如何使用卡尔?费休试剂对酒石酸钠二水合物进行标定 一、准备工作 1.仪器准备 首先要准备好滴定装置,这就像准备好做饭的锅碗瓢盆一样重要。卡尔?费休滴定仪得检查好,确保它能正常工作。比如滴定管要畅通无阻,而且刻度要清晰准确,这样才能精确量取卡尔?费休试剂。还有搅拌装置也要正常运转,因为在滴定过程中,良好的搅拌能让反应更充分。 2.试剂准备 卡尔?费休试剂得是新鲜配制或者妥善保存且未过期的。酒石酸钠二水合物要确保是干燥、纯净的。称取酒石酸钠二水合物的天平也要精准,就像我们称东西的时候得用个准秤一样。天平要提前校准好,精确到小数点后几位,这可关系到标定结果的准确性呢。 3.环境控制 标定的环境很关键。要在一个相对湿度较低的环境中进行,因为卡尔?费休试剂很容易吸收空气中的水分,如果环境湿度大,就会干扰标定结果。就好比在潮湿的天气里晒东西,东西很难晒干是一个道理。理想的相对湿度最好在 40% - 60% 之间。 二、具体标定步骤 1.称取酒石酸钠二水合物 用经过校准的天平准确称取一定量的酒石酸钠二水合物。这个量要合适,不能太多也不能太少。比如说可以称取 0.2 - 0.3 克左右(具体量可以根据实际情况和仪器的精度调整)。把称好的酒石酸钠二水合物小心地放入滴定容器中,就像把宝贝小心翼翼地放进盒子里一样。 2.开始滴定 然后往滴定容器中加入适量的溶剂,这个溶剂要能溶解酒石酸钠二水合物,并且不会和卡尔?费休试剂发生反应。开启搅拌装置,让酒石酸钠二水合物充分溶解。 接下来就可以开始用卡尔?费休试剂进行滴定了。慢慢打开滴定管的阀门,让卡尔?费休试剂一滴一滴地滴入滴定容器中。一边滴一边观察滴定仪上的读数或者颜色变化(如果是用有颜色指示的卡尔?费休试剂的话)。这个过程要特别仔细,就像给花浇水一样,一滴一滴地浇,不能一下子倒很多。 3.确定滴定终点 当达到滴定终点时,就要停止滴定。如果是用电位滴定仪,会有电位的突变来指示终点;如果是用目视法,可能会看到颜色的明显变化。这个终点的判断要准确,一旦判断失误,整个标定结果就错了。就像跑步比赛,冲线的那一刻判断错了,比赛结果就不对了。 4.计算卡尔?费休试剂的浓度 根据酒石酸钠二水合物的质量、它里面结晶水的含量(酒石酸钠二水合物中结晶水的摩尔质量是固定的,可以查出来)以及滴定所消耗的卡尔?费休试剂的体积,就可以计算出卡尔?费休试剂的浓度了。计算的时候要仔细,可不能算错数哦。
买了硝酸钯二水合物,如何溶解啊?
哪些因素会影响酒石酸钠二水合物的溶解平衡 一、温度方面 1.温度高低 温度就像一个调皮的小助手,对溶解平衡影响可大了。如果温度升高,就像给酒石酸钠二水合物的溶解加了把劲儿。因为温度高了,分子运动就变得更活跃了,溶剂分子就更容易把溶质分子(酒石酸钠二水合物)包围起来,让它溶解。就好比天气热的时候,糖在水里溶解得更快更多一样。反过来,如果温度降低,分子运动就慢下来了,酒石酸钠二水合物的溶解能力也会跟着下降,可能本来溶解了不少,温度一低就有一部分析出来了。 二、溶剂的性质 1.溶剂种类 不同的溶剂就像不同的小房子,对酒石酸钠二水合物的容纳能力不一样。如果是一种和酒石酸钠二水合物 “合得来” 的溶剂,那它就容易溶解。比如说,在水里酒石酸钠二水合物能溶解得挺好,但是如果换一种有机溶剂,像乙醇之类的,可能溶解的量就少多了,甚至几乎不溶解。这是因为酒石酸钠二水合物分子和水分子之间的相互作用力比较强,能让它很好地分散在水中,而和乙醇分子的相互作用力就弱很多。 2.溶剂的量 溶剂的量也很关键。如果溶剂很多,就像有很大的空间来容纳酒石酸钠二水合物,那它就能溶解更多的溶质。就像你有一个大杯子装水,能放很多糖溶解在里面;如果杯子很小,水少,能溶解的糖也就少了。不过呢,这里有个极限,就是达到饱和状态后,再增加溶剂也不能再溶解更多的酒石酸钠二水合物了。 三、溶质的状态 1.溶质的颗粒大小 酒石酸钠二水合物本身颗粒的大小也会影响溶解平衡。如果颗粒很大,就像一个大石块,溶剂分子要把它慢慢 “啃碎” 才能溶解,溶解的速度就慢。要是颗粒很细小,就像沙子一样,溶剂分子就能很快把它们包围起来溶解掉。不过这主要影响的是溶解的速度,当时间足够长的时候,最终达到的溶解平衡状态是一样的,只是颗粒小的时候会更快达到平衡。 2.溶质的纯度 纯度高的酒石酸钠二水合物溶解起来比较单纯。如果里面混有杂质,这些杂质就像捣乱的小坏蛋。比如说,杂质可能会占据溶剂分子和酒石酸钠二水合物分子结合的位置,或者改变溶液的性质,从而影响酒石酸钠二水合物的溶解平衡。可能会使它溶解得少一些,或者达到平衡的速度变慢。 四、搅拌情况 1.搅拌与否 搅拌就像给溶液做按摩一样。如果搅拌溶液,就能让酒石酸钠二水合物周围的溶剂不断更新,这样溶剂分子就能更快地接触到溶质分子,加快溶解的速度。不搅拌的话,溶质周围的溶剂很快就饱和了,新的溶剂分子过不来,溶解就慢。不过搅拌不会改变最终的溶解平衡状态,只是影响达到平衡的快慢。
用户如果购买了氯唑青霉素钠水合物(氯唑西林钠,邻氯青霉素钠) 标准品,进行定性分析时没有问题,但是里面没有明确是一水化合物还是二水化合物等,只是 氯唑青霉素钠xH2O,如题,这个标准品配成溶液后如何进行定量分析?
如何判断酒石酸钠二水合物是否已经完全溶解 一、直接观察法 1.整体观察 你就眼睛直勾勾地看着装着酒石酸钠二水合物和溶剂的容器呗。如果溶液看起来清清爽爽的,没有那种白花花的小颗粒或者小块块在里面晃悠,那很可能就已经完全溶解了。就像你冲一杯白糖水,白糖完全化了的时候,水就是均匀的,看不到白糖颗粒了。不过呢,你得仔细看,有时候小颗粒可能躲在角落里或者附着在容器壁上,可别把它们给漏掉了。 2.透光观察 把容器拿到有光的地方,从侧面看过去。如果溶液是均匀透明或者半透明的,光线能很顺畅地穿过,没有被什么东西挡住,那也说明溶解得差不多了。要是还有没溶解的东西,光就会在那些颗粒上散射,看起来就会有一些浑浊或者暗影。这就好比你透过干净的玻璃看东西很清楚,但是玻璃上有脏东西的时候,看东西就模糊了。 二、搅拌相关的判断法 1.搅拌时的观察 在搅拌溶液的时候,你看溶液里的情况。如果搅拌了一会儿,溶液里一直都没有出现那种顽固的小颗粒或者沉淀,而且溶液的流动很顺畅,就像水一样平滑,那可能是完全溶解了。要是还有没溶解的东西,搅拌的时候它们就会像调皮的小豆子一样在溶液里滚来滚去,很容易被发现。 2.搅拌停止后的观察 搅拌停下来之后,这时候最能看出问题了。等个一小会儿,大概一分钟左右,看看溶液里有没有东西又冒出来了。如果溶液安安静静的,没有沉淀或者小颗粒从溶液里跑出来,那基本可以确定是完全溶解了。这就像你搅和一盆沙子和水,一停搅拌,沙子就会沉下去,如果不沉,就说明沙子已经均匀地分散在水里了。 三、时间检验法 1.延长观察时间 有时候溶解得比较慢,你不能太着急。可以多等几分钟,就这么盯着溶液看。如果好几分钟过去了,溶液还是老样子,清清爽爽、安安静静的,那肯定就是完全溶解了。这就像炖肉一样,你得给它足够的时间才能炖烂,酒石酸钠二水合物溶解也需要一点时间来确定是不是真的完全溶解了。
如何确定酒石酸钠二水合物的溶解平衡是否已经建立 一、观察溶液外观 1.颗粒状态 首先呢,还是用眼睛好好瞅一瞅溶液。如果溶液里已经好长一段时间看不到酒石酸钠二水合物的小颗粒了,不管是在溶液中间,还是在容器的底部或者壁上,都没有那种白花花的东西,这是个好迹象。就像你等一杯咖啡里的糖完全溶解一样,看不到糖粒了,才有可能达到溶解平衡。不过这还只是第一步,光看这个还不能完全确定。 2.溶液的均匀性 除了看有没有颗粒,还要看溶液是不是均匀的。你可以把溶液轻轻晃一晃,如果溶液看起来就像水一样清澈、均匀,没有哪里浓哪里淡的感觉,也没有什么浑浊的地方,那说明它很可能接近或者已经达到溶解平衡了。就像你调果汁,调好之后果汁是均匀的颜色和浓度,没有沉淀或者分层,那这个果汁就调好了,酒石酸钠二水合物的溶液也是这个道理。 二、持续观察一段时间 1.短时间内无变化 你得盯着溶液看一会儿,比如说看个 5 - 10 分钟。如果在这几分钟里,溶液的样子没有任何变化,既没有新的小颗粒冒出来,也没有溶液变浑浊或者变清澈,那就有可能是达到溶解平衡了。这就像你在等一个蛋糕烤熟,你得等一会儿看它有没有变化,如果一直保持一个样子,那可能就烤好了。 2.延长观察时间 为了更保险,你可以观察得更久一点,比如 30 分钟甚至一个小时。如果这么长的时间里溶液还是老样子,那基本上就可以确定溶解平衡已经建立了。因为如果还没有达到平衡,这么长的时间里溶液的状态肯定会发生变化的,要么有更多的酒石酸钠二水合物溶解,要么会有一些已经溶解的重新析出来。 三、进行简单测试(如果可能) 1.取少量溶液测试 如果条件允许的话,你可以用个小滴管取一点点溶液出来。然后用一些简单的方法来检测溶液里酒石酸钠二水合物的含量是不是固定的。比如说,如果有办法检测溶液里钠离子或者酒石酸根离子的浓度,你可以做个简单的测试。要是你取几次溶液,每次检测出来的浓度都一样,那这也能说明溶解平衡已经建立了。不过这种方法可能需要一些特殊的仪器或者试剂,不是所有情况都能做的。 总之,确定酒石酸钠二水合物的溶解平衡是否建立,要多方面观察,不能只看一点就下结论。
[size=3][size=4]我需要做乙酰丙酸的标准曲线,看别的文章后,按他写的方法试过,完全不是他说的结果,请问哪位做过乙酰丙酸的标准曲线,用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url],可否将方法相告!不胜感激![/size][/size]
埃索美拉唑镁(esomeprazole magnesium),化学名(S)-(-)5-甲氧基-2--1H-苯并咪唑镁盐,由瑞典AstraZeneca公司研发,2004年在我国上市。临床主要用于治疗胃酸分泌过多引起的胃溃疡、十二指肠溃疡及反流性食管炎等消化系统疾病。 本文拟采用湿法敞开消解法进行样品前处理,利用火焰原子吸收光谱法测定埃索美拉唑镁二水合物的Mg含量。1 仪器与试剂1.1 仪器与条件美国Perkin Elmer原子吸收光谱仪(型号:AAnalyst 800);镁空心阴极灯(北京有色金属研究总院);石墨炉原子吸收光谱仪测试条件见表1;MS105DU型Mettler Toledo电子天平;Advantage A10 Milli-Q纯水器。表1 原子吸收光谱仪主要测试条件 测定参数 设定值 测定波长 285.2 nm 狭缝宽度 0.7 nm 工作灯电流 6 mA 空气流量 17 L×min-1 乙炔流量 2 L×min-1 1.2 试剂盐酸(MOS级,天津市风船化学试剂科技有限公司);GSB G 62005-90镁标准储备溶液(1000 mg×mL-1,国家钢铁材料测试中心钢铁研究总院,中国);Milli Q超纯水(18.2 MW×cm);50 mg×mL-1的La溶液由光谱纯La2O3(99.99 %,上海试剂厂,上海)配制而成。2 方法与结果2.1 标准工作溶液的配制及测定从镁标准储备溶液(1000 mg×mL-1)移取一定的量,利用2 %盐酸溶液逐级稀释分别配制成浓度为0、0.1、0.3和0.5 mg×mL-1的标准工作溶液(都含有2 mL的La溶液)。按照浓度从稀到浓的顺序,利用火焰原子吸收光谱仪分别测定其吸光度值。仪器自动绘制工作曲线,获得的标准曲线的线性方程为:Y=0.00618+0.01563*X,R=0.99901上式中:Y:代表吸光度; X:代表待测溶液的浓度; R:代表工作曲线的线性系数。2.2 消解法称取0.0500 g左右的样品到玻璃烧杯中,加入3 mL的MOS级HCl,加盖置于电热板上加热消解。样品很快溶解完全,取下、冷却,定容到100 mL容量瓶中,再50倍稀释,同时加入2 mL的La溶液(50mg×mL-1),定容到50 mL容量瓶中待测,同时做样品空白。2.3 测试结果样品前处理完成之后,仪器先预热稳定半小时,再按照仪器的操作规程进行样品的分析。样品分析结果见表4。表4 样品中镁的分析结果及样品加标回收率 样 品 样品中镁的含量(%) 回收率(%) 1 3.44 102.5 2 3.42 101.0 3 3.43 98.9 结论:通过上面的实验结果可以看出:采用直接敞开湿法消解法和火焰原子吸收光谱法测定埃索美拉唑镁中镁的含量,方法简单、准确、灵敏,可用于埃索美拉唑镁原料药有关物质及含量的测定以及质量控制。
求助:最近要检测5-氨基乙酰丙酸(固体)含量,可是找不到药典中的检测方法,希望高手帮忙,没有药典,有企业标准也行谢谢找了一篇国标,测尿液中的5-氨基乙酰丙酸,不知道用来测定固体的含量可不可靠,有没有可信度谢谢
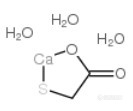
巯基乙酸钙盐三水合物 CAS号:5793-98-6 分子式:C2H8CaO5S 分子量 184 结构式http://ng1.17img.cn/bbsfiles/images/2017/10/2016042817011772_01_1490617_3.png 《化妆品安全技术规范》(2015年版)当中,3.9巯基乙酸第三法——化学滴定法的反应方程如下:https://ng1.17img.cn/bbsfiles/images/2017/01/201701191701_670059_1490617_3.png 原理是https://ng1.17img.cn/bbsfiles/images/2016/04/201604281715_591808_1490617_3.png 该方法的适用范围中这样描述:本方法适用于脱毛类、烫发类和其他发用类化妆品中巯基乙酸及其盐类和酯类含量的测定。客户委托了一款产品,要求按照巯基乙酸钙含量出报告,含量计算公式中有一个系数0.184,描述是1mmol碘溶液相当于巯基乙酸钙的克数,这样显然其指的巯基乙酸钙不是CAS:814-71-1 分子式C4H6CaO4S2(分子量222.3),不知道巯基乙酸钙盐三水合物是否依然按照上述原理与碘反应。 求高手指教,前辈指点!谢谢
用HPINNOWAX 19091N-236检测乙酰丙酸和乙酰丙酸乙酯,两个物质在同一个时间出峰,用原料和产物的标品打了也是这样的结果。请问是什么原因?

[color=#444444]用的[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url],乙醇和乙酰丙酸反应生成乙酰丙酸。乙醇和乙酰丙酸乙酯的峰分不开。柱前压0.15mpa.柱温200检测260注样260应该怎么调才能把峰分开?乙酰丙酸沸点245,乙酰丙酸乙酯沸点203[/color][color=#444444][img=,600,800]https://ng1.17img.cn/bbsfiles/images/2019/07/201907100951384017_3086_1843534_3.jpg!w600x800.jpg[/img][/color]
我们最近做方法确认,用的是GB/T 5009.252的方法,除了柱子用的是DB-624,其他全部按照国标要求来的,但是做出来乙酰丙酸的峰有拖尾,而且峰面积没有线性可言。有没有做过的大神,可以帮忙解答一下是哪里的原因。
[color=#444444]用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]时柱箱230 进样270检测器300出不了乙酰丙酸的峰,拖尾很严重,有朋友知道这是什么原因吗?或者有做过的可以分享一下反应条件吗[/color]
打开能源的“牢笼”在冰的天然气水合物矿床中,可以发现大量的天然气,但是将这些天然气开采出来却是一个严峻的挑战。一万亿立方英尺 (tcf) 有多大? 尽管我们知道这个体积非常大,但是要想像其具体的大小将会相当困难。这里有一种方法。假定我们站在足球场或橄榄球球场一端的球门附近。在另一端俯视球场,设想一条长度为 30 倍球场长度的直线。(这一距离大概为 3 公里(约 1.9 英里)或相当于 3500 步。)现在右转 90 度,然后按照该方向设想一条相同距离的直线。最后,直视前方,设想一条长度相同并且垂直于地面向天空方向延伸的直线。那么,这个立方体的三条边所包含的体积就大约为一万亿立方英尺!平均而言,地球上的每人每月大约消费七万亿立方英尺天然气! 燃烧的冰地球上的人使用天然气(甲烷,CH4)这种矿物燃料提供日常所用能源的 45%。目前,每年的天然气燃烧量约为 2.4 万亿立方米(85 万亿立方英尺)。不幸的是,按照这一速度,我们所发现的地球天然气储量只能使用 60 年。这意味着按照目前所知的情况,对于今天正在上高中的学生而言,他们的子孙就没有可用的天然气了。对于这一暗淡的前景也有一些好的消息。看起来还有另外一个天然气资源的世界,足以满足我们当前以及将来 2000 年的能源需求。这完全可以惠及我们子子孙孙!不幸的是,我们还没有找到开采这一天然气的经济方式。我们目前正在研究。 这些特殊的天然气储量称为天然气水合物,它们由其甲烷(天然气)分子中类似小鸟笼一样的冰结构构成。基本的水合单元是中空的水分子晶体,其中包含一个天然气单分子。这些晶体以紧密的网格结构相互联接在一起。如果这些天然气水合物的联接程度紧密上几倍,那么它们看起来将更象是冰。但是其属性和冰不同:它们在适当的条件下可以燃烧!这是 21 世纪一个相当热门的话题。全球天然气水合物的储量丰富,因此有些国家已经开始研究和探索计划,致力于理解水合物的行为、确定其精确储量并开发可行的开采方法。日本、印度、美国、加拿大、挪威和俄罗斯等国家都在进行天然气水合物的勘测。 天然气水合物是一个晶体结构。这一天然气水合物的每个单元小室都包含 46 个水分子,构成两个较小的十二面体和 6 个较大的十四面体。天然气水合物只能承载较小的气体分子,例如甲烷和乙烷。在常温常压(STP)下,一体积的饱和甲烷水合物将包含 189 体积的甲烷气体。天然气水合物这么大的气体储量意味着重要的天然气来源。
请问食品中丙酸钙的测定应该用那种柱子?我看标准上写的是porapak Qs填充柱,但是我们单位没有这柱子,有没有那根柱可以替代它?谢谢!
植物源性多酚由于具有预防和治疗多种疾病的特性,在制药、化工和食品工业等领域引起广泛关注[1-2]。白藜芦醇(resveratrol,图1)是一种天然多酚,存在于葡萄皮、蔓越莓、可可等植物中,具有抗氧化、抗炎、保护心脏和抗癌等生物活性[3-4]。此外,白藜芦醇对阿尔茨海默病、帕金森病和癫痫等神经系统疾病也有神经保护作用[5-6]。该化合物在自然界中以反式和顺式2种异构体的形式存在,但反式异构体更丰富,生物活性更高[7]。然而,白藜芦醇较低的水溶性、生物利用度限制了其在人体中的吸收和生物利用有效性[8]。 药物共晶是活性药物成分和共晶形成物按一定化学计量比在非共价键相互作用下自组装而成的固体结晶材料[9-10],共晶中存在的氢键或其他非共价作用,会改变原药物晶体的结构,通过降低晶格能、提高溶剂的亲和力,从而改善药物在共晶中的溶解度[11]。因此,药物共晶技术成为解决药物生物利用度低的新途径、新领域。通过药物共晶技术提高药物生物利用度是今后药物开发新的研究方向。近年来,白藜芦醇共晶和多晶型用于提高其溶解度和生物利用度已有报道,如氨基苯甲酰胺[12]、异烟肼与烟酰胺[13]、乙烯基二吡啶[14]等共晶。不同共晶之间白藜芦醇的构象和分子堆积是灵活的,且白藜芦醇共晶的物理化学性质与其晶体堆积模式密切相关。基于共晶策略优势,利用高水溶性生物活性药物增强白藜芦醇的溶解度和生物利用度,同时有助于发挥2种药物在抗炎、抗病毒功效等方面协同作用,如白藜芦醇-金刚烷胺盐酸盐共晶[15]。 盐酸巴马汀(palmatine chloride,PCl,图1)又名黄藤素,是一类典型的异喹啉生物碱,主要存在于黄柏、黄连、三棵针、南天竹等天然中草药植物中[16-17]。PCl易溶于热水,具有抗菌、抗炎、抗病毒与抗肿瘤等药用价值,在临床上常用于治疗妇科炎症、菌痢、肠炎、呼吸道和泌尿道感染以及眼结膜炎等症状[16,18-19]。PCl结构中含有1个季铵盐阳离子与氯离子(Cl?),其中Cl?是一类潜在的氢键受体,不仅空间位阻小,还具有良好的空间适应性和几何延展性,可以同时接纳多个氢键给体,与氨基、羧基、羟基等官能团可形成较强的电荷辅助氢键[20-21],利用含Cl?的PCl作为共晶形成物为药物共晶开发提供了新的思路。本课题组前期系统研究了PCl作为共形成物与外消旋橙皮素的药物共晶多晶型,2种共晶均存在O-HCl?氢键相互作用,对温度、湿度和光表现出很高的稳定性,共晶的形成降低了盐酸巴马汀的溶解度,提高了橙皮素的溶解度。同时,在纯水中实现了盐酸巴马汀的缓释和增强橙皮素的释放[22]。本实验基于Cl?与羟基之间易形成O-HCl?氢键作用,研究了白藜芦醇与PCl的共结晶。采用溶剂悬浮法成功制备了一种新的白藜芦醇-盐酸巴马汀共晶水合物(RES-2PClH2O),利用单晶X射线衍射、粉末X射线衍射和傅里叶红外光谱对其结构进行表征,并利用差示扫描量热、动态水蒸汽吸附、高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]分析等对共晶水合物的稳定性、溶解度及溶出速率等进行了考察。 图片 1 仪器与材料 Smart Lab SE型粉末X射线衍射仪,日本理学公司;Super Nova CCD型单晶X射线衍射仪,美国安捷伦科技有限公司;DSC 214 Nevio型差示扫描量热仪、TG 209 F3型热重分析仪,德国耐驰仪器制造有限公司;Intrinsic Plus型动态水蒸汽吸附仪,英国Surface Measurement Systems公司;LC-20AD型高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url],日本岛津仪器有限公司;Nicolet iS 50型衰减全反射傅里叶红外光谱仪,美国赛默飞世尔科技公司;LHH-150SD型综合药品稳定性试验箱,上海一恒科学仪器有限公司;RC806ADK型溶出度测试仪,天津市天大天发科技有限公司;SHH-100GD-2型药品强光照射试验箱,重庆市永生实验仪器厂。 盐酸巴马汀三水合物(PCl3H2O)、白藜芦醇,质量分数均为97%,购自大连美仑生物技术有限公司;甲醇为色谱纯,购自上海泰坦科技股份有限公司;磷酸为色谱纯,购自上海阿拉丁试剂有限公司。其他试剂均为分析纯,购自国药集团药业股份有限公司。 2 方法与结果 2.1 样品的制备 2.1.1 白藜芦醇-盐酸巴马汀单晶的制备 取白藜芦醇(22.8 mg,0.1 mmol)与PCl3H2O(44.2 mg,0.1 mmol)混合均匀后加入20 mL甲醇溶液,加热搅拌至完全溶解后滤过。将溶液放于避光环境下缓慢蒸发,2~3 d后有橘红色块状晶体析出,即为白藜芦醇-盐酸巴马汀单晶。 2.1.2 RES-2PClH2O共晶水合物的制备 取白藜芦醇(114.0 mg,0.5 mmol)与盐酸巴马汀三水合物(442.0 mg,1 mmol)混合均匀后加入10 mL的甲醇溶液,在室温条件下密封搅拌48 h后滤过。将固体放于自然条件下干燥即可得到RES-2PClH2O共晶水合物。 2.2 固态表征 2.2.1 单晶X射线衍射(single crystal X-ray diffraction,SC-XRD) 利用Super Nova CCD单晶衍射仪测试待测样品,在100 K条件下收集晶体参数,入射光束为Cu-Kα射线(λ=0.154 184 nm),利用CrysAlisPro程序进行经验吸收校正[23]。采用SHELX程序对晶体结构进行直接法求解,通过全矩阵最小二乘方法对F2进行精修[24-25]。非氢原子在无约束位移参数下进行各向异性细化,氢原子则放置在合适的几何位置上。单晶结构解析表明,RES-2PClH2O为单斜晶系,P21/c空间群,在晶体结构中含有2个PCl分子、1个白藜芦醇分子与1个水分子。如图2所示,白藜芦醇结构中的3个酚羟基均参与了氢键的形成,其中2个酚羟基与2个Cl?形成O-HCl?氢键作用,而另1个酚羟基则与水分子形成O-HO氢键作用。水分子又同时与2个Cl?形成O-HCl?氢键作用。白藜芦醇分子、水分子与Cl?间通过上述的多种氢键作用相连接,形成了一维链状结构。形成的链与链间通过不同白藜芦醇分子间的C-HO作用相连接,进而形成二维层状结构(图3)。在分子间弱作用力下,层与层之进而形成堆积结构(图4)。RES-2PClH2O共晶水合物的晶体学数据见表1,共晶水合物中氢键的参数见表2。 图片 图片 图片 图片 2.2.2 粉末X射线衍射(powder X-ray diffraction,PXRD) 将待测样粉末均匀铺满样品槽后开始测量。入射光束为Cu-Kα射线,工作电压为40 kV,工作电流为15 mA,2θ范围取5°~45°,步长0.02°。如图5所示,RES-2PClH2O的PXRD谱图与白藜芦醇、PCl3H2O 2种原料药均不同,在10.6°、13.1°、14.0°、14.5°、16.2°、21.5°、26.7°、28.2°等处出现新的特征峰,且图谱中并未显现PCl3H2O在9.7°、17.8°等处的特征峰,表明所制备的产物形成了新的晶相。此外,RES-2PClH2O的PXRD图谱与其单晶结构的模拟图谱吻合较好,证实所制备的共晶水合物具有较高的纯度和均匀性。 图片 2.2.3 衰减全反射傅里叶变换红外光谱(attenuated total reflection fourier transform infrared spectroscopy,ATR-FTIR) 将待测样均匀铺于iD7 ATR附件上,扫描次数为32,分辨率为4 cm?1,波长范围为550~4 000 cm?1。如图6所示,RES-2PClH2O与PCl3H2O的图谱中均存在有水分子的伸缩振动峰,与单晶结构中存在的水分子相对应。在PCl3H2O中,水分子的伸缩振动峰为3 602~3 227 cm?1,而共晶水合物中水分子的伸缩振动峰为3 292 cm?1。在形成强分子间氢键时,-OH伸缩振动峰会发生红移(100~693 cm?1)[26-27]。白藜芦醇中-OH的伸缩振动峰在3 200 cm?1左右,而共晶水合物中-OH的伸缩振动峰显著红移至在3 002 cm?1,表明白藜芦醇和PCl3H2O分子间具有较强的氢键相互作用。同时,在形成共晶水合物后,白藜芦醇中-OH的弯曲振动峰从1 145 cm?1偏移至1 170 cm?1,归因于白藜芦醇上的-OH同PCl、水分子间均存在较强的氢键作用。 图片 2.2.4 差示扫描量热/热重分析(differential scanning calorimetry/thermal gravity analysis,DSC/ TGA) 称取适量白藜芦醇、PCl3H2O、RES- 2PClH2O分别放于铝制坩埚中,密封、扎孔后进行DSC测试。以同样的空坩埚作为参比,将其放置于仪器中预热、平衡至读数稳定后,将待测样品放于空坩埚中进行TGA测试,温度范围为30~300 ℃,升温速率10 K/min,通氮气作为保护气,体积流量为40 mL/min。如图7-a所示,白藜芦醇在268.1 ℃处有1个吸热熔融峰,PCl3H2O在204.2 ℃处出现吸热熔融峰。RES-2PClH2O在136℃附近存在1个宽的脱水吸热峰,在230.5 ℃附近存在熔融吸热峰。共晶水合物的熔点介于2个原料药之间,是不同于原料药的新晶型。由TGA图谱(图7-b)可知,白藜芦醇在30~150 ℃没有明显质量变化,PCl3H2O在105 ℃失重比为11.3%。相较于2原料药,RES-2PClH2O在136 ℃附近的失重比为2.8%,与其理论的失水质量比(2.8%)一致,进一步证实共晶水合物结构中存在1个水分子。 图片 2.3 物理稳定性研究 2.3.1 稳定性分析 根据《中国药典》2020年版药物稳定性试验,评价温度、湿度、光照等环境参数对所制备共晶水合物物理稳定性的影响。将RES- 2PClH2O分别储存于烘箱、湿稳定性箱及光稳定箱中,放置10 d后取出进行PXRD表征。如图8所示,在60 ℃,90%相对湿度(RH),或4 500 lx条件下储存10 d后,RES-2PClH2O的PXRD图谱保持不变,说明所制备共晶水合物在恶劣的储存条件下未发生晶型的变化,具有物理稳定性。 图片 2.3.2 动态水蒸汽吸附(dynamic vapor sorption,DVS)分析 称取适量待测样品置于动态水蒸气吸附仪中,设定温度为25 ℃,在体积流量为200 mL/min氮气下测量,模式选择为0~95%~0相对湿度吸附、脱附水蒸汽全循环,步长5%,平衡标准为粉体质量变化(dm/dt)≤0.002%/min。如图9-a所示,PCl3H2O吸湿量随着相对湿度增加而逐步增大。相比于PCl3H2O,白藜芦醇、RES-2PClH2O吸湿量基本不变,说明白藜芦醇可有效减少PCl3H2O吸湿量。根据局部放大图(图9-b),在95%相对湿度下,RES-2PClH2O共晶水合物吸湿量仅为0.16%,吸湿性极低。此外,共晶水合物的吸附与脱附曲线基本重合,表明在吸附过程中仅存在物理吸附水,共晶水合物未发生任何固态变化,具有良好的吸湿稳定性。 图片 2.4 体外溶出度研究 2.4.1 色谱条件 白藜芦醇、PCl的色谱分析采用Kristl等建立的方法[28]及《中国药典》2020年版一部黄藤素含量测定,并进行适当修改。色谱柱为中谱蓝XR-C18柱(150 mm×4.6 mm,5 μm),采用双波长模式,白藜芦醇的吸收波长306 nm,PCl的吸收波长345 nm,体积流量1 mL/min,进样量5 μL,柱温30 ℃,流动相为甲醇-0.2%磷酸水溶液(50∶50),洗脱方式为等度洗脱。 2.4.2 对照品储备液的制备 精密量取250 mg白藜芦醇置于50 mL量瓶中,甲醇定容,摇匀即得5 mg/mL白藜芦醇对照品储备液,同法制备5 mg/mL PCl3H2O对照品储备液。 2.4.3 线性关系考察 采用甲醇将“2.4.2”项下对照储备液分别稀释成5、10、20、50、100、200、500 μg/mL系列对照品溶液,按照“2.4.1”项下色谱条件测定各质量浓度(C)的峰面积(A)。方法学结果表明,PCl的线性回归方程为A=23 744 C+22 055,R2=1.000 0,结果表明PCl在10~500 μg/mL线性关系良好。白藜芦醇的线性回归方程为A=42 114 C?161.8,r=1.000 0,结果表明白藜芦醇在5~100 μg/mL线性关系良好。 2.4.4 供试品溶液的制备 精密量取5 mg RES-2PClH2O至50 mL量瓶中,甲醇定容,摇匀即得RES-2PClH2O供试品溶液。 2.4.5 专属性考察 取稀释后的对照品溶液、供试品溶液,分别按上述色谱条件进样,结果见图10,供试品溶液中白藜芦醇与PCl出峰时间与对照品溶液一致,分离度大于1.5,峰形良好,表明该色谱条件适用性良好。 图片 2.4.6 平衡溶解度实验 选用醋酸/醋酸盐缓冲液(pH 4.5)与纯水作为缓冲介质[15,29],称取过量待测样品加入少量介质溶液,得到过饱和溶液。37 ℃振荡48 h,取上层液0.45 μm滤膜滤过,纯水稀释后利用高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱仪[/color][/url]测量其质量浓度,得到待测样品的饱和平衡溶解度,平行样为3组。实验结束后,收集未溶解的残留固体,室温干燥后进行PXRD表征。结果如表3所示,在纯水中,白藜芦醇的溶解度为(55.100±0.669)μg/mL,PCl3H2O的溶解度(24.130±0.670)mg/mL。与之相比较,白藜芦醇、PCl3H2O在pH 4.5缓冲液中的溶解度基本不变。值得注意的是,共晶水合物中白藜芦醇溶解度在2种介质中均显著提高,尤其在pH 4.5缓冲液中,共晶水合物中白藜芦醇溶解度提高约10倍。而共晶水合物中PCl溶解度在2种介质中均显著降低,在pH 4.5缓冲液中,溶解度降低到(1.760±0.015)mg/mL。上述结果均表明通过白藜芦醇与PCl形成共晶策略极大提高了白藜芦醇溶解度,同时降低了PCl溶解度。此外,溶解度测定后将未溶解的固体残渣收集后进行PXRD表征,图谱结果表明2种介质处理后的残渣与RES-2PClH2O的PXRD图谱基本吻合(图11),未发现明显的相变。 图片 图片 2.4.7 溶出速率评估 实验在RC806ADK溶出测试仪上进行,采用小杯桨法,桨转速为75 r/min,温度为37 ℃。选用醋酸/醋酸盐缓冲液(pH 4.5)与纯水作为溶出介质,溶出介质体积为250 mL。精密称取100 mg的RES-2PClH2O粉末,86.5 mg的PCl3H2O粉末以及22.3 mg的白藜芦醇粉末,待介质温度稳定后往介质投料。设置不同时间点进行取样,每次取样1 mL后随即补充1 mL缓冲液。所有样品溶液均过0.45 μm膜后,使用HPLC测量其质量浓度,平行样为3组。如图12-a可知,在2种介质中,白藜芦醇原料药释放缓慢,4 h后最大累积释放仅约45%;形成共晶水合物后,RES-2PClH2O中白藜芦醇在纯水与pH 4.5缓冲液中的溶出行为基本一致,溶出速率均增加,溶出释放量较白藜芦醇原料药显著提高,在1 h附近达到最大值,分别为82.26%与83.43%。与白藜芦醇溶出不同的是,PCl3H2O在2种介质中5 min内几乎完全溶解,共晶水合物中PCl的溶出速率较PCl3H2O有效减缓,1 h后达到最大累积释放量(图12-b)。 图片 综合上述溶出结果表明,相比于白藜芦醇原料药,通过与PCl3H2O形成共晶水合物,可有效促进白藜芦醇的溶出、同时延缓PCl的释放。 3 讨论 将水溶性较高的药物与难溶性药物形成药物-药物共晶,有利于平衡两者的溶解度[11]。利用水溶性较好的PCl[(24.13±0.67)mg/mL]与难溶性白藜芦醇[(55.100±0.669)μg/mL]通过分子间相互作用形成共晶,有望优化两者溶解度和溶出速率。本研究采用溶剂悬浮法成功制备了新的RES- 2PClH2O共晶水合物。RES-2PClH2O的PXRD图谱与其单晶结构的模拟图谱吻合较好,证实所制备的共晶水合物具有较高的纯度和均匀性。 DSC测试结果显示,RES-2PClH2O的熔点介于2个原料药之间,进一步证实该共晶水合物是不同于原料药的新晶型。通过单晶结构分析,该共晶水合物存在O-HCl?氢键作用且含有水分子。白藜芦醇上的2个羟基与2个Cl?形成O-HCl?氢键,而水分子通过O-HO与O-HCl?的氢键作用分别与白藜芦醇、PCl相连并形成一维链状结构。链与链间又通过C-HO作用形成二维层状结构,层与层之间通过分子间弱作用力进而形成堆积结构。 TGA表征结果显示,RES-2PClH2O实际失水质量与理论失水质量相一致,进一步证实该共晶水合物结构中存在1个水分子。ATR-FTIR显示,RES-2PClH2O中,水分子伸缩振动峰和白藜芦醇的-OH伸缩振动峰、弯曲振动峰均发生了明显偏移,表明白藜芦醇中的-OH与PCl、水分子间均存在较强的氢键作用,2原料药间发生了相互作用。 药物稳定性测试证实,RES-2PClH2O在高温、高湿或强光照射等恶劣条件下长期储存具有较好的物理稳定性,与非吸湿性白藜芦醇共结晶后,PCl的抗湿稳定性得到显著提高。为研究PCl对白藜芦醇溶解度影响,评估了共晶水合物在纯水与醋酸/醋酸钠缓冲液介质中的平衡溶解度,并与原料药溶解度对比分析。结果显示,可溶性PCl与不溶性白藜芦醇共结晶同时影响了2种药物的溶解性能。在所制备的共晶水合物中,白藜芦醇溶解度明显提高、PCl溶解度显著降低。 为探究RES-2PClH2O共晶水合物形成后白藜芦醇、PCl溶出速率变化,对比在纯水与pH 4.5缓冲液2种介质中共晶水合物与原料药的溶出速率。溶出结果表明PCl作为白藜芦醇共晶形成的共形成物,显著促进白藜芦醇的释放同时延缓PCl的释放。本研究阐明了PCl作为白藜芦醇药物共晶形成物的可行性,为利用共结晶技术开发白藜芦醇药物共晶提供新的借鉴。
最近实验室要准备开展这方面的检测工作,看了标准,有好几个地方不明确,请有经验的童鞋给点建议!方法: 提取准确称取30g事先均质化的样品(面包样品需在室温下风干,磨碎),置于500mL蒸馏瓶中,加入1000mI,水,再用50mI。水冲洗容器,转移到蒸馏瓶中,加10mL磷酸溶液,2~3滴硅油,进行水蒸气蒸馏,将250mI容量瓶置于冰浴中作为吸收液装置,待蒸馏液约250mL时取出,在室温下放置30min,加水至刻度,吸取10mL该溶液于试管中,加入0.5mL甲酸溶液,混匀,供色谱测定用。 (2)测定色谱柱:玻璃柱,内径3mm,长1m,内装80~100目Porapak QS。 温度:柱温180℃,进样口、检测器温度为220℃。 气流条件:氮气50mL/min,氢气50mL/min,空气500mI。/min。 定量方法:取标准系列中各种浓度的标准使用液10mL,加O.5mL甲酸溶液,混匀。取5肛I。进气相色谱仪,测定不同浓度丙酸的峰高,根据浓度和峰高绘制标准曲线。同时进样品溶液,根据样品的峰高与标准曲线比较定量。问题:1 红色部分,这样做出来的结果准确吗? 2 现在做气相的话,都是用毛细管柱了,很少有用玻璃柱子的,用DB-1或者DB-5能做出来吗?
关键词:钢铁标准物质 钢铁标样 冶金标准物质 冶金标准样品 冶金标样 生铁标样 铁矿石标样 1.原理 样品酸化后,丙酸盐转化为丙酸。经水蒸馏,收集后直接进气相色谱仪,用氢火焰离子化检测器检测,与标准系列比较定量。(钢铁标准物质) 2.试剂 磷酸溶液(取10mL 85%磷酸加水至100mL)、甲酸溶液(取1mL 99%甲酸加水至50mL)、硅油。 丙酸标准储备溶液(10mg/mL):准确称取250mg丙酸于25mL容量瓶中,加水至刻度。丙酸标准使用液:将储备液用水稀释成10~250mg/mL的标准系列。(钢铁标样) 3.仪器 水蒸气蒸馏装置、气相色谱仪(具有氢火焰离子化检测器,FID)。(冶金标准物质) 4.操作步骤 . (1)提取准确称取30g事先均质化的样品(面包样品需在室温下风干,磨碎),置于500mL蒸馏瓶中,加入1000mI,水,再用50mI。水冲洗容器,转移到蒸馏瓶中,加10mL磷酸溶液,2~3滴硅油,进行水蒸气蒸馏,将250mI。容量瓶置于冰浴中作为吸收液装置,待蒸馏液约250mL时取出,在室温下放置30min,加水至刻度,吸取10mL该溶液于试管中,加入0.5mL甲酸溶液,混匀,供色谱测定用。(冶金标准样品) (2)测定色谱柱:玻璃柱,内径3mm,长1m,内装80~100目Porapak QS。 温度:柱温180℃,进样口、检测器温度为220℃。 气流条件:氮气50mL/min,氢气50mL/min,空气500mI。/min。 定量方法:取标准系列中各种浓度的标准使用液10mL,加O.5mL甲酸溶液,混匀。取5肛I。进气相色谱仪,测定不同浓度丙酸的峰高,根据浓度和峰高绘制标准曲线。同时进样品溶液,根据样品的峰高与标准曲线比较定量。(冶金标样) 5.分析结果的表述 z一巡×1000 (9—5) z一——天l (9一b) Ⅲ 式中,z为样品中丙酸含量,g/kg;A为测定样品中丙酸含量,mg/mL;m为样品质量,g。 丙酸钠含量一丙酸含量×1.2967;丙酸钙含量===丙酸含量×1.2569。 6.技术参数 相对标准差在6%以下。二次平行测定相对允许误差的绝对值在10%以下,平行测定结果用算术平均值表示,保留两位有效数字。本方法最低检出量:面包、糕点O.05g/kg,酱油、醋O.02g/kg。(生铁标样)
[color=#444444]如题,主要测定水解液中乙酰丙酸的含量,水解液里含硫酸,咨询了[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]柱厂家,说有无机酸的话容易损坏柱子,但我看很多文献都用的FFAP毛细管柱,也没提及柱子寿命问题。 用液相色谱柱国产的不行,国外的又太贵,求这方面的大神给指条明路,感激不尽[/color]
[color=#444444]如题,主要测定水解液中乙酰丙酸的含量,水解液里含硫酸,咨询了[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]柱厂家,说有无机酸的话容易损坏柱子,但我看很多文献都用的FFAP毛细管柱,也没提及柱子寿命问题。 用液相色谱柱国产的不行,国外的又太贵,求这方面的大神给指条明路,感激不尽[/color]
弱弱的问一下:gb/t5009.120-2003 丙酸钙、丙酸钠的测定中,上机前在已定容的样液中加0.5mL的甲酸溶液的作用?如果是保持一定的酸性环境,那为什么一定要加甲酸,其他酸行不行?(第二个问题撇开标准规定来讨论)
各位老师好,本人实验室需要同时测定多种有机酸,目前前期验证遇到了困难,就是乳酸、乙酸和乙酰丙酸的峰互相离的很近,无法完全分开,老师们有没有好的手段,麻烦指点下仪器:ICS-6000淋洗液:氢氧化钠溶液(梯度:1-16min:1Mm,25min:12mM,曲线:5)色谱柱:AS18-4mm
食品中的丙酸钠、丙酸钙的测定谁有用毛细管柱做过,可否用FFAP柱做,GB/T5009.120是用填充柱做,GB/T 5009.120-2003 食品中丙酸钠、丙酸钙的测定 采用的是[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]法,用水蒸汽蒸馏提取样品,直接进样,这样对柱子是不是损害很大?
[table=100%][tr][td]请问在高效液相色谱中,用甲醇和水做流动相,糠醛和乙酰丙酸乙酯的出峰位置分别在哪里[/td][/tr][/table]
[font=黑体][color=black]天然气水合物的研究、调查现状[/color][/font][align=left][font=黑体][color=black]1.[/color][/font][font=黑体][color=black]天然气水合物的研究[/color][/font][/align][align=left][font=宋体][color=black]近年来,我国对管辖海域做大量的地震勘查资料分析得出,在冲绳海槽的边坡、南海的北部陆坡、西沙海槽和西沙群岛南坡等处发现了海底天然气水合物存在的似海底地震反射层(BSR)标志。[/color][/font][/align][align=left][font=宋体][color=black]自1999年始,广州海洋地质调查局在我国海域南海北部西沙海槽区开展海洋天然气水合物前期试验性调查。完成三条高分辩率地震测线共543.3km。2000年9-11月,广州海洋地质调查局"探宝号"和"海洋四号"调查船在西沙海槽继续开展天然气水含物的调查。共完成高分辩率多道地震1593.39km、多波束海底地形测量703.5km、地球化学采样20个、孔隙水样品18个、气态烃传感器现场快速测定样品33个。获得突破性进展。研究表明:地震剖面上具明显似海底反射界面(BSR)和振幅空白带。"BSR"界面一般位于海底以下300-700m,最浅处约180m。振幅空白带或弱振幅带厚度约80-600m,"BSR"分布面积约2400km'。根据ODP184航次1144钻井资料揭示,在南海海域东沙群岛东南地区,l百万年以来沉积速率在每百万年400-1200m之间,莺歌海盆地中中新世以来沉积速度很大。资料表明:南海北部和西部陆坡的沉积速率和已发现有丰富天然气水合物资源的美国东海岸外布莱克海台地区类似。南海海域水含物可能赋存的有利部位是:北部陆坡区、西部走滑剪切带、东部板块聚合边缘及南部台槽区。本区具有增生楔型双BSR、槽缘斜坡型BSR、台地型BSR及盆缘斜坡型BSR等四种类型的水合物地震标志BSR构型。从地球化学研究发现南海北部陆坡区和南沙海域,经常存在临震前的卫星热红外增温异常,其温度较周围海域升高5-6℃,特别是南海北部陆坡区,从琼东南开始,经东沙群岛,直到台湾西南一带,多次重复出现增温异常,它可能与海底的天然气水会物及油气有关。[/color][/font][/align][align=left][font=宋体][color=black]综合资料表明:南海陆坡和陆隆区应有丰富的天然气水合物矿藏,估算其总资源量达643.5-772.2亿吨油当量,大约相当于我国陆上和近海石油天然气总资源量的1/2。[/color][/font][/align][align=left][font=黑体][color=black]2 [/color][/font][font=黑体][color=black]有关天然气水合物的现状调查[/color][/font][/align][align=left][font=宋体][color=black]西沙海槽位于南海北部陆坡区的新生代被动大陆边缘型沉积盆地。新生代最大沉积厚度超过7000m,具断裂活跃。水深大于400m。基于应用国家863研究项目"深水多道高分辨率地震技术"而获得了可靠的天然气水合物存在地震标志:1)在西沙海槽盆北部斜坡和南部台地深度200-700m发现强BSR显示,在部分测线可见到明显的BSR与地层斜交现象。2)振幅异常,BSR上方出现弱振幅或振幅空白带,以层状和块状分布,[/color][/font][font=宋体]厚度80-450m。3)BSR波形与海底反射波相比,出现明显的反极性。4)BSR之上的振幅空白带具有明显的速度增大的变化趋势。资料表明:南海北部西沙海槽天然气水合物存在面积大,是一个有利的天然气水合物远景区。[/font][/align][align=left][font=宋体][color=black]2001[/color][/font][font=宋体][color=black]年,中国地质调查局在财政部的支持下,广州海洋地质调查局继续在南海北部海域进行天然气水合物资源的调查与研究,计划在东沙群岛附近海域开展高分辨率多道地震调查3500km,在西沙海槽区进行沉积物取样及配套的地球化学异常探测35个站位及其他多波束海底地形探测、海底电视摄像与浅层剖面测量等。另据我国台大海洋所及台湾中油公司资料,在台西南增生楔,水深500-2000m处广泛存在BSR,其面积2×104km[sup]2[/sup]。并在台东南海底发现大面积分布的白色天然气水合物赋存区。[/color][/font][/align][font=黑体][color=black]3.[/color][/font][font=黑体][color=black]天然气水合物的意见与建议[/color][/font][align=left][font=宋体][color=black]鉴于天然气水合物是21世纪潜在的新能源,它正受到各国科学家和各国政府的重视,其调查研究成果日新月异,故及时了解、收集、交流这方面的情况、勘探方法及成果尤为重要,为赶超国际天然气水合物调查、研究水平,促进我国天然气水会物的调查、勘探与开发事业,为我国经济的持续发展做出新贡献,建议每两年召开一次全国性的"天然气水合物调查动态、勘探方法和成果研讨会"。[/color][/font][/align][align=left][font=宋体][color=black]我国南海广阔的陆坡及东海部分陆坡具有形成天然气水含物的地质条件,建议尽快开展这两个海区的天然气水含物的调查研究工作,为我国国民经济可持续发展提供新能源。[/color][/font][/align][align=left][font=宋体][color=black]天然气水合物的开采方法目前主要在热激化法、减压法和注人剂法三种。开发的最大难点是保证井底稳定,使甲烷气不泄漏、不引发温室效应。针对这一问题,日本提出了"分子控制"开采方案。天然气水合物矿藏的最终确定必须通过钻探,其难度比常规海上油气钻探要大得多,一方面是水太深,另一方面由于天然气水合物遇减压会迅速分解,极易造成井喷。日益增多的成果表明,由自然或人为因素所引起温压变化,均可使水合物分解,造成海底滑坡、生物灭亡和气候变暖等环境灾害。因而研究天然气水合物的钻采方法已迫在眉捷,建议尽快开展室内外天然气水合物钻采方法的研究工作。[/color][/font][/align]
[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]测定甲酸乙酸丙酸和丁酸该如何做?具体参照什么标准
[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]测定甲酸乙酸丙酸和丁酸该如何做?具体参照什么标准?
取3.48g[color=#333333]五水合柠檬酸钠,实验室无此试剂,改用[color=#333333]二水合柠檬酸钠,应该取多少克?[/color][/color][color=#333333][color=#333333]谢谢大家![/color][/color][color=#333333][/color]
[color=#444444]质谱可以打出水合物中的水吗,[color=#444444]比如五水合物质谱上最大的峰是含水的还是不含水的呀,真心求问。[/color][/color]







 我要推广仪器
我要推广仪器
 下载APP
下载APP