
作者:李昂; 胡翮; 郭丙炎;(湖南省湘潭市药品检验所;)摘要:目的用反相高效液相色谱(RP-HPLC)法测定盐酸丁卡因的含量。方法色谱柱为Diamonsil C18柱(150mm×4.6mm,5μm),流动相为水-甲醇-乙腈(45∶45∶10,v/v,含0.02%庚烷磺酸钠,0.34%磷酸二氢钾,三乙胺调pH至7.0),流速为1.0mL/min,检测波长为314nm,柱温为室温。结果盐酸丁卡因质量浓度在5.034~161.1μg/mL范围内与峰面积线性关系良好,回归方程为Y=5.6572×105X-9.2039×105(r=0.9999),平均回收率为100.76%,RSD为0.41%(n=6)。结论RP-HPLC法简便、可靠、准确,可作为盐酸丁卡因注射液的含量测定方法。谱图:http://ng1.17img.cn/bbsfiles/images/2012/08/201208271103_386360_1606903_3.jpg
[color=black]复方盐酸阿替卡因注射液为复方制剂,是盐酸阿替卡因与肾上腺素的灭菌水溶液,作为口腔用局部麻醉剂,适用于涉及切骨术及粘膜切开的外科手术过程。[/color][color=black] [/color][img=,156,99]https://ng1.17img.cn/bbsfiles/images/2019/03/201903211036489419_4502_2297_3.jpg!w156x99.jpg[/img][align=center][/align][align=left][b][color=black]盐酸阿替卡因(Articaine hydrochloride M.W.:320.84)[/color][/b][/align][align=center][b][color=black] [/color][/b][/align][color=black]在现有国家药品标准(YBH17082004-2015Z)分析方法中,流动相添加了离子对试剂-庚烷磺酸钠,并在pH为2.0的强酸条件下进行相应分析,不利于色谱柱的使用寿命。大曹三耀实验室参考USP方法,以冰醋酸水溶液-乙腈作为流动相,选用CAPCELL PAK C18 MGII色谱柱,实现了复方盐酸阿替卡因注射液中盐酸阿替卡因的定量和有关物质的良好分析(复方盐酸阿替卡因注射液由客户提供)。[/color][color=black]CAPCELLPAK C18 MGII[/color][color=black]液相色谱柱,其采用高纯度硅胶作为基质,通过减少硅胶微细孔的数量来增大有效比表面积;并且采用新包被技术Ultimate Polymer Coating,实现了对硅醇基极大程度的封锁,兼具分离性能和普适性能,通用性非常好。[/color][align=left][b][color=#0070c0]实验方法[/color][/b][/align][align=left][img=,500,358]https://ng1.17img.cn/bbsfiles/images/2019/03/201903211037456969_5082_2297_3.jpg!w730x523.jpg[/img][/align][align=left]图1[color=black]盐酸阿替卡因[/color]对照品及供试品溶液[/align][align=left][img=,500,248]https://ng1.17img.cn/bbsfiles/images/2019/03/201903211038541919_2603_2297_3.jpg!w572x284.jpg[/img][/align][align=center][/align][align=center][/align][color=black]为进行有关物质分析,该实验将注射液样品以流动相稀释100倍,作为有关物质供试品溶液,再将该有关物质供试品溶液以流动相进一步稀释100倍,作为自身对照溶液。以冰醋酸水溶液-乙腈作为流动相,选用CAPCELL PAK C18 MGII色谱柱,通过调整流动相比例及柱温,最终在18%乙腈、柱温30℃条件下实现了盐酸阿替卡因供试品溶液及对照品的良好分析。[/color]如图2、3,使用CAPCELL PAK C18 MGII色谱柱进行分析,盐酸阿替卡因和有关物质均能得到良好分析结果,主峰与峰前杂质得到了良好分离,分离度为1.90(见表1)[img=,400,311]https://ng1.17img.cn/bbsfiles/images/2019/03/201903211045536855_9516_2297_3.jpg!w574x447.jpg[/img][img=,400,295]https://ng1.17img.cn/bbsfiles/images/2019/03/201903211045540875_8483_2297_3.jpg!w698x516.jpg[/img][align=left] 图2 [color=black]盐酸阿替卡因[/color]有关物质供试品溶液及空白 图3 自身对照溶液[/align][align=center][/align][align=left]表1 有关物质结果详表[/align][align=left][img=,600,323]https://ng1.17img.cn/bbsfiles/images/2019/03/201903211042513275_6690_2297_3.jpg!w786x424.jpg[/img][/align][align=center][/align]综上实验结果,使用CAPCELL PAK C18 MGII S5 4.6mm i.d.×250 mm色谱柱,以冰醋酸水溶液-乙腈为流动相体系,在30°C柱温条件下,能够实现复方盐酸阿替卡因注射液中盐酸阿替卡因的定量和有关物质的良好分析。[color=black] [/color]
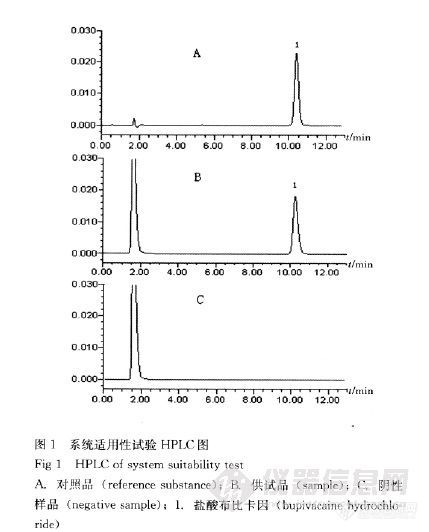
作者:兰文; 杨汉初; 黄莉;(湖南省药品检验所;)摘要:目的建立反相高效液相色谱法测定复方明矾布比卡因注射液的有关物质及盐酸布比卡因的含量。方法采用Diamonsil C18柱,以乙腈-磷酸盐缓冲液(取磷酸二氢钾1.94g,磷酸氢二钾2.48g,加水溶解并稀释至1 000mL,调节pH 6.8)(65∶35)为流动相;流速为1.0mL.min-1;有关物质检测波长为215nm,含量测定检测波长为263nm;柱温:30℃。结果在该色谱条件下,杂质峰与主峰均能有效分离,盐酸布比卡因在44.31~177.26μg.mL-1与峰面积线性关系良好(r2=0.999 7);平均回收率为99.7%(n=9),RSD=0.4%。结论本法简便、快速、准确,专属性好,可用于复方明矾布比卡因注射液的质量控制。谱图:http://ng1.17img.cn/bbsfiles/images/2012/08/201208061043_381724_1606903_3.jpg
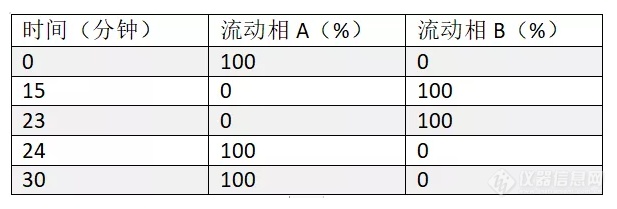
如题,俺第一次测盐酸左氧氟沙星,做有关物质时杂质A与左氧保留时间完全重叠,排除了乙酸铵、高氯酸钠等试剂滴原因,实在没辙咧,请教大虾帮忙。盐酸左氧氟沙星有关物质测定方法(来源:中国药典2010年版第一增补本): 有关物质 取本品,精密称定,加0.lmol/L盐酸溶液溶解并定量稀释制成每1ml中约含1.2mg的溶液,作为供试品溶液,精密量取适量,用0.1mol/L盐酸溶液定量稀释制成每1ml中含2.4ug的溶液,作为对照溶液。另精密称取杂质A对照品约18mg,置100ml量瓶中,加6mol/L氨溶液1ml与水适量使溶解,用水稀释至刻度,摇匀,精密量取2ml,置100ml量瓶中,用水稀释至刻度,摇匀,作为杂质A对照品溶液。照高效液相色谱法(附录V D)测定,用十八烷基硅烷键合硅胶为填充剂;以醋酸铵高氯酸钠溶液(取醋酸铵4.0g和高氯酸钠7.0g,加水1300ml使溶解,用磷酸调节pH值至2.2)-乙腈(85 :15)为流动相A,乙腈为流动相B;按下表进行线性梯度洗脱。柱温为40°C;流速为每分钟1ml。称取左氧氟沙星对照品、环丙沙星对照品和杂质E对照品各适量,加0.1mol/L盐酸溶液溶解并稀释制成每1ml中约含左氧氟沙星1.2mg、环丙沙星和杂质E各6ug的混合溶液,取10ul注人液相色谱仪,以294nm为检测波长,记录色谱图,左氧氟沙星峰的保留时间约为15分钟。左氧氟沙星峰与杂质E峰和左氧氟沙星峰与环丙沙星峰的分离度应分别大于2.0与2.5。量取对照溶液10ul注人液相色谱仪,以294mn为检测波长,调节检测灵敏度,使主成分色谱峰的峰高约为满量程的20%。精密量取供试品溶液、对照溶液和杂质A对照品溶液各10ul,分别注人液相色谱仪,以294nm和238nm为检测波长,记录色谱图。供试品溶液色谱图中如有杂质峰,杂质A(238nm检测)按外标法以峰面积计算,不得过0.3%。其他单个杂质(294nm检测)峰面积不得大于对照溶液主峰面积(0.2%),其他各杂质(294nm检测)峰面积的和不得大于对照溶液主峰面积的2.5倍(0.5%)。供试品溶液色谱图中任何小于对照溶液主峰面积0.1倍的峰可忽略不计。时间(分钟) 流动相A(%) 流动相B(%) 0 100 0 18 100 0 25 70 30 39 70 30 40 100 0 50 100 0
大家有测过盐酸利多卡因原料含量的吗?(2005版中国药典二部中的方法),在测定含量时颜色的变化明显吗?
副产盐酸中含有大量的杂质成分,我估计了一下,肯能含有硫酸根,乙酸,氯乙酸,二氯乙酸等成分,硫酸根可以用氯化钡定量,但其他杂质怎么测定呢?有比色法或滴定法可以进行检验的吗?请各位大侠帮忙解决一下,谢谢!
2010版药典中盐酸利多卡因注射液含测项下结果乘以1.156是什么意思,这个数据是根据什么公式得来的呢?
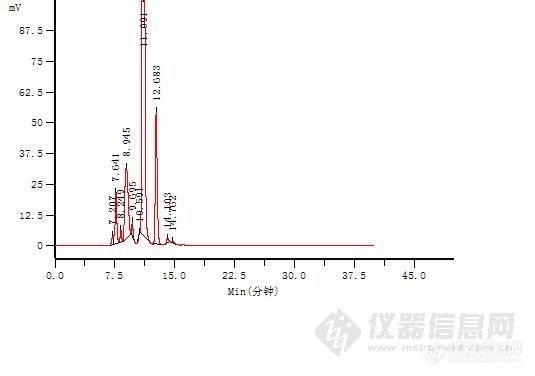
高效分子排阻色谱法测定注射用盐酸头孢替安高分子杂质头孢替安是杀菌性头孢菌素类广谱抗生素,头孢替安不但对革兰氏阳性菌有效,而且对革兰氏阴性菌。如流感嗜血杆菌,大肠杆菌、克雷白氏菌、奇异变形杆菌等的作用更强。对肠杆菌,枸橼酸杆菌、吲哚阳性变形杆菌等,也有抗菌作用头孢替安在肺中药物浓度较高,其它脏器和肌肉也有一定的浓度。临床应用于敏感菌所导致的感染,如肺炎、支气管炎、胆道感染、腹膜炎、尿路感染以及手术后或外伤引起的感染和败血症等。其基本结构同已上市的的头孢菌素类抗生素一样,头孢替安也会形成高分子聚合物,也会在临床使用中引发速发型过敏反应。对患者危害极大。已有的注射用盐酸头孢替安国家药品标准未将盐酸头孢替安高分子聚合物列为检定项目,国内的药学研究也未见头孢替安高分子聚合物的研究和报道。从临床用药安全性考虑,根据中国药典2010年版二部附录凝胶色谱原理。采用常用的葡聚糖凝胶G-10检测聚合物时由于头孢替安分子结构自身的原因,头孢替安不能完全缔合,因些我们采用高效分子排阻色谱法,以球状蛋白色谱用亲水硅胶为填充剂 TOSOH TSKgelG2000SW(7.5*300mm),测定注射用盐酸头孢替安高分子杂质1.仪器与试剂(1)仪器:岛津LC-10ATvp泵 岛津SPD-10AVP紫外可见光多波长检测器 浙大2010色谱数据工作站 色谱柱:TOSOH TSKgelG2000SW(7.5*300mm) (2)试剂: 乙腈 (色谱纯,天津市四友生物医学技术有限公司) 磷酸氢二钠(分析纯,北京化学试剂公司) 磷酸二氢钠(分析纯,北京化学试剂公司)双蒸水 (自制)2 色谱条件色谱柱:TOSOH TSKgelG2000SW(7.5*300mm)流动相:磷酸盐缓冲液(p H:6.8[/color
本人在做氨基酸分析,用的是黄酒中氨基酸的测定方法行业标准编制说明 标准,十八种氨基酸混标进样,由于仪器限制,使用的是二元梯度,标准是三元,两者的区别只是峰的分离度。但是实验中总是有杂质峰,而且重复性很好,保留时间、峰面积是一致的,怀疑是衍生副产物。单标进样仍然有,而且几乎是一个单标对应一个杂质峰。并不是所有杂质峰都出现,说明不是样品、流动相的问题。认为问题就出在衍生过程中,有做过的是否也由这些杂质峰?
[B][center]药物中杂质的来源及杂质限量检查[/center] [/B]药物只有合格品与不合格品;一般化学试剂分为4个等级(基准试剂、优级纯、分析纯、化学纯) [B]药物中一般杂质检查 [/B][B]氯化物为一指示性杂质。[/B] 通过对氯化物的控制,可同时控制与氯化物结合的一些阳离子以及某些同时生成的副产物。可从氯化物检查结果显示药物的纯度,间接考核生产、贮藏过程是否正常。 1. 原理 药物中微量的氯化物在硝酸酸性条件下与硝酸银反应,生成氯化银的胶体微粒而显白色浑浊,与一定量的标准氯化钠溶液在相同条件下产生的氯化银浑浊程度比较,判定供试品中氯化物是否符合限量规定。 Ag+ + Cl- → AgCl ↓ [B]硫酸盐检查法 [/B] 1. 原理 药物中微量的硫酸盐在稀盐酸酸性条件下与氯化钡反应,生成硫酸钡的微粒而显白色浑浊,与一定量的标准硫酸钾溶液在相同条件下产生的硫酸钡浑浊程度比较,判定供试品中硫酸盐是否符合限量规定。 [B]铁盐检查法 [/B]硫氰酸盐法 巯基醋酸法 砷盐检查法 1. 古蔡氏法 1. 原理 金属锌与酸作用产生新生态的氢,与药物中微量砷盐反应生成具挥发性的砷化氢,遇溴化汞试纸产生黄色至棕色的砷斑,与同条件下一定量标准砷溶液所生成的砷比较斑,判断砷盐的含量。 [B]硒、氟及硫化物检查法 [/B]1. 氧瓶燃烧法 适用于以共价键结合的卤素、硫、硒的有机药物。 本法系将有机药物防入充满氧气的密闭燃烧瓶中进行燃烧,将燃烧所产生的欲测组分吸收于适当的吸收液中,然后根据欲测组分的性质,选用合适的分析方法进行鉴别、检查或含量测定。 [B]注意事项及讨论 [/B]1. 根据被燃烧分解的样品量选用适宜大小的燃烧瓶。 2. 测定氟化物时应改用石英燃烧瓶。 1. 硒检查法 (1). 操作方法 样品与对照品液,调节Ph2.0±0.2,加盐酸羟胺,二氨基萘,比色。 [B]硫化物检查法 [/B] 方法同砷盐检查第一法,不装醋酸铅棉花,以醋酸铅试纸代替溴化汞试纸。 标准液取1ml 5/ml [B]澄清度检查法 [/B]将一定浓度的供试品溶液与浊度标准液分别置于配对的比浊用玻璃管,同置黑色背景上,在漫射光下观察。浊度标准液 硫酸肼与乌洛托品溶液混合分五个等级,未超过0.5等级即为澄清。BP98规定未超过1等级即为澄清。 [B]溶液颜色检查法 [/B]CHP2000 [B]1. 比色法[/B] 色调标准贮备液 黄色液 重铬酸钾液(BP98用氯化铁) 红色液 氯化钴液 蓝色液 硫酸铜液 配成各种色调色号标准比色液共50种。 [B]2. 分光光度法 [/B] [B]易碳化物检查法 [/B]检查药物中含有的遇硫酸易碳化或易氧化而呈色的有机杂质。 对照品液 样品液 加硫酸5后,加供试品。 [B]炽灼残渣检查法[/B] 取供试品1.0~2.0g或个药品项下规定的重量,置已炽灼至恒重的坩埚中,精密称定,缓缓炽灼至完全碳化,放冷至室温;除另有规定外,加硫酸使湿润,低温加热至硫酸蒸气除尽后,在700~800炽灼使完全灰化,移至干燥器内,放冷至室温,精密称定,再在700~800炽灼至恒重,即得。残渣限量一般为0.1~0.2% 一般应使炽灼残渣量为1~2mg 若需将炽灼残渣留作重金属检查时,炽灼温度必须控制在500~600。 [B]干燥失重测定 [/B]1. 常压恒温干燥法 2. 干燥剂干燥法 3. 减压干燥法 [B]水分测定法 [/B][B]费休氏法 [/B] 本法是根据碘和二氧化硫在吡啶和甲醇溶液中能与水起定量反应的原理以测定水分。 [B]甲苯法[/B] 在加热状态下,甲苯夹带着水分蒸出,收集蒸出的水分测定。 [B]药物中特殊杂质检查 [/B] [B]一、物理法 [/B] [B]二、化学反应法 [/B](一)容量分析法 (二)重量分析法 (三)比色法和比浊法 [B]三、色谱法 [/B]1.纸色谱法 薄层色谱法 TLC是药典中最常用的特殊杂质限量检查方法。 1.在一定供试品及检查条件下,不允许有杂质斑点存在 2.以待测杂质对照品检测 3.将供试品稀释到适当浓度作为杂质对照品溶液 4.选用质量符合规定的与供试品相同的药物作为杂质对照品 [B]高效液相色谱法 [/B] [B][url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法 [/B] 1.面积归一化法 2.主成分自身对照法 3.内标法测定 4.内标法加校正因子法 5.外标法 有机溶剂残留量测定法 [B]分光光度法 紫外分光光度法 比色法 [url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]分光光度法[/B]
第十章 胺类药物的分析掌握盐酸普鲁卡因、盐酸利多卡因、盐酸丁卡因和对乙酰氨基酚的鉴别、杂质检查和含量测定方法。 掌握肾上腺素、盐酸去氧肾上腺素及其制剂的鉴别、杂质检查和含量测定方法。 第一节 盐酸普鲁卡因的分析有芳伯氨基特性,显重氮化-偶合反应。含酯键易水解,产物主要为对氨基苯甲酸(PABA)。脂烃胺侧链为叔胺氮原子,具有弱碱性。盐酸盐系白色结晶性粉末,具有一定的熔点,易溶于水和乙醇,难溶于有机溶剂。一、鉴别1.重氮化-偶合反应 分子结构中具有芳伯氨基或潜在芳伯氨基的药物,均可发生重氮化反应,生成的重氮盐可与碱性β-萘酚偶合生成有色的偶氮染料。 2.水解反应 取本品约0.1g,加水2ml溶解后,加10%氢氧化钠溶液1ml,即生成白色沉淀,加热变为油状物(普鲁卡因);继续加热,产生的蒸汽(二乙氨基乙醇)能使湿润的红色石蕊试纸变为蓝色;热至油状物消失后(生成可溶于水的对氨基苯甲酸钠),放冷,加酸酸化,即析出白色沉淀。此沉淀能溶于过量的盐酸。3.氯化物反应沉淀反应 在硝酸酸性条件下与硝酸银生成白色沉淀,沉淀加氨试液即溶解。氧化还原反应 加二氧化锰混匀,硫酸润湿,加热产生氯气,能使湿润淀粉碘化钾试纸显蓝色。4.红外光谱法二、特殊杂质检查普鲁卡因分子结构中有酯键,易发生水解反应。其注射液制备过程中受灭菌温度、时间、溶液pH值、贮藏时间以及光线和金属离子等因素的影响,可发生水解反应生成对氨基苯甲酸和二乙氨基醇。其中对氨基苯甲酸随贮藏时间的延长或高温加热,可进一步脱羧转化为苯胺,而苯胺又可被氧化为有色物,使注射液变黄,疗效下降,毒性增加。故中国药典90年版规定,本品注射液应检查水解产物对氨基苯甲酸,其限度不得超过1.2%。检查方法 薄层色谱法。供试品溶液2.5mg/ml与对照品溶液30μg/ml分别点于硅胶H薄层板上,展开后用对二甲氨基苯甲醛溶液喷雾显色。供试品溶液如显与对照品溶液相应的杂质斑点,其颜色与对照品溶液主斑点比较,不得更深。三、含量测定 分子结构中具有芳伯氨基,可用亚硝酸钠滴定法测定含量。
一阶导数分光光度法测定盐酸普鲁卡因溶液的含量刘素琴(江苏省金坛市人民医院,江苏 金坛 213200)联系电话:0519-2266680 E-mail:Liusuqin666@163.com 文章编号:04040399摘要 目的:改进盐酸普鲁卡因溶液的含量测定方法。 方法:以一阶导数光谱在308.0nm波长处谷—零间的振幅为定量依据,测定盐酸普鲁卡因溶液的含量。结果:盐酸普鲁卡因溶液浓度在5~30μg/mL范围内与一阶导数谷—零间的振幅呈良好的线性关系,r=0.9999,平均回收率为100.04%,RSD=0.43%。结论:方法简便、准确,可用于测定盐酸普鲁卡因溶液的含量。关键词 一阶导数; 分光光度法; 盐酸普鲁卡因 ;含量[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=32338]一阶导数分光光度法测定盐酸普鲁卡因溶液的含量[/url]
[align=center][b]盐酸雷尼替丁系统适用性试验-2015中国药典[/b][/align]色谱条件色谱柱:Kromasil 100-5-C18, 4.6*250mm货号:M05CLA25流动相A:酸盐缓冲液(取磷酸6.8ML置1900ML水中,加入50%氢氧化钠溶液8.6ML,加水至2000ML,用磷酸或50%氢氧化钠溶液调节pH 值至7.1±0.05):乙腈=98:2流动相B:磷酸盐缓冲液-乙腈(78 : 22)梯度程序:[img=,617,209]https://ng1.17img.cn/bbsfiles/images/2018/11/201811131416404930_2830_2428063_3.png!w617x209.jpg[/img]流速:1.5ML/min柱温:35℃波长:230nm进样量:10μL[img=,534,227]https://ng1.17img.cn/bbsfiles/images/2018/11/201811131416589880_2883_2428063_3.png!w534x227.jpg[/img]结论:1. 出峰顺序为杂质I,雷尼替丁2. 杂质I的相对保留时间约为0.853. 雷尼替丁峰和杂质I峰的分离度大于4.0以上指标都符合药典要求。本应用来源于Kromasil微信公众号
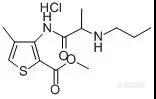
第一章 麻醉药 第一节 全身麻醉药 1、 吸入麻醉药 氟烷:2-溴-2-氯-1,1,1-三氟乙烷 起效、苏醒快、作用弱,全麻及诱导麻醉 性质:1、氧瓶燃烧 2、加入硫酸,沉于底部。 甲氧氟烷浮于硫酸上层。 甲氧氟烷:麻醉作用和肌松作用比氟烷强,诱导期长。 恩氟烷:新型高效吸入麻醉药,麻醉肌松作用强,起效快,临床常用。 异氟烷为异构体 乙醚:氧化后生成过氧化物对呼吸道有刺激作用。 2、 静脉麻醉药 盐酸氯胺酮:2-(2-氯苯基)-2-(甲氨基)环已酮盐酸盐 2个旋光异构体,用外消旋体 作用快、短、副作用小,诱导期短。 分离麻醉 羟丁酸钠: 作用弱、慢、毒性小。 --OH 1、三氯化铁红色 2、硝酸铈铵橙红色 第二节 局部麻醉药 一、 对氨基苯甲酸酯类 构效关系:1、苯环上增加共他取代基时,因增加空间位阻酯基水解减慢,局麻作用增强。 2、苯环上氨基的烃以烷基取代,增强局麻作用。 丁卡因 3、改变侧链氨基的取代基,有些作用增强。 布他卡因 4、羧酸中的氧原子若以电子等排体硫原子替代(硫卡因),脂溶性增大,作用增强。 盐酸普鲁卡因:4-氨基苯甲酸-2-(二乙氨基)乙酯盐酸盐 不宜表面麻醉 性质:1、加氢氧化钠有油状普鲁卡因析出。 干燥稳定,避光 PH=3-3.5最稳定。 2、酯键:水溶液水解失活:对氨基苯甲酸及二乙氨基乙醇,前者氧化变色 3、叔胺结构:碘、苦味酸等呈色 4、芳伯氨反应: 盐酸丁卡因:4-(丁氨基)苯甲酸-2-(二甲氨基)乙酯盐酸盐 作用:用于粘膜麻醉,与普鲁卡因一起成为应用最广的局麻药。 二、酰胺类: 盐酸利多卡因:N-(2,6-二甲基苯基)-2-(二乙氨基)-乙酰胺盐酸盐-水合物 性质:酰胺键较酯键稳定,酸碱中均较稳定 。作用强,可用于表面麻醉 布比卡因:1-丁基-N-(2,6-二甲苯基)-2-哌啶甲酰胺盐酸盐 长效局麻药,用于浸润麻醉。 三、氨基酮类及氨基醚类
各位高手可有盐酸罗哌卡因红外谱图啊?还有顺便问下,药典上说药品1-2mg氯化钾200mg,其中[color=#d40a00]1-2mg[/color]和[color=#d40a00]200mg[/color]只是个用质量来描述固体样品量的方式吧,概数,无需用天平来称量、量化吧?正如液体样品可用体积的量来描述,L,ml或滴。是这样理解的吧?
[B]杂质对钛白粉有何影响及提高其白度的方法[/B] 对于颜料钛白粉,除了生产上采取一些措施来提高白度以外,关键还在于杂质的去除,杂质对白度的不良影响是很大的,杂质去除得越彻底,产品白度就越高,这对用在涂料中的意义就更大。 在钛白粉生产过程中,如果杂质去除不彻底,当用高温煅烧时,很多杂质元素如铁、锰、钒、铅、铬、钴、铈铜、镉、镍、钼等以氧化物状态存在,这些带色的氧化物就表现出各种色相,使整个钛白粉的色相受到影响而不纯白,以致大大影响了产品的质量。因此,必须采用多种方法除去杂质。 1选矿除杂质 要除去杂质,首先就要选矿。因为任何钛铁矿一般都混杂有不少脉石和共生、伴生、复合的其它矿物。选矿就是利用矿物不同的物理化学性质,采用各种有效方法,将钛铁矿与它们分离。例如摇床的重力选矿,可以除去铸铁矿中的大部分脉石,再用磁选机进行磁选,让矿物通过磁场,由于钛铁矿是导磁率高、能被磁铁吸引而本身不能吸铁、可磁化又可去磁的顺磁性矿物,而能磁盘吸引,其它导磁率低的非钛铁矿,不能被磁盘吸引而得到分离。 2除不溶性杂质 硫酸法生产钛白粉,由酸解浸取得到的是浑浊不清的钛液,其不溶性杂质主要是颗粒较大的机械杂质和颗粒较小的胶体杂质。机械杂质是未起反应的钛铁矿物,属于粗分散状态,很容易沉析下来;胶体杂质主要是硅酸铝酸盐等,由于颗粒小,吸附H而带有相同的正电荷,由于同种电荷相斥,胶粒很难接近成比较大的颗粒而沉淀下来,因而具有较高的稳定性。解决的办法是用带负电荷的改性的聚丙烯酰胺胶体进行电性中和,使胶体粒子凝聚沉降而除去。但是由于胶体沉降不完全,经过硫酸亚铁的过滤后,仍有一些穿滤而存在于钛液中,必须用带有木炭粉为助滤层的板框压滤机进行压滤,直到检测滤液的澄清度合格为止. 3除铁杂质 在钛铁矿中,非钛杂质最多的是铁,并以二价和三价两种状态存在。将钛铁矿与硫酸作用,即生成FeSO和(SO)。由于FeSO只有在pH值大于6.5时才开始水解,因此在钛液水解过程中,因钛液的酸性较大,FeSO就始终保持溶解状态,在偏钛酸洗涤时得以除去。而Fe(SO)在pH值为1.7的酸性溶液中即开始水解生成Fe(OH)沉淀,其混杂在偏钛酸中,煅烧时即生成红棕色的FeO而使成品不够纯白。所以应用铁屑把Fe(SO)还原为FeSO。为了保证钛液中的三价铁全部还原为二价铁,还原反应还应略为过度,此时钛液中就有小部分四价钛还原为三价钛。三价钛的存在就可保证三价铁还原完全,可避免三价铁水解生成Fe(OH)进而影响产品白度。经过还原,钛液中全部是FeSO,此时冷冻钛液,Fe即达到过饱和状态而大量结晶析出,过滤即可除去大部分铁钛液中剩下的未结晶的FeSO,待水解生成偏钛酸进行水洗时,用水洗除去。由于滤饼的FeO质量分数超过90×10时,产品白度将受到影响。所以可采取漂白措施使FeO质量分数降低到30×10,并进一步除去痕量的钒、铬、铜等杂质。
液相分析时,对照浓度一般是多少,根据什么来定?杂质对照呢?
电子级别的试剂如盐酸是如何做到杂质很低的,工艺上有什么联系?
现在有一课题,是做氨基酸原料药的杂质研究,这到底是指氨基酸类杂质,还是所有可能出现的杂质,有点蒙圈,感觉好难
本人在做氨基酸分析,用的是黄酒中氨基酸的测定方法行业标准编制说明 标准,十八种氨基酸混标进样,由于仪器限制,使用的是二元梯度,标准是三元,两者的区别只是峰的分离度。但是实验中总是有杂质峰,而且重复性很好,保留时间、峰面积是一致的,怀疑是衍生副产物。单标进样仍然有,而且几乎是一个单标对应一个杂质峰。并不是所有杂质峰都出现,说明不是样品、流动相的问题。认为问题就出在衍生过程中,有做过的是否也由这些杂质峰?应该如何处理这些杂质峰?
不知哪位做过半胱氨酸盐酸盐的其他氨基酸。供试品溶液的制备 :取本品,加2%N-乙基顺丁烯二酰亚胺溶液制成每1ml中含4mg的溶液,作为供试品溶液;对照液的制备:另精密称取胱氨酸对照品10mg,加0.1mol/L盐酸溶液3ml,超声,加水适量使溶解,并加水使成100ml,摇匀,精密量取1ml,加2%N-乙基顺丁烯二酰亚胺溶液4ml,摇匀,作为对照品溶液。这里要问为什么对照液不直接拿供试品溶液稀释呢?而用胱氨酸对照品呢??不明白[em06] [em06]
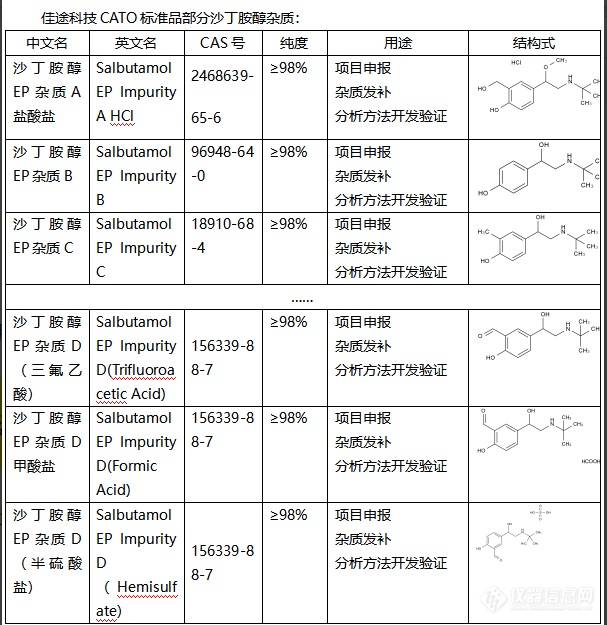
沙丁胺醇是一种药物,主要用于治疗哮喘和慢性阻塞性肺病等呼吸系统疾病。在药品制备过程中,可能会产生一些不同结构的化合物,这些化合物就被称为杂质。杂质可能会对药品的品质、安全性、效力和稳定性等产生影响。根据产生的差异,杂质的作用可以具体表现如下:1. 影响药品的稳定性:杂质可能导致药品在贮存过程中发生化学变化,从而影响药品的稳定性。2. 影响药品的安全性:如果杂质对人体有毒性,那么杂质的存在可能降低药品的安全性。3. 影响药品的效力:杂质可能与药品的有效成分发生竞争,导致药品的效力降低。4. 影响药品的品质:杂质可能改变药品的外观、颜色、溶解性等物理性质,影响药品的品质。CATO标准品制药行业对药品的杂质进行严格控制,保障药品的质量和患者的用药安全。[img=,607,625]https://ng1.17img.cn/bbsfiles/images/2024/02/202402021631332927_8650_6381668_3.png!w607x625.jpg[/img]
请问论坛里的专家、朋友用ICP-OES如何检测浓盐酸(GR)、浓硫酸(GR)中的杂质元素,有人检测过吗?
请问论坛中的各位专家、朋友,你们用ICP-OES检测过浓盐酸(GR),浓硫酸(GR)中的杂质元素吗?如何检测的?

盐酸可乐定的测定和盐酸丁咯地尔片含量的测定http://ng1.17img.cn/bbsfiles/images/2009/11/200911021833_180179_1896702_3.jpg
水质固定剂包括了盐酸和硝酸,特别是盐酸,因挥发性太强,导致外包的封口带(玻璃试剂瓶用封口带封装)内滞留了一些黄色冷凝液,味道特别呛。放这些固定剂的整理箱,每次开盖后,味道也很难受。大家是怎么处理的?
复方盐酸阿替卡因注射液为复方制剂,是盐酸阿替卡因与肾上腺素的灭菌水溶液,作为口腔用局部麻醉剂,适用于涉及切骨术及粘膜切开的外科手术过程。[img=,144,61]https://ng1.17img.cn/bbsfiles/images/2019/03/201903191545418661_4518_2222981_3.jpg!w144x61.jpg[/img][color=black] [/color][color=#3e3e3e]肾上腺素 L(-)-Epinephrine M.W. : 183.2 [/color][b]在国家药品标准(YBH17082004-2015Z)[/b]中,在对复方盐酸阿替卡因注射液中肾上腺素进行分析时,使用[b]甲醇-水[/b]进行梯度洗脱,但由于[b]肾上腺素极性较强[/b],即使初始梯度为纯水相条件,肾上腺素仍紧邻死时间出峰,[b]保留不佳,易受到溶剂峰干扰,无法进行准确定量。[/b]我们分别尝试使用反相柱CAPCELL PAK C18 MGII加离子对试剂,以及直接使用离子交换色谱柱CAPCELL PAK SCX UG80两种方式,对复方盐酸阿替卡因注射液中肾上腺素和硫酸肾上腺素进行保留分析(复方盐酸阿替卡因注射液由客户提供)。CAPCELL PAK C18 MGII液相色谱柱,其采用高纯度硅胶作为基质,通过减少硅胶微细孔的数量来增大有效比表面积;并且采用新包被技术Ultimate Polymer Coating,实现了对硅醇基极大程度的封锁,兼具分离性能和普适性能,通用性非常好。CAPCELL PAK SCX UG80是强阳离子交换柱,使用高纯度硅胶,填料中金属杂质很少,使配位化合物的吸附得到了极大程度抑制,兼具聚合物和硅胶填料的优点。[b][color=#0070c0]实验方法[/color][color=#0070c0]方法一[/color][color=#0070c0]使用[/color][color=#0070c0]CAPCELL PAK C18MGII[/color][color=#0070c0]色谱柱[/color][color=#0070c0]+[/color][color=#0070c0]离子对试剂[/color][/b]如图1,对肾上腺素对照品溶液进行分析,肾上腺素主峰保留时间为5.69 min,拖尾因子为1.19,理论塔板数为12538。在相同色谱条件下,尝试对亚硫酸肾上腺素标准品及注射液中的亚硫酸肾上腺素进行分析。如图3,亚硫酸肾上腺素标准品溶液能够得到良好分析结果,注射液(客户提供的样品)中未明显见亚硫酸肾上腺素出峰,保留时间为3.55min,拖尾因子为1.14,理论塔板数为14955。[b][color=#0070c0]方法二[/color][color=#0070c0] CAPCELL PAK SCX UG80[/color][color=#0070c0]色谱柱[/color][/b][color=#000000]考虑到使用离子对试剂的流动相条件具有流动相配制麻烦、有损色谱柱寿命、平衡时间长等缺点,我们也尝试使用键合磺酸基团的强阳离子交换柱 ——CAPCELL PAK SCX UG80进行分析。[/color][color=#000000]如图4,在流动相中添加磷酸二氢铵,通过对盐浓度进行调整,在5 mmol/L磷酸二氢铵(磷酸调pH=2.5)条件下,亚硫酸肾上腺素保留时间为3.32 min,然而出现峰形拖尾现象,拖尾因子为2.0,不如CAPCELL PAK C18 MGII色谱柱添加离子对试剂所得分析结果好。[/color][align=center][/align][align=left][img=,400,284]https://ng1.17img.cn/bbsfiles/images/2019/03/201903191547381421_926_2222981_3.jpg!w584x416.jpg[/img] [img=,400,276]https://ng1.17img.cn/bbsfiles/images/2019/03/201903191548310561_8067_2222981_3.jpg!w572x395.jpg[/img][/align][align=left][img=,400,166]https://ng1.17img.cn/bbsfiles/images/2019/03/201903191547576748_522_2222981_3.jpg!w612x254.jpg[/img] [img=,400,167]https://ng1.17img.cn/bbsfiles/images/2019/03/201903191548525761_4184_2222981_3.jpg!w624x262.jpg[/img][/align][align=left]图1 MGII分析肾上腺素对照品溶液结果(离子对条件) 图2 MGII分析注射液结果(离子对条件)[/align][align=center][/align][img=,400,258]https://ng1.17img.cn/bbsfiles/images/2019/03/201903191550354951_2539_2222981_3.jpg!w644x416.jpg[/img] [img=,400,250]https://ng1.17img.cn/bbsfiles/images/2019/03/201903191555486850_3396_2222981_3.jpg!w644x403.jpg[/img][img=,400,147]https://ng1.17img.cn/bbsfiles/images/2019/03/201903191551260881_9828_2222981_3.jpg!w696x256.jpg[/img] [img=,400,164]https://ng1.17img.cn/bbsfiles/images/2019/03/201903191556163846_9739_2222981_3.jpg!w632x260.jpg[/img][align=left]图3 MGII分析亚硫酸肾上腺素标准品和注射液结果(离子对条件) 图4 SCX UG80分析亚硫酸肾上腺素对照品溶液和供试品溶液[/align][align=left][/align][align=left]综上实验结果,使用中等极性色谱柱CAPCELL PAK C18 MGII S5 4.6 mm i.d. × 250 mm,在流动相中添加5 mM辛烷磺酸钠、30°C柱温条件下进行梯度洗脱,能够实现复方盐酸阿替卡因注射液中肾上腺素和亚硫酸肾上腺素的良好保留与分析。[/align][b][color=#0070c0][/color][/b][align=left][b][color=#0070c0] [/color][color=#0070c0] [/color][/b][/align]
提供盐酸曲美他嗪欧洲药典杂质标准品Trimetazidine for system suitabilityImp. A (EP) as Dihydrochloride: 1-(3,4,5- Trimethoxybenzyl)piperazine DihydrochlorideImp. B (EP): 1,4-Bis(2,3,4-trimethoxy-benzyl)piperazineImp. C (EP): 2,3,4-TrimethoxybenzaldehydeImp. D (EP): (2,3,4-Trimethoxyphenyl)methanolImp. E (EP) as Dihydrochloride: 1-(2,4,5- Trimethoxybenzyl)piperazine DihydrochlorideImp. F (EP) as Dihydrochloride: 1-(2,4,6-Tri- methoxybenzyl)piperazine DihydrochlorideImp. G (EP) as Hexahydrate: Piperazine HexahydrateImp. H (EP): Ethyl 4-(2,3,4-Trimethoxybenzyl)- piperazine-1-carboxylate1-Formyl-4-(2,3,4-trimethoxybenzyl)piperazine Hydrochloride
对于硝酸,盐酸,双氧水等试剂,我该如何稀释做测其中空白杂质含量?
标定盐酸标准滴定溶液的不确定度分析 作者:吴文英 张春雨 唐惠兰 来源:中华医学研究杂志 在理化分析过程中,一切测量结果都不可避免地具有不确定度。盐酸标准溶液是常用化学定量参比物质,其标定值的准确性直接影响常规分析质量。笔者以GB/T601《滴定分析(容量分析)用标准液的制备》为依据配制并标定盐酸根据JJF1059-1999《测定不确定度评定与表示》分析其测量不确定度。简述由标定过程中得到的不确定度。 1 实验部分 1.1 测定方法[1,2] 准确称量270℃~300℃干燥至恒重的基准碳酸钠(99.95%~100.05%)约0.2g左右,电子分析天平(精度为0.1mg),置于三角瓶中,加入50ml水使之溶解,加指示剂,用盐酸标准液滴定至终点同时作试剂空白实验。 1.2 主要计量仪器与试剂 电了分析天平:AG204;酸式滴定管:50ml A级。 1.3 建立数学模型 C=m (V1-V2)×0.05300 式中 C:盐酸标准滴定溶液的浓度(mol/L);m:基准无水碳酸钠的质量(g);V1:盐酸标准滴定溶液用量(ml);V2:试剂空白实验中盐酸标准滴定溶液用量(ml);0.05300:与1.00ml盐酸标准溶液[C(HCl)=1.000mol/L]相当于以克表示的无水碳酸钠的质量。 1.4 盐酸标准滴定溶液的标定结果 为获得标准溶液重复测量的不确定度分量,对同一标准溶液进行8次独立的标定。测定数据见表1。 表1 盐酸标准滴定溶液的标定结果 略 2 测量不确定度来源 从检测过程和数学模型分析,标定盐酸标准溶液的不确定度主要来源,由四个方面所引起。(1)测量的重复性(A类不确定度);(2)基准无水碳酸钠的纯度;(3)测量使用的电子分析天平及量具;(4)其他相关常数。 3 测量不确定度分析 3.1 A类不确定度的分析 利用表1中的测量结果,按照A类评定测量重复性的标准不确定度。具体计算过程:重复测量的平均值计算式:=1 n∑8 i=1xi=0.09951mol/L 单次测量的标准差按贝塞尔公式计算s(x)为 s(x)=∑8 i=1(xi-)2 n-1=0.0001555mol/L 的标准差s()为 s()=s(x) n=0.000155 8=0.0000548mol/L=5.48×10-5mol/L 由测量重复性引起的相对标准不确定度为U(x):0.0000548/0.09951=0.055%。 3.2 B类不确定度分析 3.2.1 基准碳酸钠的纯度 基准碳酸钠的纯度为1.0000±0.0005,视为矩形分布0.00053=0.00029,则标准不确定度为:由基准碳酸钠的纯度引入的相对不确定度u(p)为:0.029%。 3.2.2 天平称量所引入的标准不确定度 干燥器与天平称量仓内均放置同质硅胶,视为相同湿度,称量时无吸潮。电子天平检定证书标出线性为上0.2mg;可视为矩形分布,则标准不确定度为:因为称量采用的是减量法,故称量的标准不确定度为0.2mg /3=0.12mg:因为称量采用的是减量法,故称量的标准不确定度为:2×0.122=0.17mg,则由称量引入的相对标准不确定度u(m)为:0.17mg/0.2018g=0.084%。 3.2.3 标定体积的不确定度 (1)滴定管的校准:滴定使用50ml酸式滴定管(A级),按照检定规程,其最大允许误差为±0.05ml,相对允许误差为±0.1%,按照矩形分布,则滴定体积的相对标准不确定度u(V)为:u(V)=0.1%/3=0.0577%。(2)环境温度:实验环境在空调条件下,室温近似20℃。温度在20℃左右,标准溶液的温度补正值非常小,对实验结果影响可忽略不计,所以在不确定度分析中不把一温度影响引起的不确定度列入考虑范围。(3)滴定终点的判断:终点时的误差±0.05ml(1滴的体积),两点分布,现由终点分布判断引入的标准不确定度为0.05ml:相对标准不确定度为0.05ml/38.32ml=0.13%标定体积的影响引入相对标准不确定度U(V)为0.0572+0.132=0.142%。 3.2.4 其他常数 基准无水碳酸钠摩尔质量引起的标准不确定度很小,可以忽略。 4 合成标准不确定度 测量重复性、基准无水碳酸钠的纯度、天平称量、标定体积等的不确定度相互独立,故将上述数据合成得盐酸的相对合成标准不确定度U(C)为0.0552+0.0292+0.0842+0.1422=0.176%。 5 扩展不确定度 实验测得盐酸标准溶液浓度为0.09951mol/L,则测量结果的合成标准不确定度U(C)=0.09951mol/L×0.176%=0.000175mol/L。若取包含因子K=2,得测量结果的扩展不确定度U=2U(C)=0.00035mol/L。 6 测量结果的表示 盐酸标准滴定溶液的浓度可表示为:(0.09951±0.00035mol/L,K=2)。 【参考文献】 1 姚正堂,将已峰.奶制品中蛋白质测定的不确定度分析.中华医学研究杂志,2005,5(6):6. 2 国家技术监督局.JJF1059-1999测量不确定度与表示.北京:中国计量出版社,1997,81. 作者单位: 214171 江苏无锡,无锡市惠山区疾病预防控制中心







 我要推广仪器
我要推广仪器
 下载APP
下载APP