如题:乙酰甲胺磷的标准溶液怎么存放?我们的是用甲醇稀释到50PPM在冰柜保存,6月配标,9月测试时响应挺高的,10月测试发现响应低了很多,仪器条件没变,这是为什么?
食品中甲胺磷和乙酰甲胺磷农药残留量的测定方法1.适用范围本方法适用于谷物、蔬菜和植物油中甲胺磷和乙酰甲胺磷的残留量分析,其最小检出限分别为7.79×10-12g和1.79×10-11g。2.原理概要含有机磷的样品在富氢焰上燃烧,以HPO碎片的形式,放射出波长526nm的特征光,这种特征光通过滤光片选择后,由光电倍增管接收,转换成电信号,经微电流放大器放大后,被记录下来,样品的峰高与标准品的峰高相比,计算出样品相当的含量。3.主要试剂和仪器3.1.主要试剂丙酮;二氯甲烷:重蒸;无水硫酸钠;活性炭:用3mol/L盐酸浸泡过夜,抽滤,用水洗至中性,在120℃下烘干备用;甲胺磷(methamidophos):≥99%;乙酰甲胺磷(acephate):≥99%;甲胺磷和乙酰甲胺磷标准溶液的配制:分别准确称取甲胺磷和乙酰甲胺磷的标准品,用丙酮分别制成0.1mg/mL的标准储备液。使用时用丙酮稀释配制成单一品种的标准使用液(1mg/mL)和混合标准工作液(每个品种浓度为1mg/mL)。贮藏于冰箱中。3.2.仪器气相色谱仪:具有火焰光度检测器;电动振荡器;K-D浓缩器或旋转蒸发器;离心机。4.试样的制备取谷物实验样品经粉碎机粉碎,过20目筛后,制成谷物试样。取蔬菜实验样品洗净,晾干,去掉非食部分后剁碎或经组织捣碎机捣碎,制成蔬菜试样。5.过程简述5.1.提取和净化蔬菜:称取蔬菜试样10g,精确至0.001g,用无水硫酸钠(因蔬菜含水量不同而加入量不同,约50~80g)研磨呈干粉状,倒入具塞锥形瓶中,加入0.2~0.4g活性炭(根据蔬菜色素含量)及80mL丙酮,振摇0.5h,抽滤,滤液浓缩定容至5mL,待气相色谱分析。谷物:称取谷物试样10g,精确至0.001g,置于具塞锥形瓶中,加入40mL丙酮,振摇1h,抽滤,浓缩,定容至5mL,待气相色谱分析。小麦:称取小麦试样10g,精确至0.001g,置于具塞锥形瓶中,加入0.2g活性炭及40mL丙酮,振摇1h,抽滤,浓缩,定容至5mL,待气相色谱分析。植物油:称取植物油试样5g,用45mL丙酮分次洗入50mL的离心管内,加入5mL水,混匀,在3 000r/min下离心5min,吸取上清液,下面油层再加10mL水和10mL丙酮,离心5min,吸取上清液,合并两次上清液,用K-D浓缩器浓缩近干,残渣和水加入40g无水硫酸钠,研磨呈干粉状,倒入具塞锥形瓶中,加入0.3g活性炭、60mL二氯甲烷,振荡0.5h,抽滤,定容至5mL,待气相色谱分析。5.2.色谱条件色谱柱:玻璃柱,内径3mm,长0.5m,内装2%dEGS/Chromosorb W AWdMCS,80~100mesh。气流:载气,氮气70mL/min,空气0.7kg/cm2,氢气1.2kg/cm2。温度:进样口200℃,柱温180℃。5.3.测定定性:以甲胺磷和乙酰甲胺磷农药标样的保留时间定性。定量:用外标法定量,以甲胺磷和乙酰甲胺磷农药已知浓度的标准样品溶液作外标物,按峰高定量。6.结果计算Xi=hi•Esi•V1hsi•V2•m式中:Xi——样品中i组分有机磷含量,mg/kg;Esi——注入标样中i组分有机磷的含量,ng;hi——样品的峰高,mm;hsi——标样中i组分的峰高,mm;V1——浓缩定容体积,mL;V2——注入色谱样品的体积,μL;m——样品的质量,g。7.方法的精密度添加回收试验中甲胺磷和乙酰甲胺磷的变异系数分别为2.36%和3.95%。8.甲胺磷和乙酰甲胺磷的保留时间在5.2的气相色谱条件下,甲胺磷的保留时间为0.9min,乙酰甲胺磷的保留时间为1.9min。9.来源:GB 14876—94
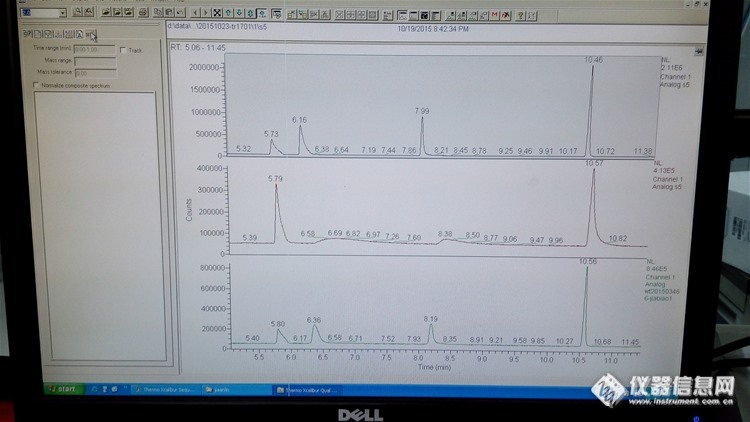
http://ng1.17img.cn/bbsfiles/images/2015/11/201511111107_573060_2547863_3.jpg如图,第一个谱图是1个月前做的标准曲线点,峰分别是敌敌畏,甲胺磷,乙酰甲胺磷,乐果,用的柱子是35柱第二个谱图是前天做的标准曲线点,其中甲胺磷,乙酰甲胺磷峰塌下去了第三个谱图是前天做的红枣加标样品,其中甲胺磷,乙酰甲胺磷峰很正常。如今往红枣样品上机液(四个峰均不出峰)添加混标液,得出的谱图和第三个一样;重新配了混标,甲胺磷和乙酰甲胺磷也是超级拖尾,但是一旦把混标加入样品液里面,两个出峰都很正常求解决方法(已经切割过柱子前端,进样瓶都是新的,除了溶液一个是丙酮定容的样品上机液,一个是色谱纯丙酮不一样外,其他条件都一样)
用气相色谱做甲胺磷和乙酰甲胺磷的回收率,过程:取1ppm的混标5ml,用离子水定容到10ml。将这10mL试样溶液倒入100mL的具塞试管中,加入30ml的丙酮,40ml的二氯甲烷,摇匀,加入4g氯化钠,5g无水硫酸钠,摇匀,静置1h,分别经无水硫酸钠漏斗过滤收集30 mL装入 50 mL圆底烧瓶中,旋转蒸发仪上浓缩近干;3mL丙酮蜗旋复溶,定容后取1 mL入气相进样小瓶中,待测。计算方法:回收率=测定浓度/计算浓度 计算浓度=(1ppm*5ml)*30ml/70ml/3ml 结果甲胺磷回收在110%,乙酰甲胺磷在129%。各容器都洗干净,且最后定容时已经尽量减少丙酮挥发了。但结果仍不理想。实在想不出哪里有问题,已经困扰快1个月了,求助各位大神帮忙
用NY761方法做乙酰甲胺磷与甲胺磷的回收率,标液用丙酮配制,衬管用高惰性衬管,不是新的,是用过的,添加量为0.16ug/mL,甲胺磷实际测得为0.20ug/mL,乙酰甲胺磷为0.35ug/mL,回收率为125-216%,为何回收率这么高,是因为高惰性衬管受到污染了吗?还是必须用基质配标液。
GC450瓦里安气相,PFPD检测器,DB1701(30*0.25*0.25)柱,进样口温度250℃,检测器300℃;程序升温:80℃保持1min,20℃/min升至130℃,5℃/min升至200℃,15℃/min升至250℃,保持1min,共21.83min。乙酰甲胺磷标液是新打开的,浓度100ug/mL,用丙酮配制成1.0ug/ml进样,乙酰甲胺磷不出峰,但配制的二唪农0.1ug/mL出尖峰。谁做过乙酰甲胺磷,用什么溶剂,它在0.1ug/mL时就出峰?
岛津的2014C[url=https://insevent.instrument.com.cn/t/Mp]气相[/url],FPD检测器,1701柱子,高惰性衬管,之前做有机磷混标的时候用丙酮溶剂0.1ug/ml敌敌畏,甲胺磷,乙酰甲胺磷,氧乐果,都出峰,除了乙酰甲胺磷出峰峰型都还好,可是这次做,这四种就是不出峰了,衬管也换了,柱子也裁了,还是不出峰,又新开标液重配还是不出峰,然后开始大浓度5ug/ml,单标进样,都出峰了,但是敌敌畏和氧乐果的峰面积偏小,直至低浓度的敌敌畏和氧乐果的1ug/ml的出峰,然后0.5ug/ml,就不出峰了。然后用黄瓜基质配做溶剂配的混标,敌敌畏0.2,甲胺磷和乙酰甲胺磷、氧乐果0.1ug/ml竟然都出峰了,而且除了敌敌畏峰小点,其他出峰都还可以,连续进了四五针峰面积也差不多,同时用丙酮配四种一样浓度的混标还是不出峰,这是啥原因造成的的?(PS:新换衬管我连续进了七八针大浓度的,然后再进的小浓度的都不出峰)
最近在做气质分析,在相同的程序升温条件下,做全扫描时出的乙酰甲胺磷峰比较好,但是在采用SIM扫描0.1倍浓度的乙酰甲胺磷后,发现基线有漂移(基线原来是在0,现在变成了10-8之间,是一条下降的弧线,出峰位置在此弧线范围中),这样在定量时,就不好积分了。我就想问问,在检测一组物质含量,如果我只是用SIM扫描混标中每一种物质用来定量(前提是已经做过FS),突然出现在走某一种物质时发生基线漂移,该如何改善?我的溶剂是正己烷或者是丙酮。
小弟最近在做乙酰丙酮分光光度法分析甲醛 按照GB/T 5009.49-2003 发酵酒卫生标准的分析方法做的。但是由于本人是第一次做,不知道标准曲线具体是什么样的,数据貌似不太对头。0.00 , 0.50 , 1.00 , 2.00 , 300 , 4.00 , 8.00 mL , l μg/mL的甲醛标准溶液于25mL比色管中,加水至10mL各加人2mL乙酰丙酮溶液,摇匀后在沸水浴中加热10 min ,取出冷却,于分光光度计波长420nm 处测定吸光度,绘制标准曲线。居然甲醛越高吸光度越低。而且做了好几次都是这样的!请求指点。哪位前辈也有做过这个标准曲线的发给我看看。谢谢! humangest@163.com
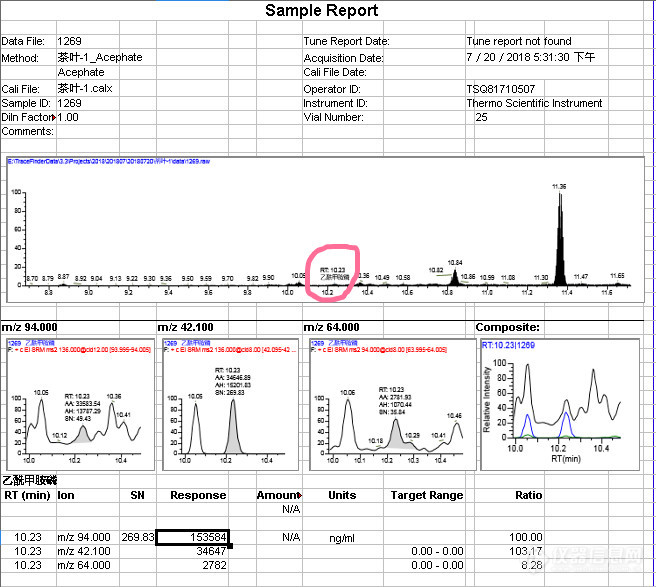
用安捷伦[url=https://insevent.instrument.com.cn/t/Mp]gc[/url]MS与热电TSQ [url=https://insevent.instrument.com.cn/t/Mp]gc[/url]MSMS测定茶叶中乙酰甲胺磷数据比较仪器:Agilent 7890B-5977A [url=https://insevent.instrument.com.cn/t/Mp]gc[/url]MS参照检测方法:GB/T23376-2009 ;检测项目:乙酰甲胺磷样品前处理:提取:取样1.0g(±0.01g)于50mL塑料离心管中,加入1mL饱和氯化钠水溶液(称取36g氯化钠溶于100mL水中)浸泡十分钟左右,加入15mL乙酸乙酯先均质,再加入一勺无水硫酸钠和2勺无水硫酸镁均质30s,用15mL乙酸乙酯洗均质头合并提取液,盖上盖子振摇一会,超声5min。把提取液和残渣一起直接过加有2勺无水硫酸镁的漏斗入鸡心瓶中,2×5mL乙酸乙酯洗离心管,振摇,合并提取液于鸡心瓶中。再用20mL乙酸乙酯冲洗漏斗上的残渣合并洗液,35℃旋转蒸发至剩2mL左右,待净化。净化:10mL丙酮+正己烷(1+1, V+V)先活化TPT(10mL,2g)柱子(填料上加1cm左右高的无水硫酸钠)下接15mL玻璃离心管,将上述大约剩2mL左右的乙酸乙酯先吸出直接过活化后的TPT柱,再用丙酮+正己烷(1+1, V+V)2.5 mL×2次洗涤鸡心瓶(必要时超声波),洗液继续过TPT柱子,加丙酮+正己烷(1+1, V+V)继续淋洗柱子共收集12mL淋洗液,35℃左右氮气吹干,丙酮+正己烷(1+1, V+V)定容1mL上机测试。前处理流程图:[img=,631,496]https://ng1.17img.cn/bbsfiles/images/2018/08/201808251507014002_4723_2166779_3.png!w631x496.jpg[/img]仪器条件:进样量:1μL 柱流量:1.0 mL/min色谱柱: DB-1701 30 m × 320 μm × 0.25 μm HP-5MS: 30 m ×320 μm × 0.25 μm 进样口温度:220 ℃ ;程序升温:60℃(1 min),以20℃/min 升至150℃ ,再以15℃/min 升至 230℃ ,最后以25℃/min 280℃(5 min)MS条件:EI源;离子源温度:230℃;SIM扫描,采集离子碎片:94、95、136、142(100:49.60:211.70:22.50);采集时间段:(8~11)min乙酰甲胺磷标液(1000ng/mL)的SIM图:[img=,690,465]https://ng1.17img.cn/bbsfiles/images/2018/08/201808251521491270_2063_2166779_3.png!w690x465.jpg[/img]茶叶样品SIM图:[img=,690,434]https://ng1.17img.cn/bbsfiles/images/2018/08/201808251523135810_173_2166779_3.png!w690x434.jpg[/img]样品的测定结果:1.2mg/kgGB2763-2016标准规定茶叶的乙酰甲胺磷要小于等于0.5mg/kg,这么说这个茶叶的乙酰甲胺磷超标了?不放心,决定换一台热电的TSQ 8000来试下:将[url=https://insevent.instrument.com.cn/t/Mp]gc[/url]MS检测后的样液用TSQ 8000 [url=https://insevent.instrument.com.cn/t/Mp]gc[/url]MSMS,MRM模式来测下:乙酰甲胺磷MRM采集参数:[img=,690,20]https://ng1.17img.cn/bbsfiles/images/2018/08/201808251534001660_9521_2166779_3.png!w690x20.jpg[/img]500ng/mL乙酰甲胺磷标液测定的MRM图:[img=,658,575]https://ng1.17img.cn/bbsfiles/images/2018/08/201808251536181610_8309_2166779_3.png!w658x575.jpg[/img]样品中乙酰甲胺磷标液测定的MRM图:[img=,656,587]https://ng1.17img.cn/bbsfiles/images/2018/08/201808251553115924_5737_2166779_3.jpg!w656x587.jpg[/img]样品加标同时同样处理所得的MRM图(加标量为400ng/mL):[img=,654,584]https://ng1.17img.cn/bbsfiles/images/2018/08/201808251554584500_6400_2166779_3.png!w654x584.jpg[/img]用热电的TSQ 8000 [url=https://insevent.instrument.com.cn/t/Mp]gc[/url]MSMS MRM模式的测定结果为0.3ng/mL,用二级质谱的测定结果相比单级质谱的测定结果应该会可靠些。看来可能是因为茶叶的基质效应的影响,用[url=https://insevent.instrument.com.cn/t/Mp]gc[/url]MS单级质谱测定结果(用94离子作为定量离子)有基质干扰吧,遇到不合格有条件还是尽量更换仪器,更换检测方法进行复测。
我在测试甲醛含量时发现对同一组浓度不同的甲醛溶液,用放置时间不同的乙酰丙酮测试时甲醛浓度的变化趋势截然相反。我很困惑...用乙酰丙酮分光光度法测试甲醛含量时,乙酰丙酮溶液是不是需要稳定数小时才可以使用的?应该稳定几个小时呢?谢谢
761做农残,之前做了大半年,柱子氧乐果不出峰了,没办法截了快两米,终于好了。可是发现做了两批样一百多个样,出峰面积又小了。走丙酮溶剂发现有小杂峰(面积很小,50左右。破罐子,破摔。各种折腾。用丙酮洗(进4微升),甲醇洗(进4微升)。老化的时候,进溶剂,甲醇,正己烷,丙酮轮着。结果更惨了,本来氧乐果1微升能出2000多的。现在就八百了。乙酰甲胺磷本来好好的,也少了很多。之前也发现,柱子老化后。本来不拖尾的峰,拖尾了。问各位老师,[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]柱,走溶剂空白,能起到清洗作用吗?还是我用的溶剂体积太大了,能用甲醇吗?以后这种情况要每次做了,截了柱子后,再老化吗?
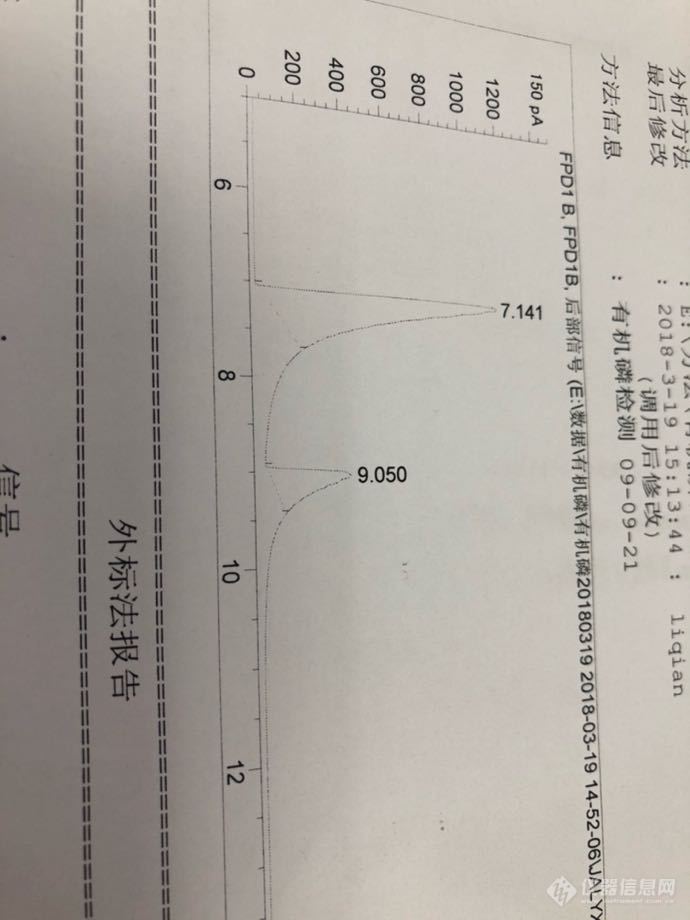
甲胺磷 乙酰甲胺磷拖尾严重…请问有啥解决办法吗?衬管也是新换的,柱头也割过一截了。方法也是之前用的。标准品也是新开的…请问有啥解决办法吗?[img=,690,920]http://ng1.17img.cn/bbsfiles/images/2018/03/201803191602167875_8255_3379628_3.png[/img]
分光光度计测试甲醛用的乙酰丙酮溶液,大家直接放在容量瓶保存吗?
[font='微软雅黑','sans-serif'][size=12pt]空[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质[/color][/url]量甲醛的测定乙酰丙酮分光光度法[/size][/font][font='Times New Roman','serif'][size=12pt]GB/T15516[/size][/font][font='微软雅黑','sans-serif'][size=12pt]中乙酰丙酮溶液的配制,新蒸馏的[/size][/font][font='Times New Roman','serif'][size=12pt]0.25ml[/size][/font][font='微软雅黑','sans-serif'][size=12pt]乙酰丙酮该怎么蒸馏可不可以不蒸馏直接用?[/size][/font][font='微软雅黑','sans-serif'][size=12pt][color=#ff0000]注:问题来自水质分析交流群。[/color][/size][/font]
(一)原理在磷酸酸性条件下进行蒸馏,使样品中的甲醛分解出来,被水吸收,吸收液中的甲醛与乙酰丙酮反应,生成黄色物质与标准比较定理。(二)试剂(1)20%磷酸。(2)乙酰丙酮溶液:取乙酸铵150g溶于水中,加入3mL冰乙酸和2mL乙酰丙酮(重蒸馏过),用水定容至1000mL。(3)甲醛标准溶液:称取特级六次甲基四胺O.3112g,溶于水后,定容至l000mL,此液甲醛浓度为400μg/mL。(4)甲醛标准使用液(临用时现配):取甲醛标准溶液2.5mL加水至100mL,此溶液每毫升相当于lOμg甲醛。(三)仪器(1)水蒸气蒸馏水装置。(2)水浴。(3)紫外一可见分光光度计。(四)操作方法1.样品处理取切碎的面制食品2g,加30mL蒸馏水于蒸馏瓶中,加20%磷酸3mL,先在接收瓶中加10mL蒸馏水,冷凝管下端浸入水中,通水蒸气蒸馏至蒸馏液约200mL时,停止蒸馏,准确加水定容至200mL。2.测定取5mL上述试液,同时取甲醛标准使用液0.0O、O.20、0.40、O.60、O.80、1.O0、2.00mL,分别置于25mL比色管中,加水至5mL,各管分别加5mL乙酰丙酮溶液,混匀,置沸水浴中加热10min后,于波长415nm处测量。(五)注意事项本法最低检测质量为0.52 μg;对甲醛含量1.5~2.0μg的6份样品,每份样品连续测定6次,平均相对标准偏差(RSD)为3.3%;取含吊白块的样品为本底样品,分别加入5.Oμg、10.0 μg甲醛标准使用液,其回收率在86.8%~100.6%。样品中存在的NaHSO3是否来自于甲醛次硫酸氢钠的分解产物。虽然目前尚未找到允许在面制品制作过程中使用亚硫酸氢钠作为漂白剂的国家标准,但亚硫酸氢钠、亚硫酸钠、硫磺可被应用在饼干、干果、干菜、粉丝等数类食品的加工制作过程,从而也有可能被应用在面制食品的加工过程中。故采用以上定量、定性两方法结果综合起来判定面制食品中是否存在吊白块时,应将亚硫酸氢钠和甲醛的测定结果综合起来判定,若同时测定面制食品中亚硫酸氢钠(以SO2计),以了解二氧化硫与甲醛实测值之比是否较接近理论上的质量比即相对分子质量之比2.1:1.O。若接近此比值,更可断定吊白块的存在,这样得出的结论应是比较科学、准确的。
茶叶中的甲胺磷与乙酰甲胺磷的检测,各位都是采用哪些方法来检测,再检测过程中有没有遇到什么问题?
有那位大侠知道,甲胺磷、乙酰甲胺磷、水胺硫磷在牛奶中的最大限量值,或者在其他标准中最大限量值呀?希望能够得到大家的帮助[em0713]
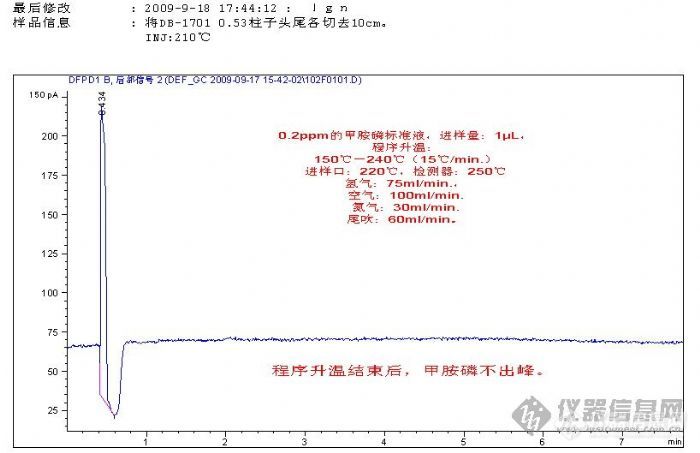
最近开展茶叶中有机磷项目的检测。购回甲胺磷、乙酰甲胺磷、毒死蜱、杀螟松4个单体标准溶液,浓度是100μL/mL.。仪器:Agilent 7890A.配双FPD检测器(DFPD),以下用谱图说明实验进程。[img]http://ng1.17img.cn/bbsfiles/images/2009/09/200909191600_171983_1620184_3.jpg[/img]
RT:甲胺磷已经禁用很久了,还是有检出甲胺磷。乙酰甲胺磷中的甲胺磷一般要求在0.8-1%,实际情况是什么样的?
最近做绿茶提取物甲胺磷,乙酰甲胺磷,对硫磷的检测,回收率做不出,甲胺磷乙酰甲胺磷才4-5%,对硫磷有70-80%。我使用的是安捷伦的6890N,检测器NPD,不分流进样,进标准品1.0ug/ml峰面积大概是300左右,但是不是很稳定。感觉问题出在前处理,分别用正己烷,乙酸乙酯,乙腈做过过加样回收,加2ml浓度1.0ug/ml。乙酸乙酯,乙腈提取浓缩后为粘稠红棕色液体,用无水硫酸镁和活性炭分散固相萃取后没有改善,正己烷提取后浓缩液无色。浓缩前进过仪器是可以测出来的,浓缩后没有了,回收液液没有,估计分解了。浓缩使用的是步其的平行定量浓缩仪,水浴45度,真空度250Mbar,冷凝水10-13度。我认为是浓缩步骤出的问题,大家帮分析一下,到底是哪里出的问题?有没有跟好的前处理分享一下,要方法简单的哦
利用waters液相分析甲胺磷和乙酰甲胺磷时,色谱柱为反相C18,流动性为水和乙腈。但通过改变流动相比例(水:乙腈=97:3,90:10,80:20,梯度:99:1 1min,98:2,97:3),改变流速(1,0.5),柱温(30,35 ,40),两种农药一直分不开。求助各位大神帮忙,急!急!!!!
请问大家知道100ppm的甲胺磷、乙酰甲胺磷、氧化乐果的保存期限是多久吗?最近做农残标准曲线,使用的是去年8月份配的100ppm的标样,这三种农残0.1ppm的浓度基本没峰,1ppm的峰面积也明显较去年的小很多,不知是不是分解了。
求助,在使用乙酰丙酮方法测甲醛时,为何配制好的乙酰丙酮溶液颜色很黄?注:所用的乙酰丙酮原本无色,但乙酸铵有点回潮。

乙酰丙酮分光光度法测定水中甲醛的不确定度分析1.方法原理及操作流程1.1原理在过量氨盐存在下,甲醛与乙酰丙酮生成黄色化合物,该有色化合物在414nm波长处有最大吸收。1.2操作流程量取100ml试样,经预蒸馏除去干扰物质;用100ml容量瓶接受馏出液并定容。用25ml具塞比色管量取25ml试样,加入2.5ml乙酰丙酮溶液,摇匀。于45-60℃水浴中加热30min,取出冷却。用10mm比色皿在波长413nm处,以水为参比测量吸光度。同时绘制校准曲线:取6支25ml具塞比色管,依次加入0.00、0.50、1.00、3.00、5.00、8.00ml 10.0ug/ml甲醛标准使用液,再分别加水至25ml。按样品测试步骤测定出标准系列的吸光度。由校准系列所测得的吸光度减去零管的吸光度值,绘制吸光度-甲醛含量的曲线;根据测得得试样中甲醛的吸光度(试样的吸光度扣除零空白试验的吸光度)从校准曲线上查得试样中甲醛的质量(ug),再除以试份的体积(ml),即得到试样中甲醛的浓度。1.3甲醛标准使用液的配制直接购买有证标准物质37.25mg/L的甲醛标准,用15ml分度吸管准确吸取13.4ml甲醛标准溶液至50ml容量瓶中,用水稀释至刻度,摇匀,此溶液即为10.0ug/ml甲醛标准使用液。1.4样品测量通过校准曲线拟合,用乙酰丙酮分光光度法测量样品中甲醛的浓度。得数据:http://ng1.17img.cn/bbsfiles/images/2013/03/201303311454_433120_2139979_3.jpg其中u(C)—C的标准测量不确定度u(m)—m的标准测量不确定度 u(V)—V的标准测量不确定度3.测量m的标准不确定度分量测量m的标准测量不确定度分量由三部分构成,其一是由标准溶液的质量-吸光度拟合的直线求得m时所产生的不确定度,记为u1(m);其二是由甲醛标准溶液配制成不同浓度的标准溶液系列时所产生的测量不确定度,记为u2(m)。3.1 u1(m)的计算甲醛校准曲线方程表示为:y=bx+a (3)式中,x—溶液中甲醛的质量,ug y—甲醛质量为x时对应的吸光度 b—校准曲线的斜率,b=0.00996(本次样品考核数据) a—校准曲线的截距,a=-0.001(本次样品考核数据)http://ng1.17img.cn/bbsfiles/images/2013/03/201303311456_433121_2139979_3.jpg3.2 u2(m)的计算绘制校准曲线的标准系列,其甲醛的质量可用下式来表示:mi=C0×V标 (5)式中,C0—为甲醛标准使用液的浓度,10.0ug/mlV标—为标准曲线标准系列中某一浓度点对应的加入甲醛标准使用液的体积,mlmi—为校准曲线标准系列中某一浓度点对应的甲醛的质量,ug10.0ug/ml的甲醛标准使用液是由标准贮备液经过稀释得到,用公式表示为:C0=C贮/(f)(6)式中,C贮—为甲醛标准贮备液的准确浓度,ug/mlf—为稀释因子,f代表甲醛贮备液稀释倍数。直接购买37.25 ug/ml甲醛标准物质作为贮备液稀释得到10.0ug/ml甲醛使用液。标准溶液的稀释是采用15ml的分度吸管和50ml的容量瓶来完成。http://ng1.17img.cn/bbsfiles/images/2013/03/201303311457_433122_2139979_3.jpg式中,[font=Times New Roma
这两天读了苏建峰 老师的溶剂转移-气相色谱-质谱法和选择洗脱-气相色谱法测定大蒜中289种农药多残留 的文章其中有不明白的地方 就是甲胺磷、乙酰甲胺磷、氧化乐果等几个强水溶性(强极性)的农药不能进行分析 这些农药的水溶性强 为何不能再法一种直接用乙腈提取 反而要使用乙酸乙酯 ?
前处理就是用无水硫酸钠研磨然后加丙酮活性炭振摇最后悬蒸至尽干,但是很不稳定有时候出峰有时候不出峰,后来换乙酸乙酯做溶剂,敌敌畏的回收率大概20~40,而乙酰甲胺磷达到500多,各位老师这是什么原因呢,通过改电流信号或者改变程序升温会有所改善吗
测涂料中甲醛,乙酰丙酮分光光度法,乙酰丙酮溶液配制时乙酰丙酮要用重蒸馏的,怎么蒸馏
用串接质谱(包括单极)检测乙酰甲胺磷时离子碎片比不稳定,也就是说走标准,离子碎片比例都不一致。为什么?大家有没有发现过。希望专家给予答复。当然19648中也没有乙酰甲胺磷这个目标物。
最近做乙酰甲胺磷发现一个奇特的现象。方法如下,乙腈提取提取,浓缩后过某大品牌氨基柱,乙腈甲苯3:1淋洗,浓缩后甲醇水定容,上LCMSMS,定量用基质标。基质为普通白菜。上机试液理论值100ppb,结果乙酰甲胺磷没有出来,开始以为氨基柱的问题,后来一个突发想法,把试样稀释了十倍,再上机分析,乙酰甲胺磷回来了,而且值为6~8ppb,符合回收率。LCMSMS为watersuplc,TQD,C18液相柱。请问各位同行,有用液质做乙酰甲胺磷的大侠吗?基质效应那么大?







 我要推广仪器
我要推广仪器
 下载APP
下载APP