阿拉丁小分子抑制剂、激动剂、拮抗剂--血管生成信号通路(上)
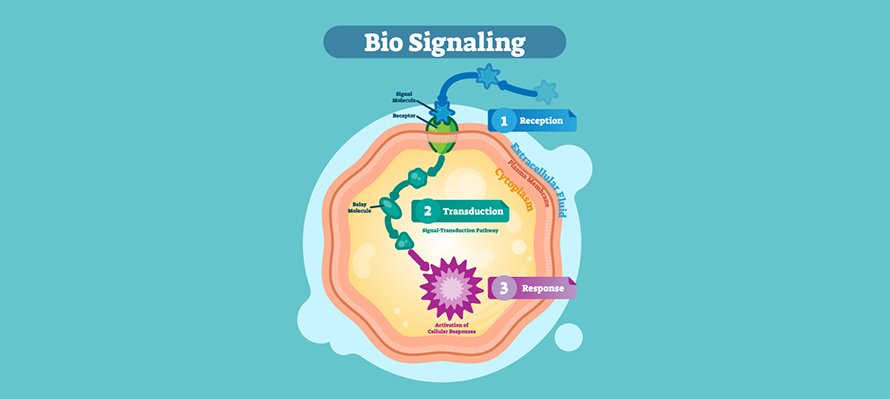
血管生成血管生成,即从已存在的血管中生成新血管。此通路是通过人体中存在的诸多互补和复杂的信号途径调节的。正常情况下,血管生成的相关诱导剂和抑制剂之间保持平衡状态。但对于创伤、缺氧或炎症继发等微环境破坏的条件下,此通路能做出迅速的应答。在各种慢性病理和肿瘤的情况下,血管生成的平衡状态被打破,导致异常的血管生成或新生血管。血管生成信号通路转导过程血管生成的激活导致促血管生成生长因子(vegf、pdgf、fgf和tgf等)的释放,这些因子将其受体结合到已有血管内的内皮细胞上,从而诱导pi3k/akt、erk1/2、smad和notch等多种途径的信号转导,引起内皮细胞增殖和迁移。内皮细胞利用基质金属蛋白酶和整合素来消化细胞外基质,迁移到新的区域,在那里它们延长并形成管子,产生新的血管。在肿瘤血管生成过程中,癌细胞刺激新血管的形成,为肿瘤输送氧气和营养。随着肿瘤的生长,位于肿瘤中心的细胞缺氧,使得转录因子hif-1α(缺氧诱导因子-1)稳定表达。该转录因子与hif-1β结合上调几种促血管生成基因的表达。此外,生长因子信号还刺激hif-1活性,以维持生长细胞的氧稳态。血管生成信号通路图 产品列表 *flt项目号产品名称规格cas包装细胞靶点ic50kis129593sorafenib tosylate≥99%475207-59-150mg,100mg,250mg,1g,5g无细胞raf-16 nmb-raf22 nmvegfr-290 nmvegfr-320 nmpdgfr-β57 nmflt-359 nmc-kit68 nmt127762tozasertib≥98%639089-54-625mg,100mgflt330 nms125267sp600125≥98%129-56-625mg,100mg,500mg,1g,5g无细胞jnk140 nmjnk240 nmjnk390 nmf127011foretinib (gsk1363089)≥98%849217-64-75mg,25mg,100mg无细胞met0.4 nmkdr0.9 nmq127558quizartinib (ac220)≥99%950769-58-15mg,10mg,50mg,500mgmv4-11flt3(itd)1.1 nm/4.2 nm rs4 11flt3(wt)1.1 nm/4.2 nmd126778dovitinib (tki-258, chir-258)≥99%405169-16-610mg,50mg,250mg无细胞flt31 nmc-kit2 nmt126330fedratinib (sar302503, tg101348)≥98%936091-26-85mg,25mg,100mg无细胞jak23 nml126993linifanib (abt-869)≥99%796967-16-35mg,10mg,50mgkdr4 nmcsf-1r3 nmflt-1/33 nm/4 nmpdgfrβ66 nmc127776crenolanib (cp-868596)≥99%670220-88-95mg,10mg,50mgpdgfrα2.1 nmpdgfrβ3.2 nmr129910r406 (free base)≥99%841290-80-05mg,10mg,25mg,50mg,100mgsyk41 nmm127412amuvatinib (mp-470)≥98%850879-09-35mg,25mg,100mgc-kit10 nmpdgfα40 nmflt381 nmt125150tandutinib (mln518)≥98%387867-13-225mg,100mg,500mgflt30.22 μmt127523tg10120998%936091-14-45mg,25mg,100mg无细胞jak26 nmflt325 nmret17 nmk127169kw-2449≥98%1000669-72-65mg,25mg,100mgflt36.6 nme126318enmd-2076≥99%934353-76-15mg,10mg,50mgaurora a14 nmflt31.86 nml127618ldk378≥99%1032900-25-65mg,25mg,100mg,250mg无细胞alk0.2 nmigf-1r8 nminsr7 nmstk22d23 nmflt360 nmp129908prt062607 (p505-15, biib057) hcl≥98%1370261-97-45mg,25mg无细胞syk1 nms125098sorafenib≥99%284461-73-0250mg,1g无细胞raf-16 nmb-raf22 nmvegfr-290 nmvegfr-320 nmpdgfr-β57 nmflt-359 nmc-kit68 nmc129757cabozantinib malate (xl184)≥99%1140909-48-310mg,25mg,50mg,100mg,250mgvegfr20.035 nm无细胞c-met1.3 nmret4 nmkit4.6 nmflt-1/3/412 nm/11.3 nm/6 nmtie214.3 nmaxl7 nmt129806tcs 359≥99%301305-73-710mg,25mg,50mgflt343 nme129946enmd-2076 l-(+)-tartaric acid≥99%1291074-87-75mg,25mgaurora a14 nmvegfr(flt3)1.86 nml127298ly2801653-1206799-15-65mg,10mg,50mgmet2 nmc168062crotonoside95% (hplc)1818-71-95mg,10mg,25mgflt3hdac3/6g172979gilteritinib97%1254053-43-45mg,100mgflt30.29 nmaxl0.73 nmp171724pexidartinib97%1029044-16-35mg,100mgcsf-1r20 nmkit10 nmflt3160 nm *dna alkylator项目号产品名称规格cas包装细胞靶点ic50ec50kis129593sorafenib tosylate≥99%475207-59-110mg,50mg,250mg,1g,5g无细胞raf-16 nmb-raf22 nmvegfr-290 nmvegfr-320 nmpdgfr-β57 nmflt-359 nmc-kit68 nme129728sunitinib malate≥99%341031-54-7100mg,500mg,1g无细胞vegfr2 (flk-1)80 nmpdgfrβ2 nml125046lenalidomide≥99%191732-72-650mg,250mg,1g,5gpbmcstnf-α13 nmc126195cabozantinib (xl184, bms-907351)≥98%849217-68-15mg,10mg,50mg,100mg,250mg无细胞vegfr20.035 nmc-met1.3 nm ret4 nmkit4.6 nmflt-1/3/412 nm/11.3 nm/6 nmtie214.3 nmaxl7 nmp127550ponatinib (ap24534)≥99%943319-70-810mg,50mg,250mg无细胞abl0.37 nmpdgfrα1.1 nmvegfr21.5 nmfgfr12.2 nmsrc5.4 nmk125585ki20227≥98%623142-96-110mg,50mgkdr2 nmvegfr-212 nmc-kit451 nmpdgfrβ217 nma129732axitinib≥99%319460-85-010mg,50mg,250mg,1g猪主动脉内皮细胞vegfr10.1 nmvegfr20.2 nmvegfr30.1-0.3 nmpdgfrβ1.6 nmc-kit1.7 nmf127011foretinib (gsk1363089)≥98%849217-64-75mg,25mg,100mg无细胞met0.4 nmkdr0.9 nmn129725nintedanib (bibf 1120)≥98%656247-17-55mg,10mg,25mg,50mg,100mg,500mg无细胞vegfr134 nmvegfr213 nmvegfr313 nmfgfr169 nmfgfr237 nmfgfr3108 nmpdgfrα59 nmpdgfrβ65 nmv125180vandetanib (zd6474)≥99%443913-73-325mg,100mg,500mg无细胞vegfr240 nmvegfr3110 nmegfr500 nmr127804regorafenib (bay 73-4506)≥99%755037-03-75mg,10mg,25mg,100mg无细胞vegfr113 nmvegfr24.2 nmvegfr346nmpdgfr-β22 nmkit7 nmret1.5 nmraf-12.5 nmp129722pazopanib hcl (gw786034 hcl)≥98%635702-64-625mg,100mg,250mg,1g无细胞vegfr110 nmvegfr230 nmvegfr347 nmpdgfr84 nmfgfr74 nmc-kit140 nmc-fms146 nmc125911cediranib≥98%288383-20-010mg,50mgvegfr(kdr)5 nm/≤3 nmp125865pd173074≥99%219580-11-75mg,10mg,50mgfgfr125 nmvegfr2100-200 nmd126778dovitinib (tki-258, chir-258)≥99%405169-16-610mg,50mg,250mg无细胞flt31 nmc-kit2 nml126993linifanib (abt-869)≥99%796967-16-35mg,10mg,50mgkdr4 nmcsf-1r3 nmflt-1/33 nm/4 nmpdgfrβ66 nmv125857vatalanib (ptk787) 2hcl≥99%212141-51-010mg,50mg无细胞vegfr2/kdr37 nmr127906raf265 (chir-265)≥98%927880-90-81mg,5mg,10mg,50mgvegfr230 nmb-raf3-60 nmt126012tivozanib (av-951)≥98%475108-18-05mg,25mg,100mgvegfr10.21 nmvegfr20.16 nmvegfr30.24 nmm129736motesanib diphosphate (amg-706)≥98%857876-30-35mg,10mg,50mgvegfr12 nmvegfr23 nmvegfr36 nml125518lenvatinib (e7080)≥99%417716-92-85mg,10mg,50mg,100mgvegfr2(kdr)4 nmvegfr3(flt-4)5.2 nmb127317brivanib (bms-540215)≥98%649735-46-65mg,10mg,50mgvegfr225 nmm127064mgcd-265≥98%875337-44-31mg,5mg,10mg,50mgc-met1 nmvegfr1/2/33 nm/3 nm/4 nma126830aee788 (nvp-aee788)≥97%497839-62-05mg,25mg,100mgegfr2 nmher2/erbb26 nme126318enmd-2076≥99%934353-76-15mg,10mg,50mgaurora a14 nmflt31.86 nmo126155osi-930≥99%728033-96-31mg,5mg,25mg,50mgkit80 nmkdr9 nmcsf-1r15 nmc126929cyc116≥99%693228-63-61mg,10mg,50mgaurora a8.0 nmaurora b9.2 nmvegfr244 nmk125876ki8751≥98%228559-41-95mg,25mg,100mgvegfr20.9 nmt129747telatinib≥99%332012-40-51mg,5mg,10mg,50mgvegfr2/36 nm/4 nmc-kit1 nmpdgfrα15 nmp126419pp121≥98%1092788-83-410mg,50mgpdgfr2 nmhck8 nmmtor10 nmvegfr212 nmsrc14 nmabl18 nmdna-pk60 nmp125184pazopanib≥99%444731-52-625mg,100mg,500mggfr110 nmvegfr230 nmvegfr347 nmpdgfr84 nmfgfr74 nmc-kit140 nmc-fms/csf1r146 nmk125907krn-633≥97%286370-15-85mg,25mg,100mgvegfr1170 nmvegfr2160 nmvegfr3125 nm




























 我要推广仪器
我要推广仪器
 下载APP
下载APP