醇啊?看标准上说用水,甲醇的溶解性不是更好么?各位一般标准品是用苯甲酸钠还是苯甲酸啊?
求教苯甲酰氯和二乙二醇二苯甲酸酯的分析方法,谢谢
戚平始料未及的是,自己的一篇学术文章竟然会在近来颇为敏感的中国乳品安全问题上引起轩然大波。戚平是广州市食品工业卫生检测所的研究人员。英国专业杂志《食品管理》近日发表了他和同事们的论文《中国液态奶中的苯甲酸水平评估》(Assessment of benzoic acid levels in milk in China)。 2月18日,一篇题为《中国奶制品大多被检出苯甲酸》的文章称,根据戚平的论文,“2006年10月至2007年1月期间,研究人员从广州商店和超市购买了142份乳制品,其中包括巴氏消毒奶、超高温灭菌奶、普通奶粉和婴幼儿配方奶粉。142份乳制品样本中,有109份检出苯甲酸,含量从0.51毫克/千克到110毫克/千克不等。”“普通奶粉和婴幼儿配方奶粉的检出率,分别高达87.1%和85.7%。”文章称,戚平指出,“苯甲酸对婴幼儿等特殊人群的长期健康影响值得关注。” 报道迅速传开,甚至被国内的新闻网站刊载于显著的位置上,激起了网友们对中国乳品安全性的又一轮热议。 2月19日晚间,记者联系了两天未果的戚平主动向本报发来声明。戚平说,报道是断章取义地从论文里攫取了一些观点。“我的文章在2008年4月已经投稿,纯粹是一篇学术论文,其发表的时间不是我所能左右的。由于目前中国乳制品深受各种事件困扰,文章刚好在这个敏感时期被发表。” 苯甲酸本身广泛存在于自然界,是食品工业中常见的一种防腐剂。戚平说,各国对苯甲酸的使用范围和添加量都有规定。根据我国《食品添加剂使用卫生标准》(GB2760-2007),在冰棍类、风味(果味、乳味等)饮料、果蔬汁中,允许的苯甲酸最大使用量是1000mg/kg。另外,GB2746-1999《酸牛奶》标准也针对苯甲酸的限量做出过规定,纯酸牛奶为30 mg/kg,风味酸牛奶为230 mg/kg。 “苯甲酸是不能添加在乳制品中的。因此,人们往往认为乳制品中不得检出苯甲酸。但这是一个错误的观念,‘不得添加’并不意味‘不得检出’。”戚平说,乳制品中一般都含有苯甲酸,因为牛乳的成分非常复杂,特别是一些微量成分会随着季节、饲料、饮水及原料乳中微生物变化而变化,乳制品中往往自身就含有苯甲酸。世界卫生组织(WHO)对苯甲酸和苯甲酸钠的风险评估为“自然界中许多动植物本身含有苯甲酸,因此可以认为苯甲酸是食品(包括牛奶)中的一种天然成分”。 戚平说,早在十多年前,国外就对发酵的乳制品做过苯甲酸的调查,但是关于液态奶中苯甲酸的含量似乎是一个空白。因此,他萌生了研究液态奶中苯甲酸含量的念头,“如果了解到一袋液态奶在正常情况下,自身能产生的苯甲酸的含量范围,那么就可以正确判断哪些是不良厂商‘非法添加’的,哪些是本身具有的。” 《中国奶制品大多被检出苯甲酸》一文说,“此前,国外也有研究在液态奶中检测出苯甲酸。与之相比,中国液态奶的苯甲酸水平要高一些。”对此,戚平解释,1989年一份关于对乳制品的调查研究数据表明,牛奶中苯甲酸的含量为 “6 mg/kg”。而在研究中他发现,7%仅5个样品中苯甲酸含量高于6 mg/kg。“这是研究数据与已有数据的对比,是时间上的对比,而非中国液态奶和国外液态奶的对比,样品中也包含国外品牌。” 苯甲酸的产生与人类活动有很大关系。WHO在“简明国际化学品评估文件26”中介绍,目前,木材产品、铸造废水、市政焚毁炉灰、汽车尾气,甚至香烟中都监测到了苯甲酸。另外,苯甲酸酯类香料在光催化下也会分解产生苯甲酸。通常来说,在各种环境介质中,包括空气、雨水、雪、地表水、土壤中都存在着游离和非游离态的苯甲酸和苯甲酸钠。“与30年前相比,由于人类的作用,环境中苯甲酸的含量肯定会增高。因此,即使乳制品本身的苯甲酸的含量升高也可以理解。”戚平说。 戚平强调,自己在文章中并没有说“对包括婴幼儿在内的一些特殊人群而言,长期摄入苯甲酸也可能带来哮喘、荨麻疹、代谢性酸中毒等不良反应”。“论文原文是‘对敏感人群,低剂量的苯甲酸会引起过敏反应’,‘高剂量地摄入苯甲酸也可能带来哮喘、荨麻疹、代谢性酸中毒等不良反应’。这也是WHO在‘简明国际化学品评估文件26’中的论述,而敏感人群是指类似于花粉过敏等体质敏感的人群,并不是指婴幼儿。”目前乳制品中的苯甲酸含量,只要正常食用,其摄入量远低于权威组织规定的安全值,因此是安全的。 “目前,人们对物质本身所产生的衍生物的检测还很缺乏。”中国农业大学教授姜微波说,不仅是中国,世界各地在这方面都存在空白。理论上,衍生物在很多情况下都会存在,它的危害性不那么强,而实际操作中要检测它们却非常困难,“因为要找到这种衍生物究竟会在哪种食品中产生,就要将这种衍生物与所有的食品进行试验。” 中国农业大学教授何计国将这场虚惊归因于我国对食品行业的管理比较混乱、不够专业,消费者很难放心自己喝的是安全乳品。“在国外,食品行业会有一个介于政府和个人企业的第三方组织定期到社会取样、调查,并且由单一的部门管理,一旦出了事就能责任明晰。但在中国,不同的食品企业很可能由不同的部门主管,而一个技术监督局同时负责监管化肥、食品等十几种行业。”他建议从标准更新、立法准确入手,对食品生产和原料经营做好监督。来源:北京科技报
液相色谱法测定乳制品中苯甲酸含量摘要: 使用液相色谱仪检测乳制品中苯甲酸防腐剂含量。关键词:乳制品;液相色谱;苯甲酸;防腐剂参考文献:GB 21703-2010苯甲酸是食品工业中常见的一种防腐保鲜剂,但乳制品中是禁止添加的。一般情况下,苯甲酸被认为是安全的,但对包括婴幼儿在内的一些敏感人群而言,长期摄入苯甲酸也可能带来哮喘、荨麻疹、代谢性酸中毒、抽搐等不良反应。实验部分1试验材料、仪器及试剂1 样品:新疆奶牛基地生鲜乳2 仪器:液相色谱仪-Waters 2695配紫外检测器;天平(0.01g)(奥豪斯(上海)有限公司);3试剂:氢氧化钠、浓硫酸、亚铁氰化钾、乙酸锌(分析纯,天津盛奥化学试剂厂);甲醇(色谱纯,Therm Fisher)2 标准品配制:苯甲酸标准贮备液:500mg/L;标准工作液:10mg/L。3 样品前处理 称取乳制品液体试样20g±0.01置于100mL的容量瓶中,加入25mLNaOH(0.1mol/L),混匀振荡15min,用H2SO4(0.5mol/L)调节pH≈8,再加入2mL亚铁氰化钾(0.25mol/L)和2mL乙酸锌(1mol/L),混匀振摇,静置后用甲醇-水(v/v=1/1)定容至100mL,上清液过0.45μm滤膜过滤后装进样瓶待测。4 仪器条件色谱柱:C18柱(Waters);流动相:甲醇-磷酸盐缓冲溶液=1:9;流速:10mL/min;检测波长:227nm;进样量:10uL。5 结果与分析 http://ng1.17img.cn/bbsfiles/images/2014/07/201407071107_505678_1634341_3.bmp[
请问雌二醇的稳定性怎样,大家是怎样储存标准品储备液的
就是我在做液相色谱分析的时候,分析3.5二甲基苯甲酸,在主峰出现之前出的杂质峰比以前大,这是请问这是什么原因?
求助:对羟基苯甲酸丁酯钠和对羟基苯甲酸丙酯钠的相关行业标准,国内、国外的标准都可以。
包括婴幼儿配方奶粉在内,中国奶制品中大多检出了苯甲酸(benzoic acid)。 研究人员表示,尽管苯甲酸含量尚未超过世界卫生组织的每日允许摄入量推荐值,但其对婴幼儿等特殊人群的健康影响值得关注。 在最新(2009年2月)出版的国际期刊《食品管理》(Food Control)上,广州市食品工业卫生检测所戚平等研究人员对中国乳制品的苯甲酸含量进行了分析。 苯甲酸是食品工业中常见的一种防腐保鲜剂,但乳制品中不允许添加。一般情况下,苯甲酸被认为是安全的。但对包括婴幼儿在内的一些特殊人群而言,长期摄入苯甲酸也可能带来哮喘、荨麻疹、代谢性酸中毒等不良反应。 2006年10月至2007年1月期间,研究人员从广州商店和超市购买了142份乳制品,其中包括巴氏消毒奶、超高温灭菌奶、普通奶粉和婴幼儿配方奶粉。 经过高效液相色谱分析,这142份乳制品样本中,有109份检出了苯甲酸,其含量从0.51毫克/千克到110毫克/千克不等。 其中,情况最好的是巴氏消毒奶,24份产品仅有11份检出苯甲酸。而普通奶粉和婴幼儿配方奶粉的检出率,分别高达87.1%和85.7%。 研究人员认为,这或许是因为巴氏消毒奶在低温下储存,且储存时间短,不利于苯甲酸的出现。 戚平告诉《财经》记者,他们检测的所有乳制品中,苯甲酸含量都不高,“说明不是厂家人为添加的”。 他推测说,乳制品中苯甲酸的来源可能与环境因素、生产过程等有关。例如,汽油不完全燃烧等环境污染,最终也会在自然环境中形成苯甲酸;奶牛养殖中饲料和兽药的滥用,也可能带来苯甲酸。 此前,国外也有研究在液态奶中检测出苯甲酸。与之相比,中国液态奶的苯甲酸水平要高一些。 戚平强调,根据他们检测的情况,中国乳制品的苯甲酸不会对公众健康产生影响。即使是食用苯甲酸含量最高的那种婴幼儿配方奶粉(88毫克/千克),也不会超过世界卫生组织推荐的每日允许摄入量(5毫克/每千克体重)。 不过,他同时指出,苯甲酸对婴幼儿等特殊人群的长期健康影响值得关注。 根据农业部2007年底发布的推荐标准《绿色食品 乳制品》,苯甲酸含量不超过30毫克/千克的乳制品,才可称为绿色食品。而此次抽样分析发现,婴幼儿奶粉的平均苯甲酸含量达到了36毫克/千克,普通奶粉更是达到了49毫克/千克。 戚平及其同事在论文中建议,奶牛养殖户和乳制品厂家应该在饲养、生产和储存等环节尽力避免苯甲酸的出现。例如,确保饲料的质量,以减少奶牛对苯甲酸的摄入;增加对苯甲酸含量的监控;寻找影响乳制品中苯甲酸含量的因素等。
在做国标 GB5009. 28 — 2016 食品安全国家标准食品中苯甲酸、山梨酸和糖精钠的测定对照品配制疑惑 标准溶液配制:苯甲酸、山梨酸和糖精钠(以糖精计)标准储备溶液( 1000mg / L ):分别准确称取苯甲酸钠、山梨酸钾和糖精钠 0.118g 、 0. 134g 和 0.117g (精确到 0.0001g ),用水溶解并分别定容至 100mL 。于 4℃贮存,保存期为 6 个月。当使用苯甲酸和山梨酸标准品时,需要用甲醇溶解并定容。红色这句话的意思是想用适量的甲醇溶解,然后在用水定容,还是用甲醇定容,感觉怪怪的?有那位老师可以指导一下, 共享一下经验,谢谢。
对羟基苯甲酸酯,俗称尼泊金酯是一类安全高效的防腐剂, 广泛用于食品、饮料、化妆品、医药等许多方面。在化妆品行业中,由于其安全、高效和广谱的特点,长期以来被用于各类产品中。 在中国,对羟基苯甲酸酯是经过卫生部批准的化妆品组分中的限用防腐剂,中国《化妆品卫生规范》(2007版)规定,单一酯和混合酯的限量分别为0.4%和0.8%。在规定的范围内适量添加防腐剂对于保护化妆品的产品质量是非常必要的,防腐剂的功能是使化妆品免受微生物的污染,确保消费者的使用安全。 此外,对羟基苯甲酸酯也是我国允许使用的食品添加剂,广泛用于酱油、醋、果酱、糕点馅、果汁和碳酸饮料等食品中,我国国家标准——《食品添加剂食用卫生标准GB 2760-2007》中规定最高使用浓度可以达到0.5g/kg (相当于500ppm)。 对羟基苯甲酸酯类在世界范围内得到了各国及国际性官方组织的认可。美国食品药品管理局(US FDA)批准对羟基苯甲酸酯类可以作为化妆品防腐剂使用,据统计截至到2006年,在美国仍有很高的使用频度。美国独立的化妆品原料安全性评价专家小组(Cosmetic Ingredient Review, CIR)也对对羟基苯甲酸酯类在化妆品中使用情况作了权威性结论,即按目前的使用惯例,对羟基苯甲酸酯类作为化妆品防腐剂成分是安全的。欧盟委员会也批准对羟基苯甲酸酯类作为化妆品防腐剂使用。联合国食品法典委员会(CODEX)批准其作为允许添加的食品防腐剂。 消费者应该对于化妆品、食品等日常消费品中的防腐剂有正确的认识,防腐剂对于保护产品、防止有害微生物滋生和最终保护消费者的健康是非常必要的,只要防腐剂添加的含量符合相关法规标准的要求,产品就是安全的,消费者尽可以放心使用。
气相测3.5-二甲氧基苯甲酸甲酯要用什么标准物做外标
各位老师,GB5009.31-2016是检测食品中对羟基苯甲酸酯。标准中检测原理是:试样酸化后,对羟基苯甲酸酯类用乙醚提取,浓缩近干用乙醇复溶,并利用氢火焰离子化检测器[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法进行分离测定,保留时间定性,外标法定量。我想请教的是为什么要酸化,酸化的作用是什么?是让目标成分都以酯的形式存在吗?

为什么甲氨基阿维菌素苯甲酸盐标准品会有这么多杂质峰?[img]http://ng1.17img.cn/bbsfiles/images/2018/04/201804231728132335_5892_3399169_3.jpg[/img]
雌二醇(17β-Estradiol)是天然雌激素的重要成分,为防止儿童性早熟等问题,食品中尤其是畜禽产品的雌二醇检测不容忽视。胶体金免疫层析技术(GICA)在检测小分子有机物方面越来越受到重视。其基本原理是胶体金颗粒能够稳定吸附蛋白质,而蛋白质的生物活性无明显变化,所以它可以取代酶标记抗体〔1,2〕。本实验采用标记有雌二醇单克隆抗体的胶体金颗粒与相对应的固定在硝酸纤维膜上的雌二醇结合物相结合,固定有雌二醇结合物的检测线处就显现明显的颜色〔3,4〕。本法具有灵敏度高、特异性强、简便快速、成本较低、结果轻易判读等优点,适用于样品的初步筛选。[b]1材料与方法[/b]11试剂与仪器氯金酸(上海化学试剂厂);牛血清白蛋白、卵清白蛋白、兔抗鼠多抗(天津联星生物公司);雌二醇标准品、雌二醇单抗、17β-Estradiol-6-one6-(o-carboxymethvyoxime)美国Sigma公司);其他试剂均为国产分析纯。硝酸纤维膜、玻璃纤维膜(美国Millipore公司);BeckmanDu530分光光度计(德国Beckman公司);低温高速离心机(军事医学科学院);点膜机(美国Bio-Dot公司)。12方法121胶体金的制备用三蒸水溶解氯金酸,使其终浓度为01g/L。先进行沸水浴,待氯金酸溶液煮沸后,每100ml加入1%柠檬酸三钠25ml,再沸水浴下快速搅拌,直到氯金酸溶液的颜色稳定,继续沸水浴10min〔5〕,冷却至室温后,用透射电镜镜检并用分光光度计检测颗粒均匀度及粒度。最后置于4℃冰箱保存备用。122最适标记蛋白量的确定用02mol/LK2CO3将胶体金溶液调至pH为82,然后取试管9支,分别加入10ml胶体金溶液。将雌二醇抗体逐级稀释后,各取等体积稀释液顺序加入上述试管中,混匀,另设一不加抗体的对照管。放置10min,在各管中加入01ml的10%NaCl,混匀后静置2h。观察各管中胶体金溶液变化,未加蛋白及加入量不足的溶液颜色由红变蓝,而加入蛋白量达到或超过时的溶液则保持不变。标记蛋白量即为最低稳定胶体金溶液不变色的蛋白量基础上再加20%。123胶体金探针的标记将抗体用0005molNaCl溶液透析过夜,离心除去蛋白沉淀,调至05mg/ml。取胶体金100ml,用0,2mol/LK2CO3将胶体金溶液调至pH为90,磁力快速搅拌下缓慢加入24ml稀释的雌二醇抗体,继续搅拌10min,加入牛血清白蛋白(BSA),使其最终浓度为1%,再搅拌10min。将初步制得的胶体金探针以4000r/mm离心20min;弃沉淀,上清以10000r/min离心60min;弃上清,沉淀用001mol/L的磷酸盐缓冲液(PBS)(含1%牛血清白蛋白)重新悬浮;同前离心洗涤2次,将沉淀用001mol/L的PBS(pH82,含1%BSA,002%NaN3)悬浮,4℃保存备用。124抗原的合成小分子物质难以固定在硝酸纤维膜上,只有使它连上大分子物质才能较好地固定。本实验采用混合酸酐法制备完全抗原。雌二醇-6-肟43mg,加入三正丁胺50μl,二氧六环4ml,冰浴冷却到10℃以下,加入氯甲酸乙酯15μl〔6〕。4~10℃下反应30min。配制卵清白蛋白(OVA)溶液(OVA10mg,3ml水,3ml二氧六环,1mol/L氢氧化钠03ml)。将配制好的OVA溶液加入反应液,4℃冰浴搅拌6h,反应过程中,用氢氧化钠维持pH值为8。反应完毕后,将反应液加入透析袋,透析2d。将透析液调至pH45,冰箱4℃,放置4天,有黄色沉淀出现。离心收集沉淀,冷冻干燥,4℃冰箱保存。将OVA溶于透析液内作为参比,用紫外光谱法对雌二醇-6肟-OVA进行定性检验。经紫外测定证实蛋白质已与雌二醇衍生物结合。125胶体金免疫层析试纸条制备测试条由玻璃纤维膜、硝酸纤维膜、加样纸、吸水纸4部分组成。将玻璃纤维膜裁成6mm的细条,然后放入含1%BSA、1%Tween-20的PB液中浸泡30min,37℃烘干,最后将胶体金探针灌注已处理好的玻璃纤维膜上,真空干燥备用。在硝酸纤维膜上用点膜机将上述抗原和兔抗鼠IgG喷成2条线,分别为检测线和对照线,经真空干燥后,用1%BSA、001mol/LPBS(pH90)封闭2h,以001mol/LPBS洗涤,再真空干燥。将30mm吸水纸、25mm硝酸纤维膜、6mm玻璃纤维膜、15mm加样纸,由顶部依次粘于PVC板上,裁成细条备用。126检测与判读样品:含已知浓度雌二醇样品的乙醇溶液分为3组,第1组不加雌二醇,第2组为02μg/ml雌二醇,第3组为04μg/ml雌二醇。将加样纸一端插入待测液,湿润后取出,水平放置,约3~5min,观察结果。如试纸条硝酸纤维膜上仅对照线呈一条紫红色带为阳性;如出现2条紫红色带为阴性;如质控线不出现紫红色带,无论检测线是否出现,测试结果均无效。[b]2结果与分析[/b]21胶体金颗粒大小选择胶体金颗粒吸附蛋白并显示检测结果,其大小是重要影响因素之一。胶体金颗粒的大小,由制备胶体金时加入的柠檬酸三钠量决定,当柠檬酸三钠加入越多,胶体金颗粒也越多,但颗粒直径越小,反之胶体金颗粒越大。胶体金颗粒小,结合蛋白量少,反应结合率低,另外难以显示明亮清楚的颜色,影响显色效果;胶体金颗粒大,其结合蛋白后不稳定,不易保存,另外其结合蛋白后难以通过膜〔7〕。本实验选用的胶体金颗粒直径约为15min,既可以通过膜,反应彻底,又易保存,反应结果显色清楚。22胶体金颗粒鉴定胶体金颗粒大小及均一程度是检测结果可靠的基础,制备完毕后须经质量鉴定才能应用。用BeckmanDu530分光光度计对胶体金进行扫描,发现其主峰宽度较小,表明制备的胶体金颗粒较均匀,λmax为520nm(图1),表明其粒度约为15nm〔8〕。用电镜观测结果(图2)与分光光度计测定结果一致。电镜观测是鉴定胶体金颗粒粒径及分布的标准,但这种方法不便用于日常工作使用,而用分光光度计测定操作简便,更适合实验室日常使用。图1胶体金的可见光谱图(略)23最适蛋白量阴性对照为未加抗体管,1~7管抗体浓度分别为10,12,14,16,18,20,22μg/ml,阳性对照为胶体金溶液。可见,本实验最适标记蛋白量为20μg/ml(图3)。24E2-OVA浓度用PBS(pH74)溶解E2-OVA浓度为005~10mg/ml,喷在硝酸纤维膜上作检测线,发现E2-OVA浓度越高,检测液中也需要越高浓度的E2与之竞争,检测灵敏度也就越低。但是,E2-OVA浓度低于05mg/ml,检测线颜色太弱,难用肉眼分辨。图2胶体金电镜图(略)阴性对照为未加抗体管;阳性对照为胶体金溶液图3胶体金与雌二醇抗体结合浓度确定实验(略)25检测限本实验的检测限是以完全看不见检测线为最低检测限〔9〕。将雌二醇标准品用甲醇配制成04,02,01,005μg/ml4个标准溶液,用上述检测方法进行检测(图4)。由图可见,最低检测线为100ng/ml。图4试纸条检测样品(略)26对比实验取30份样品,检测分为3组,第1组不加雌二醇,第2组加02μg/ml雌二醇,第3组加04μg/ml雌二醇,分别用GICA法与[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质联用[/color][/url]法(GC/MS)检测。实验结果显示,第1组2种方法均未检出;第2组GICA法阳性9粒,阴性1粒,GC/MS法阳性10粒;第3组2种方法均检测阳性。由此可见,与标准检测方法相比,GICA法准确度为9667%。27交叉反应试验实验结果显示,与己烯雌酚、炔雌酚、壬基酚、阿特拉津及双酚A等环境内分泌干扰物中的环境雌激素无交叉反应,表明该方法具有良好的特异性。[b]3小结[/b]目前,关于雌二醇的检测有气、液相色谱和光谱、质谱等方法,但其仪器昂贵、操作复杂、检测时间较长。本文建立的免疫胶体金层析法具有灵敏度高、特异性强、简便快速、成本较低、结果轻易判读等优点,且无需任何仪器,操作简单,适用于样品的初步筛选。[list][/list]







 我要推广仪器
我要推广仪器
 下载APP
下载APP