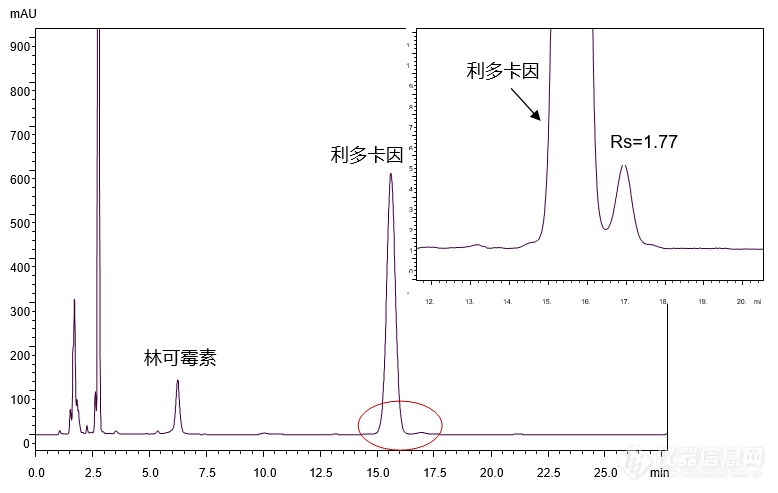
[align=center][b]【国家药品标准】林可霉素利多卡因凝胶的分析[/b][/align][align=center][b][/b][/align][align=right][b]——依据国家药品标准WS-10001-(HD-0140)-2002方法[/b][/align][b]林可霉素利多卡因凝胶[/b]为复方制剂,每克含林可霉素5毫克,利多卡因4毫克。适应症为用于轻度烧伤、创伤及蚊虫叮咬引起的各种皮肤感染。 [img=,193,127]http://ng1.17img.cn/bbsfiles/images/2018/07/201807260834522166_2994_2222981_3.gif!w193x127.jpg[/img] [img=,140,64]http://ng1.17img.cn/bbsfiles/images/2018/07/201807260834520028_3541_2222981_3.gif!w140x64.jpg[/img] 林可霉素 利多卡因 Lincomycin Lidocaine M.W.: 406.54 M.W.: 234.34客户提供林可霉素利多卡因凝胶样品,希望本实验室帮忙通过筛选色谱柱及调节分析条件,依据[color=#ff0000][b]国家药品标准WS-10001-(HD-0140)-2002[/b][/color]方法,实现林可霉素利多卡因凝胶样品的良好分析。首先,使用能在纯水条件下稳定使用的高极性色谱柱[color=#ff0000][b]CAPCELL PAK C[sub]18[/sub] AQ S5 4.6 mm i.d. × 150 mm[/b][/color],对林可霉素利多卡因凝胶样品进行分析,结果如图1所示,[color=#330099]利多卡因与其峰后杂质之间分离度为1.77[/color]。[align=center][img=,690,437]http://ng1.17img.cn/bbsfiles/images/2018/07/201807260858200006_8607_2222981_3.png!w690x437.jpg[/img][/align][align=center]图1 CAPCELL PAK C[sub]18 [/sub]AQ分析所得色谱图[/align]注:峰上标数字为分离度。[img=,528,205]http://ng1.17img.cn/bbsfiles/images/2018/07/201807260858202566_2695_2222981_3.png!w528x205.jpg[/img]为进一步提高利多卡因与其峰后杂质之间的分离度,在原条件基础上将柱温由30℃降低至25℃,并分别使用 CAPCELL PAK C[sub]18[/sub] AQ、CAPCELL PAK C[sub]18[/sub] MG及高含碳量ODS色谱柱SUPERIOREX ODS进行分析,结果如图2所示。[align=center][img=,690,490]http://ng1.17img.cn/bbsfiles/images/2018/07/201807260859201516_7229_2222981_3.png!w690x490.jpg[/img][/align][align=center]图2 25℃条件下不同色谱柱分析结果对比[/align]注:峰上标数字为分离度。[img=,637,223]http://ng1.17img.cn/bbsfiles/images/2018/07/201807260859204236_7198_2222981_3.png!w637x223.jpg[/img]如图2所示,在柱温25℃条件下使用三款色谱柱进行分析,其中,[color=#ff0000][b]CAPCELL PAK C[sub]18[/sub] AQ色谱柱分析结果最好,利多卡因与其峰后杂质分离得到最佳分离,分离度为4.23[/b][/color];[color=#330099][b]使用CAPCELL PAK C[sub]18[/sub] MG色谱柱进行分析时,利多卡因与其峰后杂质分离度为3.27[/b][/color];而使用SUPERIOREX ODS色谱柱分析时,利多卡因与其峰后杂质未得到有效分离。综上,在国家药品标准WS-10001-(HD-0140)-2002方法基础上,将色谱柱柱温由30℃降低至25℃,使用高极性色谱柱CAPCELL PAK C[sub]18[/sub] AQ及中等极性色谱柱CAPCELL PAK C[sub]18[/sub] MG进行分析,均可在25 min内完成林可霉素利多卡因凝胶样品的分析,并得到利多卡因与其峰后杂质之间的良好分离结果。[align=right][/align][align=right][/align][align=right] [/align][align=right]三耀精细化工品销售(中国)有限公司[/align][align=right]技术开发部[/align][align=right]地址:北京经济技术开发区宏达南路5号[/align][align=right]宏达利德工业园1栋418室[/align][align=right]邮编:100176[/align]
(按笔划顺序排列) 八角莲、八里麻、千金子、土青木香、山莨菪、川乌、广防己、马桑叶、马钱子、六角莲、天仙子、巴豆、水银、长春花、甘遂、生天南星、生半夏、生白附子、生狼毒、白降丹、石蒜、关木通、农吉痢、夹竹桃、朱砂、米壳(罂粟壳)、红升丹、红豆杉、红茴香、红粉、羊角拗、羊踯躅、丽江山慈姑、京大戟、昆明山海棠、河豚、闹羊花、青娘虫、鱼藤、洋地黄、洋金花、牵牛子、砒石(白砒、红砒、砒霜)、草乌、香加皮(杠柳皮)、骆驼蓬、鬼臼、莽草、铁棒槌、铃兰、雪上一枝蒿、黄花夹竹桃、斑蝥、硫磺、雄黄、雷公藤、颠茄、藜芦、蟾酥。
标准曲线法是仪器分析中最常用的方法,从标准来看GB/T22554-2010《基于标准样品的线性校准》中:1、标准曲线的浓度范围应覆盖正常操作条件下的被测量范围;2、标准样品的组分尽量与被测样品组分一致;3、标准样品的浓度值应等距离的分布在被测量范围;4、标准样品的个数至少应有3个浓度;5、每个标准点至少重复2次,这个重复是指从稀释开始;如果国家标准有相应的浓度系列推荐,尽量按国家标准。工作中我们经常采用线性校准,因为线性方程最为简洁。 标准曲线需要几个数据点,是由所检测组分的浓度范围、分析仪响应特性、干扰因素、浓度与检测信号响应类型有关的。 3个点:对于一些低浓度,特别是微量分析,并且浓度范围不是很大的,检测器响应可靠,背景干扰非常小的,则可以选用较少的工作点就行,一般有3个浓度点就足够了,有些甚至可以只用一个浓度点就行,另一个点直接用坐标原点。 4~6 个点:对于平时测量的样品浓度范围较宽,并且检测器响应不完全是一次曲线(直线)的,可以采用二次曲线或分段校正方式,以减少数据偏差,这种情况就需要多用几个数据点,如果不分段,数据点有4~6 个就够用。但如果是分段校正,则每段需要至少两个数据点。而对于一些样品无浓度范围规律的,特别是一些检测机构,如果分析仪的检测响应可靠,环境因素影响少的,可以做校正曲线。若是环境因素影响大的,则不必做校正曲线,而采用标准加入法或标准加入-一次稀释法反而简便一些。 实际上,一般是做五个点(不包括零浓度);一般以检出限的5~10倍为第一个点,以后根据1倍(或接近一倍)递增,最高浓度是最低浓度的10~20倍为宜。当然,要根据仪器的灵敏度来调整。 标曲R值是几个9才合格? 实验室应按检测标准(方法)的要求使用标准溶液或标准物质建立标准曲线。所用标准溶液或标准物质应覆盖被测样品的浓度范围。检测标准(方法)如无具体的要求,至少使用5个标样(除空白外)建立线性标准曲线,每个点重复测定1-3次,对于筛选方法,线性回归方程的相关系数不应低于0.98,对于确证方法,相关系数不应低于0.99。其实〉0.99是判断是否为线性相关的一个标准,实际应用中线性 〉0.999才是比较理想的。线性在0.99到0.999之间的监测结果只用接近最高浓度一半(中间浓度)的位置才比较准确,如果线性大于0.999的话,在整个线性范围内都会有一个比较满意的结果。如果检测的线性不好,可以减少标准的覆盖范围,将标准的浓度调整到待测样品浓度附近,这样结果也是非常准确的。例如,样品的浓度约20ppb,但在0~50ppb范围建立标准曲线,但线性非常不理想,这时可以将标准范围调整到15~25ppb之间,作五个标准。标准曲线建立后应在样品的检测中消除空白造成的影响。高于接受限的试剂空白表示与空白同时分析的这批样品可能受到污染,检测结果不能被接受。标准曲线建立后,必要时应在分析样品前加标,添加物浓度水平应接近分析物浓度或在校准曲线中间范围浓度内,加入的添加物总量不应显著改变样品基体。标准曲线有效期到底规定:(1)标准曲线属于实验室质量控制的范围,按照《实验室资质认定评审准则》中结果控制的要求:定期使用有证标准物质(参考物质)进行监控和/或使用次级标准物质(参考物质)开展内部质量控制。(2)准则中并未对“定期”进行规定,所以如果“定期”,就根据实验室的实际情况来定了。(3)既然准则上没说明,那根据一般的分析教材,当实验条件(包括药剂、人员、仪器等)发生变化时,最好重新制作标准曲线。一般来说仪器如果长期使用,并经过检定,是处于稳定状态,而人员药剂的变化往往会较大。如果说每次换个人操作都要换曲线,那工作量就太大了。每次开机都必须做标准曲线?从药品检测的要求来说,因为:1、每天所配的流动相都会有所不同,导致出峰的时间都会有一定的差异,峰面积相应都有所差异。2、检测器光能也在不断的衰弱,因此其每天的相应值也有所不同,其峰面积也有差异。基于以上原因,原则上应该是每天都要进行标准曲线校正的。标准曲线不要每次都做,但是每次必须进标准品样品;因为每次你的流动相和原来的不可能完全一样,同时仪器的状态也在变化,所以不同批次间的保留时间是不一致的,所以你必须用随行标准品来定位与定量;标准曲线不一定非要过原点,只要线性好就可以了。因为做空白的话基本上不会过原点。过原点是强制过的,实际曲线可能不过,就会造成误差。不过原点是因为有系统误差。不过系统误差的话,大家都误差也就抵消了,没有太大影响,针对是否过原点(0,0)问题,可以从原点(0,0)代表的意义来考虑:即所测组分浓度为零的时候,信号响应值(液相色谱也就是峰高或者峰面积)是否也为零。通常来说色谱分析是属于这种情况的,因而可以把(0,0)点作为一个浓度水平计入标准曲线(甚至不需要真的去配一个浓度为零的标准溶液来进样),这也是单点法定量的一个依据;标准曲线的线性范围线性范围,主要从相关系数r看,一般要求r大于等于三个九。之前做实验有时候浓度高时,线性不好,高浓度点不在标准曲线上,而是在标准曲线的下面,而且离拟合的标准曲线比较远。遇到这种情况,标准曲线的线性相关系数就很差,有时候才一个九,,如果自己用手动拟合的话,用平滑的曲线去连接所有点的话,你就会发现,如果在线性范围内,连接起来就是直线,如果超出了线性范围,连接起来就是一条弯曲的曲线。标准曲线的相关系数的有效数字该如何保留呢?GB5750.3-2006 8.2.7项有如下规定:校准曲线相关系数只舍不入,保留到小数点后出现非9的一位,如0.99989→0.9998。如果小数点后都是9时,最多保留小数点后4位。小结标准曲线法有一定的优势,也有一定的缺陷,它特别适合于大量样品的分析。但由于每次样品分析的色谱条件(检测器的响应性能,柱温,流动相流速及组成,进样量,柱效等)很难完全相同,因此容易出现较大误差。此外,标准工作曲线绘制时,一般使用欲测组分的标准样品(或已知准确含量的样品),而实际样品的组成却千差万别,因此必将给测量带来一定的误差。
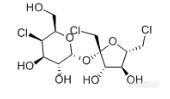
[align=center][b]GB 22255-2014 食品安全国家标准 食品中三氯蔗糖(蔗糖素)的测定——三氯蔗糖标准品分析-RI[/b][/align]三氯蔗糖(TGS),是唯一以蔗糖为原料的功能性甜味剂,甜度可达蔗糖600倍。这种[url=http://baike.sogou.com/v130009.htm][color=windowtext]甜味剂[/color][/url]具有无能量,甜度高,甜味纯正,高度安全等特点,是最优秀的功能性甜味剂之一。[align=center][img=,170,99]http://ng1.17img.cn/bbsfiles/images/2018/03/201803080920210187_4197_2222981_3.png!w170x99.jpg[/img][/align][align=center]三氯蔗糖结构式[/align]实验室前期按照《GB 22255-2014 食品安全国家标准食品中三氯蔗糖(蔗糖素)的测定》方法,使用高灵敏度气溶胶型检测器——纳克级水凝粒子计数检测器(NQAD),得到了三氯蔗糖标准品的良好分析结果。本实验按照相同条件,使用示差折光检测器(RI)对三氯蔗糖标准品进行分析。色谱柱同样选择中等极性的普适型色谱柱CAPCELL PAK C[sub]18 [/sub]MGII S5 4.6 mm i.d. × 150 mm,得到结果如图1所示。三氯蔗糖保留时间为12.400min,与标准谱图保留时间基本一致,理论塔板数为12350,不对称因子为0.95,峰形良好。[align=center][img=,690,489]http://ng1.17img.cn/bbsfiles/images/2018/03/201803080945469257_8172_2222981_3.png!w690x489.jpg[/img][/align][align=center]图1 三氯蔗糖标准品分析色谱图(0.4 mg/mL)[/align]*注:峰上标数字由下至上依次为保留时间、理论塔板数及不对称因子。[img=,472,187]http://ng1.17img.cn/bbsfiles/images/2018/03/201803080945471937_6640_2222981_3.png!w472x187.jpg[/img][align=center][img=,690,435]http://ng1.17img.cn/bbsfiles/images/2018/03/201803080946205953_7240_2222981_3.png!w690x435.jpg[/img][/align][align=center]附图:GB方法中标准色谱图[/align]接下来,按照国标要求配制三氯蔗糖工作液,0.02 mg/mL、0.05 mg/mL、0.1 mg/mL、0.2 mg/mL、0.4 mg/mL,进行线性考察实验。线性实验结果如图2所示,R[sup]2[/sup]=0.9939,得到良好线性结果。同时,由于低浓度0.02 mg/mL、0.05 mg/ mL标准品溶液均未检出色谱峰,因此根据标准曲线最高浓度的信噪比计算出检出限(以S/N=3计)约为0.17 mg/ mL。[align=center][img=,650,398]http://ng1.17img.cn/bbsfiles/images/2018/03/201803080947051037_4812_2222981_3.png!w650x398.jpg[/img][/align][align=center]图2 三氯蔗糖标准曲线图[/align]综上,按照《GB 22255-2014 食品安全国家标准食品中三氯蔗糖(蔗糖素)的测定》方法,使用示差检测器(RI)进行检测,以及CAPCELL PAK C[sub]18[/sub] MGII S5 4.6 mm i.d. ×150 mm色谱柱进行分析,可得到三氯蔗糖标准品的良好线性分析结果;但RI检测器的检测灵敏度较低。
[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]分析脂肪酸甲酯标准品,怎样处理标准品?用什么溶剂稀释?
食品分析中标准物的管理及其标准溶液的校正一、意义食品分析标准物质是分析方法质控的核心,是定性和定量的依据。标准物质以一定纯度和浓度配制的溶液称标准溶液,其稳定性受其自身降解、化学转化、溶质和溶剂挥发等内在因素的影响,又受其存放条件如温度、湿度、光线照射、存放时间、存放容器及其配制技巧等外界条件的影响。由于影响标准物及其标准液的因素较多,如果条件控制不当或管理不严密,标准物质浓度易发生变化,这是食品分析中难以进行质量控制的主要原因。以上原因也是实验室食品分析测定产生误差的最主要因素,要减小这些产生误差的主要因素,就要特别注重标准物管理,以及进行标准溶液的稳定性观察和校正工作,也是实验室质量控制关键工作之一。当标准溶液发生变化时就要重新配制,并找出变化原因,为分析工作积累经验,并可写入方法注释中。在食品分析质量控制中,准确度和精密度的提高,是以标准溶液稳定性和准确性为前提的,因此对于标准溶液稳定性和准确性的关键技术问题,是质量控制的核心问题。二、标准物质的管理 1、容量分析的基准物是标定其它标准溶液的基准,应购买基准试剂,它可以保证其纯度。另一个因素是水份的影响,用前应充分干燥和恒重,配制时量器要校正。溶剂要纯化,使用的容器要充分洗涤。最终目的都是防止基准物的化合损失。2、用于分光光度分析的标准物,分有机的和无机的标准物,无机标准稳定性好,但使用液浓度低时,极易被容器吸附,并与容器中离子进行交换,因此决定了其稀溶液使用时间短。玻璃容器在碱性介质中易溶出,塑料容器在成型时加入助剂时也含有不同金属杂质,容易溶出如Zn、Ca等金属离子。有机标准最好不放在塑料制容器中,因塑料在成型时加入的有成份比较复杂的助剂和增塑剂。标准使用液应现用现配为最佳。三、标准物存放使用1、无机的标准物要求在干燥并无化学干扰物的条件下存放,选择合适干燥剂如硅胶和分子筛。有机标准物最好分装封入安培瓶中低温避光保存。固体的多环芳烃可配制成溶液,再分装在安瓿瓶中保留溶剂封存,也可把溶剂挥掉后干燥封存,使用时再定容,后一种方法更为稳妥些。配制好一批标准溶液,再一支一支使用也是很方便的。如果液体的标准物特别是几种标准的混合物用于色谱分析同系物如醇类物质,可同时配制一批分装安瓿瓶低温存放,再一支一支使用,能避免溶剂挥发体积变化产生的误差。这种做法更适于实验室间的标准分发和校正工作。最难办的是气体,标准如氯乙烯、氟里昂,最好是钢瓶中存放,或配制钢瓶标准气。这些条件不具备时也可以选择高沸点溶剂,密封溶解这些气体,称量溶质重量,一次性使用。从这一事实出发,气体,测定误差可以稍加放宽,因为标准自身稳定性差。2、用于色谱分析的标准物要求色谱纯,其配剂溶液剂也要求色谱纯,准确配制前要在色谱上进行检查。特别是几种标准进行混合更应慎重,每种要严格检查否则给定性定量带来很多麻烦。如果纯度不够时可以纯化,再结晶或用制备色谱制备。勉强使用是无益的。四、标准溶液的校正1、从安瓿瓶分装标准溶液无论是有机的或是无机的用于校正是很方便的。如原子吸收测定金属,从安瓿瓶中取一定量配制浓度系列,再封存。每隔一段时间(1~2周)再用原溶液配制同样浓度系列,严格控制仪器条件来比较二次标准曲线的斜率,斜率下降时表明有损失。2、相同浓度同时配制的标准的几支安瓿瓶,先用打开的一支标准的测定值与间隔一段时间后打开的另一支标准的测定值进行比较,以此类推最后在一段时间内几支同时测定,其变异程度就是标准在这段时间稳定性变化程度。3、几个实验室用同一标准物分别配制相同浓度标准液,各自进行标准曲线的测定,再按规定交换该标准液再进行测定,比较测定结果差异来观察同一标准的时间和空间变异。如果标准液稳定,配制不准确的实验室很容易查出。配制都准确时,标准液若不稳定时,会使各实验室的测得值都偏低。4、同种标准物来源不同,也应采用分别配制交叉测定的办法来检查标准的纯度及配制是否准确。在食品分析中无论用何种手段分析样品,所使用的标准物应作统一的或确切的规定。例如:过硫酸铵测锰,用MnSO4·H2O作为标准使用,到底硫酸锰需要不需要烘烤呢?对于这个问题,在一部份的教科书中有规定烘烤的,也有不烘烤的。按照MnSO4·H2O的性质遇到空气可能吸潮或风化,如果直接称重计算Mn量,就有可能出现误差。用烘烤称重测得水分所含的量比理论值高1.6%,有同一硫酸锰配制锰标准溶液测一合成水样,使用烘烤后配制的锰标准溶液,测得的Mn含量为0.205mg/L,未烘烤过的则高达约2.3%,从中说明硫酸锰在配制标准溶液时应经过烘烤,使标准一致。
下午好,大家从事操作原子吸收光谱仪,我们首先是做标准曲线,一、做标准曲线就需要配制各单元素四至五个不同浓度点标准溶液;配制单元素如铅、铜、铬、汞,镉、银等,一般保质为几个月? A 、一个月期限 B、二个月期限 C、三个月期限 D、半年期限 二、原子吸收光谱仪各单元素标准溶液浓度为1000ug/ml,在配制过程中要加入不同浓度介质溶液如(10%硝酸.5%盐酸)等.最关键是各单元素生产批号, 有效期为两年,单元素有值日期;单元素标准溶液由国家标准样品证书;由国家质量监督检验检疫总批准;然而单元素两年有效已经是否如处理; A 、两年单元素有效过期就作废处理, B 、继续使用半年至一年是否合理; C 、请购新标准品;三、新购的单元素标准品买回检查生产批号已经过三个月 处理方法; A 、退换近期生产生产 批号单元素标准溶液; B 、 厂家或代理商不予退换应如何处理; C 、接收新进标准溶液,下次注意就好;四、 国家标准样品证书的标准单元素正规厂家有多少?并且获得国家质量监督检验检疫总局 批准;如国家钢铁材料测试中心、纳克标准溶液、百灵科技有限生产标准溶液等。
各位同行: 在农药残留分析中,一个讨论较多的话题就是关于基质效应的问题,一般采用添加基质的标准品来补偿基质效应,但是在具体的配制过程中,现在尚为有一个统一规范的标准,请大家都来讨论一下,各自在平时的实验中,是怎么样来操作的! 此贴只为抛砖引玉,希望大家踊跃讨论,有好的操作,我们一起学习!
急急急急急急急急急!!!!!大家谁手头有这个标准:DB45/T 607-2009 饲料中脱氧雪腐镰刀菌烯醇(呕吐毒素)的测定 竞争酶联免疫分析法,我在网上下载都无法下载,总是提示出错,所以看看大家谁有下载好的分享一下呗!谢谢。
老师同学们有无这样的经历?对已知化合物定量分析,标准品和质控样分别出自不同的厂商,皆在有效期内,定量过程线性良好,RSD合规,但是多种化合物合成的质控样一直有一个化合物无法进入合理区间范围?多次尝试无果?这个时候考虑标准品有问题还是指控样有问题?还是分析方法有问题?从哪里入手?类似问题咋解决???
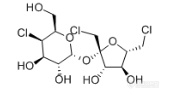
[align=center][b]GB 22255-2014 食品安全国家标准 食品中三氯蔗糖(蔗糖素)的测定——三氯蔗糖标准品分析-NQAD检测器[/b][/align]三氯蔗糖(TGS),是唯一以蔗糖为原料的功能性甜味剂,甜度可达蔗糖600倍。这种甜味剂具有无能量,甜度高,甜味纯正,高度安全等特点,是最优秀的功能性甜味剂之一。[align=center][img=,170,99]http://ng1.17img.cn/bbsfiles/images/2018/03/201803011004470313_2453_2222981_3.png!w170x99.jpg[/img][/align][align=center]三氯蔗糖结构式[/align]本实验按照[b]《GB 22255-2014 食品安全国家标准 食品中三氯蔗糖(蔗糖素)的测定》[/b]方法,使用[b][color=#ff0000]高灵敏度气溶胶型检测器——纳克级水凝粒子计数检测器(NQAD)[/color][/b]对三氯蔗糖标准品进行了分析。色谱柱选择中等极性普适型[color=#3333ff][b]CAPCELL PAK C18 MGII S5 4.6 mm i.d. × 150 mm[/b][/color],得到结果如图1所示。三氯蔗糖保留时间为12.709min,[b]与标准谱图保留时间基本一致,理论塔板数为9992,不对称因子为1.06,峰形良好。[/b][align=center][b][img=,690,497]http://ng1.17img.cn/bbsfiles/images/2018/03/201803011006155125_1559_2222981_3.png!w690x497.jpg[/img][/b][/align][align=center]图1 三氯蔗糖标准品分析色谱图[/align]*注:峰上标数字由下至上依次为保留时间、理论塔板数及不对称因子。[b][img=,633,176]http://ng1.17img.cn/bbsfiles/images/2018/03/201803011006367633_3986_2222981_3.png!w633x176.jpg[/img]附图:GB方法中标准色谱图[/b][align=center][b][img=,690,448]http://ng1.17img.cn/bbsfiles/images/2018/03/201803011007162573_9264_2222981_3.png!w690x448.jpg[/img][/b][/align][b][/b]接下来,按照国标要求配制三氯蔗糖工作液,浓度分别为0.02 mg/mL、0.05 mg/mL、0.1 mg/mL、0.2 mg/mL、0.4 mg/mL,进行线性考察实验。[b][color=#3333ff]由于NQAD检测器原理与常规蒸发光散射检测器ELSD不同,能够直接得到线性回归结果,不需要做对数方程,更加简单快捷。[/color][/b]线性结果如图2所示,R[sup]2[/sup]=0.996,得到良好线性结果。同时,我们根据标准曲线最低浓度的信噪比计算出定量限(以S/N=10计)约为3 μg/mL,[b][color=#ff0000]能够实现三氯蔗糖的高灵敏度检出[/color][/b]。[align=center][img=,658,399]http://ng1.17img.cn/bbsfiles/images/2018/03/201803011008425185_5014_2222981_3.png!w658x399.jpg[/img][/align][align=center]图2 三氯蔗糖标准曲线图[/align]

学家如今证实了考古学家和艺术史学家一直想要搞清的东西:那些来自非洲西部的古代雕塑上的光泽,可能包含源自宗教仪式上的动物祭品的血迹。这项研究利用了新的诊断技术,它有助于更好地了解古代艺术家的行为方式,同时为进一步研究这些世上最宝贵的史前古器物开辟了新的途径。 通过口述历史和图案记录,人类学家和种族学家发现了大量证据,表明古代非洲艺术家经常会在他们创作的艺术作品中涂抹宗教仪式上的动物血液,这通常是为了满足或安抚他们心目中的神灵。例如,在马里帝国(兴盛于13世纪早期至15世纪晚期),Dogon人会使用不同的颜料装饰或描绘他们的雕塑品,而这些颜料很可能就包含有动物的血液。但是由于这些古代艺术品往往年代久远,其光泽的成分很难用标准化学分析方法进行研究。 因此来自巴黎市法国博物馆研究与复原中心的一个科学家小组与法国的其他研究机构合作,使用了4种新的技术对此进行研究。通过与吉夫续尔伊凡特市法国国家研究局的质谱分析专家以及格勒诺布尔市欧洲同步加速器辐射实验室的科学家进行合作,研究人员引入了两种质谱分析方法和两种X射线扫描技术。参与该项研究的物理化学家Pascale Richardin表示,这些技术非常敏感,它们甚至能够发现几微克样品中的几个目标分子。这些技术还具有一个独特的优势,即也许一种技术并不足以发现那些科学家想要的分子,但总有另一种技术能够填补这一缺口。在这项研究中,科学家们发现了确凿无误的证据,表明铁在这些史前古器物光泽中的一种存在形式仅仅与血液有关。研究小组在即将出版的《分析化学》杂志上报告了这一研究成果。 巴黎高等师范学院的电分析化学家Christian Amatore指出,这项研究毫无疑问地表明,在宗教仪式上使用的木制器具与动物祭品有关,Amatore认为这一技术几乎是“完美的”。Richardin表示,下一步,研究人员将分析欧洲文艺复兴时期的艺术大师所使用的颜料物质——这并不是为了寻找血迹,而是为了揭示关于其作品颜色和质地的秘密[img]http://ng1.17img.cn/bbsfiles/images/2007/12/200712262201_74767_1603372_3.jpg[/img]新的诊断技术从来自古代非洲艺术品的微量样品中发现了血(小图)的痕迹。(图片提供: Werner Forman/Corbis)
核心提示:GB2760-2011与GB2760-2007标准对比分析之四——可在各类食品中按生产需要适量使用的食品添加剂卫生部在2011年第12号卫生部公告中发布了4项食品安全国家标准,其中之一即为《GB 2760-2011 食品安全国家标准 食品添加剂使用标准》(以下简称新标准),该新标准将于2011年6月20日起正式实施,新标准的前言中明确指出对2007版的《GB 2760-2007 食品添加剂使用卫生标准》(以下简称旧标准)部分内容进行了调整,食品伙伴网已经对食品分类系统、食品工业用加工助剂和食品工业用酶制剂的调整分别进行了详细的对比,详细内容见GB2760-2011与GB2760-2007标准对比分析之一——食品分类系统、GB2760-2011与GB2760-2007标准对比分析之二——食品工业用加工助剂、GB2760-2011与GB2760-2007标准对比分析之三——食品用酶制剂,本次继续对可在各类食品中按生产需要适量使用的食品添加剂进行详细的对比分析,希望通过这样的系列对比分析,食品行业人士能够对新标准的使用有更深入的了解。 本次对比分析的总体情况:旧标准中列举可在各类食品中按生产需要适量使用的食品添加剂共71种,而新标准中列举了77种,经过对比发现旧标准中有两种可在各类食品中按生产需要适量使用的食品添加剂没有在新标准中收录。新标准中新增了8种。具体结果如下: 新标准中增加的可在各类食品中按生产需要适量使用的食品添加剂名单,共8种,分别是:冰乙酸(低压羰基化法)、赤藓糖醇、甲基纤维素、抗坏血酸钠、酶解大豆磷脂、羟丙基淀粉、碳酸钠、天然胡萝卜素。 新标准中删除的可在各类食品中按生产需要适量使用的食品添加剂名单,共2种,分别是:磷酸二氢钠、磷酸氢二钠。 除了新增和删除的情况之外,分析过程中还发现名称修改及功能新增的情况,列表如下: 添加剂名称修改的情况:

[size=16px] 食品安全综合分析仪主要标准是什么 食品安全综合分析仪的主要标准包括以下几个方面: 检测项目:食品安全综合分析仪的检测项目涵盖了多个方面,主要包括营养成分、添加剂、农药残留、重金属和微生物等。这些项目的检测是确保食品安全的重要手段,可以全面了解食品的质量和安全状况。 检测精度和效率:食品安全综合分析仪需要具备高检测精度和效率,以确保测试结果的准确性和可靠性。为实现这一目标,需要选择可实现自校的仪器,并设置合理的自校时间间隔,按期校对仪器。此外,在仪器操作前也需进行校对,以及定期对仪器进行检修与保养,降低检测出现误差的可能性。 农残检测标准:食品安全综合分析仪的农残检测标准通常是由国家食品药品监管总局制定的。对于农药残留限量,一般采用“最大残留限量(MRL)”作为标准。这个限度是由卫生、环境和经济等多方面考虑制定的。在农残检测过程中,除了检测仪器的准确性外,样品的采样和制备也是非常重要的。 国家和行业标准:食品安全综合分析仪的检测方法应符合国家和行业标准。例如,对于营养成分、添加剂、重金属和微生物等的检测,应采用国家规定的检测方法,确保数据的准确性和可靠性。 仪器性能:食品安全综合分析仪应具备良好的稳定性、重复性和灵敏度等性能指标,以确保在不同环境和条件下都能获得准确的检测结果。 综上所述,食品安全综合分析仪的主要标准包括检测项目、检测精度和效率、农残检测标准、国家和行业标准以及仪器性能等方面。这些标准共同构成了保障食品安全的重要技术支持。[img=,690,690]https://ng1.17img.cn/bbsfiles/images/2024/02/202402200937557726_9841_6098850_3.jpg!w690x690.jpg[/img][/size]
1 整体框架结构GB2760-1996以及随后每年的增补品种仍然沿用1981年标准的格式,正文包括“范围”、“引用标准”、“食品添加剂品种、使用范围、最大使用量”三部分。由于对标准使用所做的解释和说明不够,往往使得食品和食品添加剂生产企业,甚至监督管理机构不能够完全正确地理解、执行。例如,某监督机构在对牛肉干制品抽检中检出微量苯甲酸就判定其产品违反GB2760规定,给予处罚,而没有充分考虑该添加剂的可能来源,如配料中使用了含苯甲酸的酱油,即没有考虑到“残留”原则而造成了错误判断。虽然这可能是由于标准的宣传贯彻不够,但客观上也说明了标准本身确实存在着缺陷,不能让标准执行者和监督管理者理解实施。CAC的GSFA则在前言中对术语定义、质量规格要求、残留原则、食品分类系统作了系统地解释、说明和要求。例如,在“进入食品中的食品添加剂残留物”章节,明确规定了食品添加剂可以因在食品配料中的残留而进入食品中的三种允许情况,很好地避免了上述问题。在列表格式方面,GB2760按照食品添加剂的功能分类排序,规定了每一种食品添加剂的使用范围和最大使用量。这种格式在七、八十年代,食品工业还不太发达,批准使用的食品添加剂范围相对狭窄的情况下是可以满足工作需要的,但是随着科学技术和食品工业的不断飞速发展,以及我国加入WTO和市场经济全球一体化进程的加速,食品添加剂使用标准的内容越来越庞杂。现有的格式在实际使用过程中存在着一些不便,例如:如果一种添加剂具有两种以上功能,就会在标准的不同段落出现,很容易查找不全 如果想查找某种食品中允许使用的食品添加剂情况,更是难上加难。GSFA的列表格式则有很多优点,表1按照食品添加剂的第一个英文字母排序,限定了每一种或一组食品添加剂允许使用的食品以及允许的最大用量,并在每种添加剂的名称后标注国际代码编号(1NS编号)和功能类别。表2与表l的资料内容相同,只不过是按照食品类别排序,便于查找。表3列出了“未规定ADI”的添加剂,当这些添加剂按照该标准前言中的良好生产规范(GMP)适量使用时,原则上可以允许在所有食品中使用。表3的附录列出了表3所不能适用的食品类别或个别食品,这些食品类别的添加剂的使用应遵循表1和表2的规定。2 食品添加剂功能类别GB2760中食品添加剂主要分为酸度调节剂、抗结剂、消泡剂、抗氧化剂、漂白剂、膨松剂、胶姆糖基础剂、着色剂、护色剂、乳化剂、酶制剂、增味剂、面粉处理剂、被膜剂、水分保持剂、营养强化剂、防腐剂、稳定和凝固剂、甜味剂、增稠剂、其他、香料共22类。GSFA将食品添加剂的功用分为酸、酸度调节剂、抗结剂、消泡剂、抗氧化剂、膨胀剂、着色剂、护色剂、乳化剂、乳化用盐、固化剂、增香剂、面粉处理剂、发泡剂、胶凝剂、被膜剂、保湿剂、防腐剂、推进剂、膨松剂、稳定剂、甜味剂、增稠剂共23类。两者的重要区别是GSFA明确指出为了保持或提高营养质量而添加的物质不包括在内,而GB2760中营养素强化剂属于其中非常重要的一大类。鉴于对营养强化剂所进行的危险性评估和标准化管理与一般的食品添加剂有所不同,建议在标准修订时,将营养强化剂的部分仍纳入GB14880之内单独修订。另外,我国与CAC对于酶制剂的管理方式也不尽相同,按照GB2760酶制剂均属食品添加剂,而GSFA是按照酶制剂的作用特点分成食品添加剂和加工助剂分别对待,这是我国应该借鉴的。3 食品添加剂品种截至2003年底,我国批准允许使用的食品添加剂近460种,其中包括120种营养素强化剂,不包括香料。目前,列入GSFA的添加剂约有370种。两者在数目上虽然相近,但在具体品种上有相当部分的差异。例如,我国允许使用的相当部分食品添加剂品种属于天然的动植物提取物,虽然有相当长时间的使用历史,但是质量规格资料较为贫乏,纯度不高或者质量不够稳定,安全性毒理学评价不够系统规范,很难为联合国粮农组织/世界卫生组织食品添加剂联合专家委员会(JECFA)所认可,自然也未被纳入GSFA。尽管经过多方努力,曾经有两种天然色素(红曲红、栀子黄)列入JECFA评价的优先名单,但是一直未能提交相关资料。甜菊糖苷也有类似问题,由于不能提供必要的质量规格资料,再次评估的时机被搁浅。JECFA的评价影响了世界上一部分国家和地区对食品添加剂的管理决策,这无形中给我国的食品添加剂工业造成了负面影响。另外,还有一部分添加剂品种的差异属于在国外或国际上已经使用多年,也在GSFA之列,但由于我国目前没有生产、使用商提出申请,也未进行暴露量评估而未纳入GB2760。对于这部分添加剂建议采取审慎态度,不宜急于采纳照搬。4 食品添加剂使用范围一食品分类GB2760没有明确的食品分类系统,对食品涵盖范围的限定不够清晰,没有给予必要的解释说明,给标准的实施执行带来一些困惑,尤其是当有些条款的规定与我国食品工业的产品分类不一致时更是如此。其次,使用范围过细和过粗的现象都有存在,例如“炸鸡调料”、“香橙果珍干粉”过细,“软饮料”过粗。另外,食品名称不规范、前后不一致的现象较多出现,如“生湿及干面条”和“生切面”的表述同时存在等等。此外,由于缺乏对食品类别的标准化编码,很难查找某种食品中食品添加剂的允许使用情况,这往往是添加剂使用和监督中最常遇到的困难。GSFA在食品分类方面提供了一个很好的模板,标准中的食品分类系统是食品添加剂使用时的定位工具。它是在欧共体食品饮料工业联盟制定的食品分类系统上修订而成的,适用于所有食品,包括那些不允许使用添加剂的食品。分为乳制品、脂肪,油和脂肪乳剂、可食冰、水果和蔬菜、糖果、谷物和谷物制品、焙烤制品、肉和肉制品、鱼和鱼制品、蛋和蛋制品、甜料、盐,调味料,汤,调味汁,沙拉,蛋白制品、特殊营养用食品、饮料、即食小食品以及复合食品品种共16大类,大类下又分若干小类,如04.1为水果,04.1.2为加工水果。需要说明的是如果允许一食品添加剂应用于一食品总类时,就理所当然地允许其应用于总类下面的所有亚类,除非另有规定。同样,如允许一添加剂应用于一亚类,也就允许其应用于该亚类下的所有次类的食品以及在此次类下所涉及的若干或个别食品。该食品分类系统简化和规范了对食品添加剂的使用规定,建议在GB2760修订时,参照GSFA食品分类系统的基本原则,分析目前GB2760涉及的食品类别品种,结合我国食品行业的有关分类,制定明确的食品分类系统框架和主要内容。5 使用规定GB2760和GSFA在使用限量规定上的差异主要表现在以下几个方面。首先,早年间GB2760的使用规定主要根据申报者提出的申请,采用危险性评估的程度不够,因此有些限量指标显得不够严谨、科学,例如规定碳酸氢钾在矿物质饮料中最大使用量为0.033g/L等。GSFA是将法典各产品标准中涉及添加剂的条款进行汇总,按照新的格式要求列入进来。同时,起草国还搜集了世界各国的几十万份数据,按照“一种添加剂如经过JECFA评价,且分别在两个国家或地区同时允许使用,便可将该产品收录”的原则,补充了新的内容,因此,它是一个相当庞杂的标准,相当部分内容不适用于我国的情况。建议在标准修订时采用危险性评估方法,结合我国消费人群的膳食暴露量以及食品中添加剂的监测数据,参考GSFA的相关内容做出规定。其次,GB2760规定某些添加剂品种可以“按生产需要适量使用”于“各类食品”,但忽略了对“各类食品”的限制,例如对于婴幼儿食品、牛乳、面粉等食品的必要限制,造成了管理上的漏洞。GSFA则将这类添加剂统一列表,规定可以按照生产需要适量使用,同时又以附件的形式列出了应该除外的食品类别和品种,从而很好地解决了这个问题。另外,对于结构、功能、安全性相同或类似的添加剂做出规定时,GB2760有时将其视为同一组添加剂统一规定,有时又分具体品种单独规定,显得凌乱。而GSFA通常将其作为一个品种规定其使用范围和最大限量,例如,GSFA在规定叶绿素铜盐类的使用时,将叶绿素铜钠盐和叶绿素铜钾盐作为一个品种规定,这样得以简化了制表的繁杂,便于使用。结语中国已经加入WTO,CAC标准对我国的食品工业发展和国际贸易将产生巨大的影响,它是目前和将来我国食品卫生国家标准制定、修订过程中的重要参考依据。因此,分析比较GB2760和GSFA的主要差异具有重要的意义。据悉,GB2760的修订工作已于今年年初启动,有关部门表示整个标准修订过程将本着科学、公开、透明的原则进行,广泛征求相关部门、行业、企业意见和建议,力争制定出既保护消费者健康又符合我国经济技术发展状况以及WTO相关要求的添加剂卫生标准。
造成样品或标准品 “溢值” 主要有以下几点原因:1)样品/标准品不正确稀释要严格按照说明书来稀释样品和标准品。2)偶合试剂不正确稀释要严格按照说明书来稀释偶合试剂,浓度过高会造成OD的 “溢值” HRP偶合试剂的不正确稀释3)所用稀释液不对只能使用相应试剂盒内的稀释液稀释样品和标准品。4)检测波长不正确要保证分光光度剂的检测波长是正确的。5)TMB底物的显色时间过长请监控显色时间。说明书中所示的显色时间可能因为温度和其它检测条件的不同而稍有变化。6)样品不适合此检测在说明书中会列出每种产品的适用标本范围,超出范围的标本可能不适于此试剂盒或者检测条件需要摸索(如调节稀释度等)7)样品/标准品污染样品或标准品的污染可能会导致检测结果 “溢值”。8)孵育时间/温度不正确要严格按照说明书来确定孵育时间和温度。9)在孵育时为盖上酶联板覆盖物在孵育时需盖上酶联板覆盖物,以防止液体蒸发或孵育期间的污染。每个试剂盒内均有足够数量的酶联板覆盖物
我一直从事血铅分析,用石墨炉[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]分光光度计。临床大夫要求我用标准血样进行质量控制,他们知道美国CDC提供这种标准血样,但他们只提供给美国的检测机构。在中国是否有机构提供或出售这种标准血样?
随着食品行业的迅速发展,食品资源的开发备受关注,而感官评价是食品最重要的评价指标,但在食品开发的过程中人为的感官评价缺乏一定的局限性和准确性,食品质构分析了研究食品在加工储藏中组织的软化与分解等,这些质构的变化会引起材料流变特性的变化。为了架起食品的感官与仪器量化之间的桥梁,质构仪、粘度计、流变仪等仪器孕育而生。 质构仪是目前对样品物性分析最常用的仪器之一,它是模拟人的触觉,分析检测触觉中的物理特征的仪器,在其主机和机械臂和探头连接处有个力学感应器,能感应样品对探头的反作用力,并将这种力学信号由专门的软件翻译成数字和图形信号,直接快速的记录样品的受力情况,从而根据受力情况分析所测定样品的硬度、脆性、弹性、回弹力、粘合性、粘结力、粘稠度、弯曲能力、破裂/断裂力、酥脆性、脆度、咀嚼性、胶粘性、拉伸强度、延展性等。不同的样品之间存在不同的物理特性,在采用质构仪分析样品时,可以针对性的选择探头和确定测定的指标,质构仪分析的样品包括面制品、烘焙食品、肉制品、米制品、乳制品、鱼、糖果及果蔬等。食品流变学的研究起步较早,但是由于食品体系的复杂性,早期流变学的研究主要是一些经验性的测定,例如产品在自身质量下其流动性、铺展性和碎裂性的测定等。近年来由于食品科学工作者为了提高对食物加工性,特别是食品的深加工性、工艺及设备设计的依据性等的需要,食品流变学的研究变得愈来愈广泛。随着研究活动的深入,研究手段亦有了较大地发展,表现在先进的流变学测试仪器的引入和开发。应用先进测试仪器,使实验与研究在建立食品物料的流变特性力学模型上更为方便。早前流变学主要采用粘度计分析食品的粘度,随着流变技术的发展,流变仪的发明成为食品流变分析较为精密的仪器。 粘度计包括毛细管式、旋转式、振动式3种,常见的旋转式粘度计是锥板式粘度计,它主要包括一块平板和一块锥板。电动机经[url=https://baike.baidu.com/item/%E5%8F%98%E9%80%9F%E9%BD%BF%E8%BD%AE][color=windowtext]变速齿轮[/color][/url]带动平板恒速旋转,依靠毛细管作用使被测样品保持在两板之间,并借样品分子间的摩擦力而带动锥板旋转。在扭矩检测器内的扭簧的作用下,锥板旋转一定角度后不再转动。此时,扭簧所施加的扭矩与被测样品的分子内部摩擦力(即粘度)有关:样品粘度越大,扭矩越大。扭矩检测器内设有一个可变电容器,其动片随着锥板转动,从而改变本身的电容数值。这一电容变化反映出的扭簧扭矩即为被测样品的粘度,由仪表显示出来。特点:1)测量轴的轴向负荷主要由吊丝,或者弹簧片、锥形宝石轴承支撑,因此精度低,摩擦力大;2)其扭矩测量系统,灵敏度低,对瞬时粘性力拒相应严重滞后;3)旋转粘度计的转速控制系统一般时开环的(无反馈控制),通过齿轮结构改变转速,很难实现低切变下的平稳运行。总的来说,旋转粘度计不具有动态特性,只能用于测量非牛顿流体在稳态下的相对粘度。粘度计的适用范围很广,包括各种油脂、油漆、油墨、涂料、塑料、浆料、橡胶、乳胶、洗涤剂、树脂、炼乳、奶油、药物、以及化妆品等各种流体的粘度,是纺织、化工、石油、机电、医药、食品、轻工、建筑等行业以及大专院校、科研单位、军工部门的实验室、分析室必备仪器。 流变仪包括毛细管式、旋转式、界面式3种,其中旋转式流变仪两种,一种是驱动一个夹具,测量产生的力矩,这种方法最早是由Couette在1888年提出的,也称为应变控制型,即控制施加的应变,测量产生的应力;另一种是施加一定的力矩,测量产生的旋转速度,它是由Searle于1912年提出的,也称为应力控制型,即控制施加的应力,测量产生的应变。旋转式流变仪相当于旋转粘度计的升级版本,流变仪的测量轴的轴向载荷和径向载荷都是采用空气轴承(或者磁力轴承)进行支撑,没有机械部件,因此测量轴无摩擦,精度高,可靠性好,寿命高。其次,由空气轴承和无摩擦传感器组成的力拒测量系统,灵敏度高,重复性好,能快速响应瞬时粘性力拒的变化。最后,流变仪采用负反馈速度控制系统可以实现无级调速,并且能够在极低的切变速度下平稳运行,因此流变仪具有良好的动态特性,不但能够准确的测量非牛顿流体的稳态流变特性(表观粘度),还能准确的测量流体的其他动态特性——触变性和粘弹性、屈服应力等指标。由上述分析不难看出,流变仪的功能远远强于传统的粘度计,不仅仅能够测量 食品的稳态特性——粘度,还能对浆料的触变性、粘弹性屈服应力等动态指标进行测量,能为我们提供更丰富的关于食品特性的数据,对于指导食品深加工具有重要的意义。总之,粘度计和流变仪在实际应用中食品的特异性决定其使用的广泛性,而质构仪在食品的物性分析方面,绝对是一台有效的、全面的分析食品的质构特性的仪器,在科研院校和企业研发部门均得到了广泛的推广。【关于保圣】上海保圣实业发展有限公司网址:[url]http://www.shbosin.com/[/url]售后服务:021-37656257销售咨询:18117403825 13564769697在线QQ咨询:3152715460 3011823639E-mail:[email]bsen001@vip.163.com[/email] [email]shbosin@163.com[/email]地址:上海市松江工业区茸梅路1108号传真:021-61769285微信公众号:保圣科技仪器
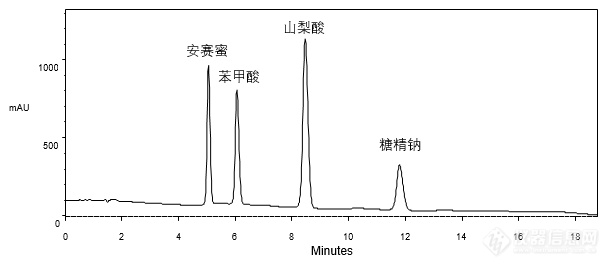
[align=center][b]GB 5009.28-2016食品安全国家标准 食品中苯甲酸、山梨酸和糖精钠的测定[/b][/align][align=center][b] ——标准品与乳品实际样品的分析[/b][/align][align=center][/align][align=left]本实验按照《GB5009.28-2016 食品安全国家标准食品中苯甲酸、山梨酸和糖精钠的测定》方法,分别对安赛蜜、苯甲酸、山梨酸、糖精钠的标准品混合溶液及加标乳品样品进行了分析。首先,使用CAPCELL PAK C[sub]18[/sub] MG S5 4.6 mm i.d. × 150mm色谱柱,对标准品混合溶液进行分析,如图1,安赛蜜、苯甲酸、山梨酸、糖精钠标准品均得到了良好的分析结果。[/align][align=left][/align][align=center][img=,611,268]http://ng1.17img.cn/bbsfiles/images/2018/03/201803221532276656_9890_2222981_3.png!w611x268.jpg[/img][/align][align=center]图1 标准品混合溶液分析色谱图[/align][img=,400,200]http://ng1.17img.cn/bbsfiles/images/2018/03/201803221532280132_6863_2222981_3.png!w400x200.jpg[/img][align=left][/align][align=left]其次,对乳品加标样品进行分析,如图2,糖精钠(Rt 12 min)与其后杂质峰之间未能取得基线分离,分离度仅为1.02。[/align][align=left][/align][align=center][img=,668,335]http://ng1.17img.cn/bbsfiles/images/2018/03/201803221533054905_2223_2222981_3.png!w668x335.jpg[/img][/align][align=center]图2 加标乳品样品分析色谱图[/align][align=left][img=,406,203]http://ng1.17img.cn/bbsfiles/images/2018/03/201803221533317202_2333_2222981_3.png!w406x203.jpg[/img][/align][align=left][/align][align=left]为改善糖精钠与杂质间的分离,在国标方法基础上,将流动相由[b]乙酸铵 / 甲醇 = 95 / 5[/b]调整为[b][b]乙酸铵 / 甲醇[/b][color=red]([/color][color=red]2 mmol/L [/color][color=red]甲酸)[/color]= 92 / 8[/b],再次对混合标准溶液和加标样品进行分析,结果如图3所示。[/align][align=left][/align][align=center][img=,690,545]http://ng1.17img.cn/bbsfiles/images/2018/03/201803221534141056_4073_2222981_3.png!w690x545.jpg[/img][/align][align=center]图3 混标与加标乳品样品分析色谱图[/align][align=left][img=,464,171]http://ng1.17img.cn/bbsfiles/images/2018/03/201803221535548985_7176_2222981_3.png!w464x171.jpg[/img][/align][align=left][/align][align=left]如图3,在酸性条件下,出峰顺序发生了变化,安赛蜜保留时间略有缩短,糖精钠保留时间明显缩短,由12 min缩短至8 min,苯甲酸和山梨酸保留时间分别延长至2 min和6 min;在分离度方面,糖精钠与苯甲酸之间分离度为2.79,苯甲酸与峰后杂质间分离度为2.04,所有色谱峰之间都达到了基线分离。[/align][align=left][/align][align=left]为使客户有更多选择,实验室又在国标原方法条件下继续筛选色谱柱,最终使用SUPERIOREX ODS S5 4.6 mm i.d. × 250 mm色谱柱时,仅微调有机相比例即可实现加标乳品样品的良好分析结果。如图4,杂质峰与糖精钠之间分离度达到2.48,达到基线分离要求。[/align][align=left][/align][align=center][img=,580,332]http://ng1.17img.cn/bbsfiles/images/2018/03/201803221537130173_1058_2222981_3.png!w580x332.jpg[/img][/align][align=center]图4 加标乳品样品分析色谱图[/align][align=left]*注:峰上标所示数字由下至上依次为分离度与不对称因子。[/align][align=left][img=,326,177]http://ng1.17img.cn/bbsfiles/images/2018/03/201803221537540634_9437_2222981_3.png!w326x177.jpg[/img][/align][align=left][/align][align=left]综上所述,按照国标《GB 5009.28-2016 食品安全国家标准食品中苯甲酸、山梨酸和糖精钠的测定》方法进行分析,使用CAPCELL PAK C[sub]18 [/sub]MG色谱柱对标准品混合溶液能得到良好分析结果,但在对加标乳品样品进行分析时,糖精钠与样品中的杂质未能实现基线分离,通过在流动相中添加甲酸可实现安赛蜜、糖精钠、苯甲酸、山梨酸及杂质的基线分离;另一方面,使用SUPERIOREX ODS色谱柱,在原条件基础上微调即可实现乳品中安赛蜜、苯甲酸、山梨酸、糖精钠及杂质间的良好分离。[/align]
国内最大最专业的国家标准物质服务平台坛墨质检-国家标准物质中心(北京坛墨质检科技有限公司),是国家质检总局指定的国家标准物质研制单位,是国内最大最专业的食品、环境、职业卫生标准物质生产商和服务商。BW6104 6-羟基芹菜素-6-O-葡萄糖-7-O-葡萄糖醛酸苷,对照品,有报告 HPLC≥98% BW6105 6-羟基山奈酚-3,6,7-三-O-葡萄糖苷,对照品,有报告 HPLC≥98% BW6106 6-羟基山奈酚-3-O-芸香糖-6-O-葡萄糖苷,对照品,有报告 HPLC≥98% BW6107 多根乌头碱,对照品,有报告 HPLC≥98% BW6108 塔拉萨敏/塔拉乌头胺,对照品,有报告 HPLC≥98% BW6109 德尔塔林,对照品,有报告 HPLC≥98% BW6110 硬飞燕草碱,对照品,有报告 HPLC≥98% BW6111 尼奥林/雪上一支蒿乙素,对照品,有报告 HPLC≥98% BW6112 10-羟基乌头碱,对照品,有报告 HPLC≥96% BW6113 13-去羟基印乌碱,对照品,有报告 HPLC≥98% BW6114 黄芪甲苷/黄芪皂苷IV,对照品,有报告 HPLC≥98% BW6115 异黄芪皂苷I,对照品,有报告 HPLC≥98% BW6116 异黄芪皂苷II,对照品,有报告 HPLC≥98% BW6117 (3β,6α,16β,20R,24S)-3-O--20, 24-环氧-16,25-二羟基-9,19-环羊毛甾烷-6-O-葡萄糖苷,对照品,有报告 HPLC≥97% BW6118 Cyclocephaloside II HPLC≥98% BW6119 罗汉果皂苷IVa,对照品,有证书 HPLC≥98% BW6120 异-罗汉果皂苷V,对照品,有报告 HPLC≥98% BW6121 罗汉果皂苷IIIA1,对照品,有报告 HPLC≥98% BW6122 罗汉果皂苷IIA2 HPLC≥98% BW6123 罗汉果皂苷IIIe HPLC≥98% BW6124 罗汉果黄素,对照品,有报告 HPLC≥98% BW6125 11-0-罗汉果苷V HPLC≥98% BW6126 罗汉果苷IV/罗汉果皂苷IVe,对照品,有报告 HPLC≥98% BW6127 赛门苷I,对照品,有报告 HPLC≥98% BW6128 罗汉果苷VI HPLC≥90% 坛墨质检现有员工79人,办公室面积450平米,实验室1650平米;销售、客服、财务及行政人员35人,实验室工作人员21人,库房14人,市场部8人。实验仪器设备:气相色谱、液相色谱、气质联用、液质联用、离子色谱、紫外分光光度计,原子吸收、ICP-OES和ICP-MS;库房面积450平米,库房工作人员12人,现货产品5万个,坛墨质检自主研发的产品近3000个,已申报国标345项,填补国内空白的产品达到65项。坛墨质检是国内唯一提供标准溶液定制服务的标准物质研制单位,定制范围:特殊浓度定制、特殊溶剂定制、混标定制。
分析检测中绘制标准曲线目的是可以根据标准曲线查出待测物质的含量,所以标曲的制定是实验室工作中必不可少的工作。做标准曲线真的那么简单?你所做的标曲真的做好了吗?导致标准曲线弯曲的原因是啥?RSD值、线性相关系数和线性方程如何?线性好与不好分别由于哪些原因造成的?是仪器、标液,还是配制的问题? 前期测定时注意: 1、仪器校验好。 2、移取液体体积要精确,保证迅速准确,尽可能用一根移液管;为减小人为误差,同一种液体要一个人操作。 3、保证标准品的纯度,所用标准品最好是新打开的,纯度较高的固体或溶液,防止污染。 4、容器要保证洁净。 5、根据标准品理化性质注意加样的先后顺序。 6、如果要测OD值,应保证测前各管液体充分混匀。 7、若还是有某个点误差较大,应舍弃。 另外: 1.样品的浓度等指标是根据标准曲线计算出来的,所以首先要把做标准曲线看作是比做正式实验还要重要的一件事,否则后面的实验结果无从谈起。 2、设置标准曲线样品的标准浓度范围要有一个比较大的跨度,并且要能涵盖你所要检测实验样品的浓度,即样品的浓度要在标准曲线浓度范围之内,包括上限和下限。而对于呈s型的标准曲线,尽量要使实验样品的浓度在中间坡度最陡段,即曲线几乎成直线的范围内。 3、最好采用倍比稀释法配制标准曲线中的标准样品浓度,这样就能够保证标准样品的浓度不会出现较大的偏离。 4、检测标准样品时,应按浓度递增顺序进行,以减少高浓度对低浓度的影响,提高准确性。 5、标准曲线的样品数一般为7个点,但至少要保证有5个点。 6、做出的标准曲线相关系数因实验要求不同而有所变动,但一般来说,相关系数r至少要大于0.98,对于有些实验,至少要0.99甚至是0.999。 分光光度法绘制标准曲线所遇问题? 1、单色光纯度不够 问题:标准曲线上端向下弯曲。 措施:光度法中要求在最大吸收峰处测定吸光度,光度计的有效谱带宽度越窄越好,有利于获得纯度高的单色光。 2.比色皿的厚度或光学性能不一致 如果装试剂空白液的比色皿较其它比色皿薄或对光的吸收和反射少一些,则曲线的延线与纵坐标相交;反之,与横坐标相交。 3.显色反应和反应条件的问题 当显色反应的灵敏度不高时,被测物低于某一浓度就不能显色,当浓度不同时,溶液对光的吸收、散射的程度不同,低浓度段往往弯曲,加上浓度高时部分胶体颗粒的聚沉,致使测定的吸光度降低,曲线上端向下弯曲。 4.操作上的原因 问题:褪色反应,显色溶液的颜色不稳定、易褪色,若比色时间超过了稳定时间,标准系列中高浓度溶液颜色减褪,致使曲线上端向下弯曲。 措施:控制好显色剂的加入时间,可以每加3份,停3~3min,再加上3份,依次进行,以使每支比色管的显色时间相近。 5、干扰物质的影响 标准曲线绘制完毕了,该如何检验? 标准曲线的检验是实际操作中最大的难点,也是工作中误区和争议最多的话题了! 先来了解下最重要的三大检验项目: 1.精密度检验 标准曲线的精密度检验:精密度检验就是看试验点距离拟合的直线的距离有无异常,所以也称线性检验(拟合检验),需用F检验,P0.05作为线性检验合格的标准。 2.截距检验 3.斜率检验 标准曲线的截距检验和斜率检验分别考察Y=a+bX中a和b与0的统计学差异,a与0有差别说明有试剂空白或系统误差,而b若与0没差别说明仪器的灵敏度根本达不到分析要求。 标准曲线的检验应该是线性检验结合失拟检验,以及残差的正态性检验结合才是统计学上比较完备的。 标准曲线常见问题汇总 问题一:标准曲线需要人为的增加(0,0)点吗? 答:不能。通常的标准系列多是配制0,1,2,3mg/L系列这样的说法,没做实验人为添加(0,0)很不妥,因为很多时候0管进入仪器可能也有响应值,这也是我们考察试剂空白的一个重要步骤。这个0管在某些时候非常重要,如,全血铅测定,我们采用牛血清来基体匹配标准系列,如果此时你用酸做空白或没做实验人为添加(0,0),那你就很难做好标准曲线,所以,标准曲线的0管也是做好标准曲线的重要考虑点。 问题二:标准曲线需要减掉试剂空白来做吗? 答:不需要。仪器测出来标准系列的响应值可以减掉试剂空白或减掉0管的响应值来做,工作中我们也常用0管来做仪器调零。其实没有必要那么麻烦,即使空白或0管有响应值,在构建标准曲线时,我们已经认为该响应值就是0浓度,也就是扣除了这个空白的。 问题三:什么时候用Y=bx和二次曲线呢? 答:标准曲线我们通常采用的是Y=a+bX,曲线拟合完必须要做统计检验且要做统计完备的线性检验和失拟检验,然后再做a与0的差别检验,如果a与0的统计学上无差异,你就可以考虑用Y=bX的拟合曲线,拟合出来后同样做线性检验和失拟检验,如果线性检验合格(P0.05)此时你就可以采用Y=bX。二次曲线的采用同样是这样的道理,如果你Y=a+bX时拟合不合格,你就考虑用Y=a+bX+cX2,同样做失拟检验,考察拟合的符合情况。如果Y=a+bX和Y=a+bX+cX2都满足拟合检验和失拟检验合格,则采用Y=a+bX形式,这样符合统计学上参数最少的统计简洁性原则。 问题四:标准曲线去查含量时是先减空白信号算样品含量还是先算出空白含量相减呢? 答:工作中我们常要减掉空白得到样品含量,现有国家标准方法有的推荐先算出空白含量,用样品含量相减,也有推荐先用样品信号减空白信号然后去标准曲线推算含量。而且这两种算法常常差距很大。其实这种差距往往是低含量水平时才出现,在低含量水平通过标准曲线推算含量时,本身不确定度就很大。这两种方法都可以。个人推荐先用样品信号减空白信号然后去标准曲线推算含量,因为这样出来的含量不确定度要小一些,而先算出空白含量再相减就增加了1次标准曲线推算含量时的不确定度,因为好的测量永远是不确定度小的测量。 结语 要想绘制出合格的标准曲线、使用好标准曲线,真心不易,必须将以上各个方面的条件都考虑进去,即对标准曲线的绘制也实行质量控制,只有这样,才能得出理想的标准曲线。
国内最大最专业的国家标准物质服务平台坛墨质检-国家标准物质中心(北京坛墨质检科技有限公司),是国家质检总局指定的国家标准物质研制单位,是国内最大最专业的食品、环境、职业卫生标准物质生产商和服务商。 产品编号 产品名称 标准值 BW61066-羟基山奈酚-3-O-芸香糖-6-O-葡萄糖苷,对照品,有报告HPLC≥98%BW6108塔拉萨敏/塔拉乌头胺,对照品,有报告HPLC≥98%BW6109德尔塔林,对照品,有报告HPLC≥98%BW6110硬飞燕草碱,对照品,有报告HPLC≥98%BW6111尼奥林/雪上一支蒿乙素,对照品,有报告HPLC≥98%BW611210-羟基乌头碱,对照品,有报告HPLC≥96%BW611313-去羟基印乌碱,对照品,有报告HPLC≥98%BW6114黄芪甲苷/黄芪皂苷IV,对照品,有报告HPLC≥98%BW6115异黄芪皂苷I,对照品,有报告HPLC≥98%BW6116异黄芪皂苷II,对照品,有报告HPLC≥98%BW6117(3β,6α,16β,20R,24S)-3-O--20, 24-环氧-16,25-二羟基-9,19-环羊毛甾烷-6-O-葡萄糖苷,对照品,有报告HPLC≥97%BW6118Cyclocephaloside IIHPLC≥98%BW6119罗汉果皂苷IVa,对照品,有证书HPLC≥98%BW6120异-罗汉果皂苷V,对照品,有报告HPLC≥98%BW6121罗汉果皂苷IIIA1,对照品,有报告HPLC≥98%BW6122罗汉果皂苷IIA2HPLC≥98%BW6123罗汉果皂苷IIIeHPLC≥98%BW6124罗汉果黄素,对照品,有报告HPLC≥98%BW6126罗汉果苷IV/罗汉果皂苷IVe,对照品,有报告HPLC≥98%BW612511-0-罗汉果苷VHPLC≥98%BW6127赛门苷I,对照品,有报告HPLC≥98%BW6128罗汉果苷VIHPLC≥90%BW6129罗汉果苷III,对照品,有证书HPLC≥98%BW6130拟人参皂苷Rh2,对照品,有证书HPLC≥98%BW6131(S型)原人参三醇,对照品,有报告HPLC≥98% 坛墨质检现有员工79人,办公室面积450平米,实验室1650平米;销售、客服、财务及行政人员35人,实验室工作人员21人,库房14人,市场部8人。实验仪器设备:气相色谱、液相色谱、气质联用、液质联用、离子色谱、紫外分光光度计,原子吸收、ICP-OES和ICP-MS;库房面积450平米,库房工作人员12人,现货产品5万个,坛墨质检自主研发的产品近3000个,已申报国标345项,填补国内空白的产品达到65项。坛墨质检是国内唯一提供标准溶液定制服务的标准物质研制单位,定制范围:特殊浓度定制、特殊溶剂定制、混标定制。
微生物学检验方法国家标准的演变, 建国五十年来,我国食品卫生微生物学检验方法的发展经历了三个阶段。 ——方法逐步建立、统一阶段(1949~1976年) ——方法标准化形成阶段(1977~1984年) ——不断修订和完善阶段(1985年至今) 上海市疾病预防控制中心 国家标准检验方法的演变 GB 4789-94主要修订内容:菌落总数的培养时间由24小时改为48小时;对小肠结肠炎耶尔森氏菌、空肠弯 曲菌、副溶血性弧菌进行了补充和改进;增加了单核细胞增生李斯特氏菌、椰毒假单胞菌酵米面亚种、罐头食品商业无菌、金黄色葡萄球菌肠毒素检验方法, 沙门氏菌、志贺氏菌和致泻大肠埃希氏菌的肠杆菌科噬菌体检验方法。新修订后的GB 4789-94共31项,其检验项目、方法水平与FDA 1992年出版的微生物分析方法手册相近,但未列入嗜水气单胞菌和霍乱弧菌的检验。
国内最大最专业的国家标准物质服务平台坛墨质检-国家标准物质中心(北京坛墨质检科技有限公司),是国家质检总局指定的国家标准物质研制单位,是国内最大最专业的食品、环境、职业卫生标准物质生产商和服务商。 BW6109德尔塔林,对照品,有报告HPLC≥98%BW6110硬飞燕草碱,对照品,有报告HPLC≥98%BW6111尼奥林/雪上一支蒿乙素,对照品,有报告HPLC≥98%BW611210-羟基乌头碱,对照品,有报告HPLC≥96%BW611313-去羟基印乌碱,对照品,有报告HPLC≥98%BW6114黄芪甲苷/黄芪皂苷IV,对照品,有报告HPLC≥98%BW6115异黄芪皂苷I,对照品,有报告HPLC≥98%BW6116异黄芪皂苷II,对照品,有报告HPLC≥98%BW6117(3β,6α,16β,20R,24S)-3-O--20, 24-环氧-16,25-二羟基-9,19-环羊毛甾烷-6-O-葡萄糖苷,对照品,有报告HPLC≥97%BW6118Cyclocephaloside IIHPLC≥98%BW6119罗汉果皂苷IVa,对照品,有证书HPLC≥98%BW6120异-罗汉果皂苷V,对照品,有报告HPLC≥98%BW6121罗汉果皂苷IIIA1,对照品,有报告HPLC≥98%BW6122罗汉果皂苷IIA2HPLC≥98%BW6123罗汉果皂苷IIIeHPLC≥98%BW6124罗汉果黄素,对照品,有报告HPLC≥98%BW6126罗汉果苷IV/罗汉果皂苷IVe,对照品,有报告HPLC≥98%BW612511-0-罗汉果苷VHPLC≥98%BW6127赛门苷I,对照品,有报告HPLC≥98%BW6128罗汉果苷VIHPLC≥90%BW6129罗汉果苷III,对照品,有证书HPLC≥98%BW6130拟人参皂苷Rh2,对照品,有证书HPLC≥98%BW6131(S型)原人参三醇,对照品,有报告HPLC≥98%BW6132(R型)人参皂苷Rh1HPLC≥98%BW6133(R型)人参皂苷Rh2,对照品,有报告HPLC≥96% 坛墨质检现有员工79人,办公室面积450平米,实验室1650平米;销售、客服、财务及行政人员35人,实验室工作人员21人,库房14人,市场部8人。实验仪器设备:气相色谱、液相色谱、气质联用、液质联用、离子色谱、紫外分光光度计,原子吸收、ICP-OES和ICP-MS;库房面积450平米,库房工作人员12人,现货产品5万个,坛墨质检自主研发的产品近3000个,已申报国标345项,填补国内空白的产品达到65项。坛墨质检是国内唯一提供标准溶液定制服务的标准物质研制单位,定制范围:特殊浓度定制、特殊溶剂定制、混标定制。
无意中看到一首情诗,通篇234个字,有124个字是药名,内藏54味药,其中一味是隐藏的。看看你能找出多少来!据说,学医的找出40味药算及格,找出45味算B等,找出50味才是A。 策海马,执虎杖。 轻登重楼,急越常山。 欲照九里明,耀出千里光, 忽闻神曲绕竹黄,白芷通草欲降香。 天冬未见仙入凡,只见佳人紫菀旁。 我心为伊小飞扬,爱将紫草换丁香。 人参没药君佩兰,寸心龙骨续断缘。 岂闻首乌换雪胆,几人能懂女贞香。 丽影轻舞西河柳,空有沉香在莲房。 三月十日蛇床上,徐长卿兮配紫萱。 苁蓉豆蔻白僵蚕,方藤牵牛欲合欢。 六月雪中一点红,留得一轮苏合香。 木棉林小鹰不泊,男儿远志在四方。 雪上一枝蒿如月,腊梅花开无需赏。 寻梦千里路路通,钩藤狼毒不需还。 功成名就便当归,王不留行速还乡。 身着彩衣桂枝冠,轻粉朱砂是新娘。 与伊连理金不换,誓欲百合到天荒。
O.999,则该校准曲线可判定为合格。 c.精密度评价 一般用高、中、低三种浓度的标准溶液,用相同的方法分别进行多次平行测定,并应分散在一段适当长的时间里进行分析,计算相对标准偏差、评价实验方法精密度。重复分析是精密度检验的一种手段,它反过来会影响结果。重复可以是一批样品中邻近的部分(检验重复性)或者随机放置的部分(检验漂移)。盲样分析是有效进行重复分析的一种方式,并且也为精密度的评价提供了一种手段。它包括可能由实验室管理人员进行的分批试样的重复测试部分。 d.准确度评价 可以用测量标准物质或将不同浓度的标准物质加到实际样品中做回收率测定来评价分析方法准确度。或者采用具有可比性的其他分析方法,根据所得结果的符合程度来估计测定的准确度。 在分析过程中用到的各种标准和化学校准物能够检验分析过程对被分析物的响应是稳定的。 用标准物质进行准确度评价时,对标准物质进行分析测定,计算平均值(;)和标准偏差(s),用£检验法将分析结果与标准物质的含量进行比较,判断分析方法的准确度。 测定样品加标回收率时,在测定成批样品时,随机抽取10%~20%的样品,加入一定量的待测组分的标准物质,与样品一起在相同条件下进行分析,并计算百分回收率 尸(%)===垒二鱼×lOO%. m式中:P——加入标准物质的回收率; m——加人标准物质的量; 27。——加标样品测定值;. 27。——样品测定值。 一般要求,被测定物质的回收率应达85%~110%。 ②质量控制图 每种分析方法都存在变异,都受时间、空间的影响,即使在理想条件下获得的一组数据,也存在随机误差。当某一结果超出了随机误差的允许范围时,统计学即可判断这一结果异常或不可信。根据一组连续测定数据,可用质控样的值z与标准偏差s,在一定的置信水平,绘制成质量控制图。从概率意义上说,测定结果有99%0的几率应在z I-3s(上、下控制线)内;95%应在x-+-2s(~、下警告线)内;68%应在at~s(上、下辅助线)内,这样质控图就确定了这种误差是否在一定允许范围内。 在常规样品分析过程中,每分析一批样品插入一个“质量控制样品”,或者在分析大批样品时,每隔10~20个样品插入一个“质量控制”,其分析方法应与试样完全相同,并至少独立分析20次以上,然后以实验测定结果为纵坐标,实验顺序为横坐标绘制而成。常用的质量控制图有精密度控制图和准确度控制图。 a.精密度控制图(均值控制图) 以测定结果的平均值z(应指明是哪一组测量的平均值)为控制图的中心线,并计算出测量值的标准偏差s,以j±2s作为上、下警告限,用虚线表示;j±3,作为上、下控制限。 这一控制图通常用来控制精密度,因此称精密度控制图。 b.准确度控制图(回收率控制图)用在分析样品中加入标准物质的百分回收率制作质控图,来控制样品分析的准确度。在理想状态下,向样品中加入已知标准物质的回收率应为100~/{o,但对于任何一种分析方法而言,由于存在各种因素的干扰,不可避免产生误差,使测得的回收率通常大于或小于100 9/6,回收率越趋近于100%,说明准确度越高。因此,利用回收率的平均值作中心线,计算回收率的标准差作为控制范围,就可绘制成百分回收率质控图。向不同浓度的样品中加入不同的已知量的标准物质,测得回收率数据,计算百分平均回收率F及其标准偏差,,,以F±2。,为E、下警告限,P±3sp为上、下控制限,绘制成准确度控制图。 , 10 I 5 序号或日期 图5.3准确度控制示意图 在进行样品分析时,将“质量控制样品”插入样品组内,在相同条件下进行分析测定,如果测定结果在控制图中的警告限内,说明测定过程处于控制状态;如果在警告限外,但仍在控制限内,则表示应进行初步检查;如超出控制限,表示测定过程失控,应找出原因并纠正;如果所有测定结果均在控制限内,但有七个结果连续在中心线的同一侧,亦为异常,应查明原因并加以纠正。 适当的质量控制级别是实验室管理的职责。在日常分析中,5%的内部质量控制级别是合理的并得到广泛承认。也就是说,每20个分析样品中应有一个样品是质量控制样品。然而,对于具有较高样品处理量的稳定日常方法,也许较低级别的质量控制更为合理。对于比较复杂的分析方法,20%o的质量控制级别是常见的,有时甚至需要50%的级别。对于不常进行的分析,在每一种情况下均应进行全面系统的方法确认。这通常涉及被分析物已被定值或浓度已知的(有证)标准物质使用,在这之后伴随着样品和添加样品(人为添加已知量被分析物的样 (2)实验室间质量控制 一个实验室根据分析质量要求与同行实验室进行比较,从而监测自身测试能力。 实验室间质量控制的一种公认方式是参加能力验证计划。能力验证结果可作为检验质量保证的一种手段。通常情况下,如果没有与实验室期望检验相关类型的能力验证计划可以利用,这时可以采用: ①用标准物质作平行测定 通常由中心实验
很多人看到这个题目可能无法理解,也无法将其联系起来,然而如果将色谱分析的样品的结果当作产品的话,或许大家会豁然开朗。 在这里我通过比喻来分析色谱分析样品的过程,这个分析结果的不同要求和档次比喻成是生产产品、制作工艺品还是创作艺术品。 我对产品、工艺品和艺术品的理解是,产品在今一般是机器批量生产的,有一定的生产标准,方法成熟,由于其往往是批量检测,价格就比较低,产品风格一致,结果容易比较,无需具体操作者更改分析过程。这种检测,前篇一律,标准化。 而工艺品就不一样,像限量版,就是小批量生产,大多由专业人员定做,同一类型的工艺品,由于是手工制作,相互之间存在一定的差异,这种特定设计,虽有规范但不标准,价格就高于产品了,但受时间限制,并不完善。这种级别,比上面低,大多作为自己的标准,而不普遍使用。当然如果标准化了,就可以升级为产品。 艺术品就不一样了,它是高层次的专业人员创作,有特定的艺术价值,其创作过程很长,倾注了作者的大量新血,而且作品一般是孤品,其市场的价格往往没有标准,因人而已。如果放到色谱分析中,就是为某个化合物开发特定的分析方法,这方法往往带有分析者的个人风格和习惯,以及自己仪器的特色。 因此,对于我平常的校内检测,其实理论上大多数应该属于艺术品的性质,应该追求完美,但是一个样品仅仅几十元到百把元的测试费,在短短1-2天内做到极致是不可能的。而大多数情况下,仅仅是较为合理的结果,对方的要求有时也是很简单,你做的过于完美,其实是无用的,他往往不需要这种完美的结果。只有一个样品到数万的费用,才能耗费精力,将其做成艺术品,一篇完整的论文。 在学校20多年的液相检测,由于时间和精力的限制,很多样品分析其实结果并不理想,达不到要求,但现实是不容许你将其做成艺术品。即使你愿意做成精品,那么你就无法完成所送的样品,完不成学校的任务,成本会高到你不可想象。但有谁能真正理解哪?
做食品中甜蜜素的检测,一直都是很正常的。最近遇到了一个新问题,请大家帮助分析一下吧!以前甜蜜素在3.7分钟左右出峰,保留时间一直很稳定。最近,甜蜜素(标准品)依然在3.7出峰,但是在3.9分钟也出了一个峰,两个峰峰形都很好,而且分离的也很好,无重叠现象。做标准曲线,随标准品浓度的增加,两个峰峰面积同时增加,只是我能确定是待测物的峰呈线性,而另外一个峰递增,但线性不如第一个峰好(R2=0.9911)。样品中,如果有甜蜜素,最后检出仍然是两个峰,如果是空白试样(没有甜蜜素)就两个峰都没有,请大家帮我分析一下,究竟是怎么回事?一直稳定的系统,常规的检测项目,为什么会出现上述问题。谢谢大家!

在分析技术的园地里发现许多朋友提到有关标准品的各种问题。其实我想主要的就是很少有人把这个题目讲解的透彻一点,让大家都能够一次彻底了解。自己在美国工作了30余年,也一直在分析的领域上,所以决定花时间把它写下来与大家共享。也算是抛砖引玉,能够给大家一个思考的机会,同时大家也互相交流,相得益彰。闲话少说,言归正传。分析的方法分成两种,一种就是绝对的方法,是不需要标准品,当然使用这种方法必须使用相当纯度的化合物。分析的方法譬如元素分析,DSC,NMR, 滴定,电化学等等,这些方法都可以用来决定化合物的纯度。如何取得相当纯度的化合物,一般我们就用色谱,首先肯定色谱的纯度(其实是均匀度homogeneity),当然在注射相当量的前提下(要能察觉到0.1%的杂质),只能看见一个主峰,也就是说均匀度接近100%(根据峰面积计算)。此时我们拿这个接近100%均匀度的化合物来定性,我们必须确认其专有性(specificity,属性)的存在,然后再定量得出此化合物的纯度。这里说的纯度是指化学组成分而言(composition)。因为这些方法没有办法测出金属盐类,或非金属盐类。因此我们要有另外的方法来决定离子的存在与其含量。当然还有其它成分如水分,有机溶剂等等。当我们把所有的成分定出来之后,总和就是我们这个化合物的组成成分。有了这些数据,自然我们可以求得物质平衡。所以,任何标准品的订定都必须经过这个程序。另外一种分析的方法就是相对的方法。顾名思义,就是需要标准品来对比。除了上述的绝对的方法外,其它的方法都是相对的。看看我们做色谱,就是一个最好的例子。我们需要标准品来定性及定量我们所要分析的样品。我以前曾经开发过新药(New Molecular Entity),标准品就需要自己来定了。经过色谱的分离,纯化干燥后,进行了如上段所叙述的定性,定量工作,我们取得了化学纯度。这个标准品,我们把它叫做原始标准品(primaryreference standard)。如果说化学纯度是95%,也就是说每次我秤了1毫克,里面含有此化合物0.95毫克。因为这种原始标准品,制备很繁杂而且获得的量也不大(在一定时间内),所以没有必要每次都使用这种原始标准品来进行色谱或其它相对分析方法分析样品。因此我们可以拿我们的化学原料药(一般就是工艺开发后的产品)来订为二级标准品(secondaryreference standard)。有人把它说成工作标准品,其实是有商酌余地的。工作标准品也可以是原始标准品,我们不需费力,可以采购而来。我们所要的做的工作,就是把这个二级标准品做色谱分析,然后用原始标准品来做工作标准品,完成定性,定量。如此,我们可以根据峰面积,及原始标准品的相应比(responsefactor = peak area / concentration)求出二级标准品的含量。以后我们做色谱的时候,就可以把这个订好的二级标准品拿来做我们的工作标准品来分析我们的样品。前面提到原始标准品的由来。可以自己定,当然也可以购买。在美国就是美国药典(USP),它不是一个官方机构。但是只要有官方承认的标准品,自然就去买,不管贵贱。如果太贵,那就买一次,然后用来做原始标准品,定出我们自己的二级标准品。一般美国药典的标准品都有规定干燥的程序。在执行干燥完成后,如果没有表明纯度,那就是100%。当下一个批次原始标准品发行的时候,前一批次自动失效。也就是有效期完全看新的批次何时出现。这样也避免了稳定性的考察。当然我们可以拿这个原始标准,来考察我们二级标准品的稳定性。国内的情形我不太清楚,我相信只要官方机构认可的标准品,都可以直接使用的。一般最常用的化学原料药的美国药典标准品,200毫克在200美元左右。如果我们使用微量天平,每次用量1.000毫克,那就可以每次使用原始标准品来做色谱,就避免再去设定二级标准品的麻烦了。另外我想大家都知道,当我们定量的时候,同一个标准品,我们必须秤两次,配置两个不同浓度的工作液。这样可以确认两个工作液是否配置正确。关于到底应该进样几针。我一向的做法就是第一个进样六针,做为系统适应度(system suitability)的检查,其它的任何样品,一律三针。有的人说两针就可以了。我要提醒的是两点决定一条线,得出的统计结论那是数学公式计算出来的。三点才能决定一个面,也就是高斯的分配图(GaussianDistribution)。所以至少在统计学上三点比两点得出的结果要好。又有人说了,如果我一个样品,要走60分钟,那不是完蛋了。首先,是否有必要要走60分钟。我认为最理想的方法是在30-45分钟(当然看功力了),这是指对杂质的分析,必须走梯度。我曾经用5厘米的柱子,梯度,30分钟分出17个化合物,包刮两个主要成分,两个抗氧剂,其它的就是杂质,分解物。如果走等梯度,一般是不超过10分钟为原则。这样就没有问题了。如果真的要走60分钟,那就必须选择好的仪器。并不是所有HPLC的仪器都可以无忧的走完全部样品。当然,柱子也十分重要。扯远了,以后有时间再分开来讨论。
背 景: 按照《中国检验检测学会 2019年检验方法类食品安全国家标准跟踪评价工作方案》(中检学12号)开展“2019年检验方法类食品安全国家标准跟踪评价”之食品中天然污染物和生物毒素检验方法标准跟踪评价。 相关标准: 目前跟踪评价的标准之一--GB 5009.190-2014 食品安全国家标准 食品中指示性多氯联苯含量的测定 目 的: 需要继续扩大标准使用意见的征集,后续会根据反馈的意见考虑是否对标准进行修订。 意见/问题侧重:1.该标准的检测范围能否满足基础标准,产品标准等标准所对应的食品基质类型要求(样品类型)2.该标准的性能指标能否满足基础标准,产品标准等标准的要求(比如化合物种类,定量限等)3.该标准的操作步骤是否规范,科学(包括操作步骤存在的问题,包括方法里参数如)4.该标准中第一法跟第二法在相同化合物上的检测结果是否有可比性5.该标准与基础标准,产品标准等标准之间的一致性是否协调6.其他(比如该标准跟其他标准尤其是国际标准在测试结果上是否存在明显差异,该标准是否具有先进性等) 意见收集时限: 暂定到2020年6月15日前。 有意见的同仁们,最好回帖反馈详细问题并留下lian xi fang shi,以便沟通。







 我要推广仪器
我要推广仪器
 下载APP
下载APP