
http://ng1.17img.cn/bbsfiles/images/2012/03/201203081038_353205_1638724_3.jpg上文中的阿莫西林系统适用性对照品就是含量测定用的对照品吗?
我在做氨苄西林钠聚合物,在以水为流动相B的时候,进样对照溶液,对照溶液严重拖尾,流速1.0。对照溶液浓度0.5mg/ml。在这个过程中调过流速0.8,但峰很宽;流速1.2只是出峰时间提前而已,拖尾问题没有改善。调过对照溶液浓度0.25mg/ml,拖尾仍然没有改善。水用的是注射用水,抽滤2遍。有关文献中又说对照溶液严重拖尾可以加0.5%葡萄糖溶液或0.01mol/l甘氨酸适量,抑制氨苄西林和葡聚糖凝胶的缔合。我两个都试过了,没有改善啊。这个适量真的是很难控制,几滴?几毫升?求求各位老师帮帮我吧,对照溶液严重拖尾啊!!!怎么办???
测甘油杂质需要做一个系统适用性,其中用到甘油浓度为400mg/ml,请问必须要对照品吗,用分析纯可以吗
HPLC测定时需要做系统适用性,那么这个系统适用性到底该用供试品溶液做还是用对照溶液做?
我做的是生物制品中的氨苄西林残留,内标选的阿莫西林。仪器:美国AB3200,液相是岛津LC-20AD。流动相条件:乙腈,甲酸水(甲酸调PH3.1)梯度程序:0.01min 乙腈5%2min 乙腈5%6min 乙腈80%7min 乙腈5%8min 乙腈5%由于基质里面含有不挥发性盐,所以用了切换阀,前三分钟打进废液,后面再进质谱。这个条件一直做的很好,内标在4min出峰,氨苄西林在5.4min出峰。现在的问题是:条件不能重复了,内标峰形很怪(峰分叉,很毛糙),而且出峰时间延迟了0.5min.而氨苄西林没有变化。如果轻微变一下梯度条件,内标峰就变的很好了,所以我认为质谱是没有问题的。现在的问题就出在液相条件上,我找了很多原因,最开始换了色谱柱,换了两根(同品牌,同规格),一根还是新的,但内标出峰还是一样怪!现在就排除了柱子的问题,那么问题是不是就出在流动相条件上?!乙腈用的牌子是默克的,应该没问题吧。水是制的超纯水,以前也一直这样用的,调PH前是校正了PH仪的,PH值应该还是没问题。但问题还是没有解决!是否是梯度程序问题呢?(但以前一直都是用的这个梯度,重复性很好啊)请教高手,帮忙找下原因啊!这个问题困扰我好久了,方法学已经做完了,现在要测样品了,确出现了这种问题,小妹真的很急啊!
氨苄西林的杂质对于其质量、效力和安全性非常重要。一些杂质可能对药物的安全性和疗效产生负面影响。杂质的存在可能引发药物不良反应,如过敏反应,或者降低药物的效力。因此,对氨苄西林的杂质进行严格的质量控制和监测是必要的。质量控制不仅包括在生产过程中对杂质的检测和去除,还包括对贮存条件的监控,以防止在贮存过程中产生新的杂质或使现有的杂质浓度升高。CATO标准品对氨苄西林杂质的研究也有利于优化制药工艺,从而提高药物的质量,并减少不良反应和副作用的危险。这对于保证药物的治疗效果和患者安全性至关重要。[img=,609,523]https://ng1.17img.cn/bbsfiles/images/2024/02/202402052057087895_2740_6381668_3.png!w609x523.jpg[/img]
求助氨苄西林红外光吸收图谱鉴别 标准规定:红外光吸收图谱应与对照的图谱一致。1、样品和对照怎么做前处理?
色谱条件与系统适用性试验 用十八烷基硅烷键合硅胶为填充剂(5~10μm);0.2mol/L硫酸盐缓冲液(取无水硫酸钠28.4g,加水溶解后,加磷酸2.7ml、水800ml,用乙醇胺调节pH值至2.3,加水至1000ml)-乙腈(74:26,或适宜比例)为流动相;柱温为40℃;检测波长为214nm。取重组人胰岛素对照品,用0.01mol/L盐酸溶液制成每1ml中含1mg的溶液,室温放置至少24小时后,取2ul注入液相色谱仪,胰岛素峰和A21脱氨胰岛素峰的分离度应不小于1.8,拖尾因子不大于1.8。请问我在工作中做系统适用性实验时,是进上文中的1mg/ml的放置24小时的溶液5针,以其做系统实用性试验;还是进上文中的1mg/ml的放置24小时的溶液1针,再进对照品5针,以5针对照品作为系统适用性试验,而一开始进的只看一下分离度?
向大家请教关于系统适用性实验的问题:同一样本中有A(A为样本),B(杂质)两种成分,且需要同时测定A,B两种物质的含量,那么在进行系统适用性实验时,是否可以用A和B的混合对照品,连续进样5针,分别计算A和B的RSD???
做系统适用性时,到底该用对照品还是供试品啊,看到很多说法都不一样......

[img=,480,596]https://ng1.17img.cn/bbsfiles/images/2018/09/201809260906045200_3378_2449119_3.png!w480x596.jpg[/img]请教各位同仁: 上述是盐酸雷尼替丁含量测定方法,其中划红线的是系统试液。可见系统适用性试液是用氢氧化钠进行破坏的溶液,因该物质极易被氢氧化钠破坏,故在进样5针后,其重复性不满足要求。故在进样过程中,只测试一针。 [b] 疑问:[/b]1.测试一针是合理的,但是否还要用对照品溶液连续测试5针来验证系统适用性?2.盐酸雷尼替丁的有关物质方法同含量测定,采用的是自身对照法,请问测试有关物质与含量不是同一天,请问有关物质方法中系统适用性试验如何测试?是继续用破坏性试液测试1针,还是用什么测试5针?目前有没有对有关物质方法中系统适用性试验测试要求?3.请问有关物质测试中,是对照溶液和供试品溶液各进1针,各配1个样?还是双样双针?单样双针?希望有比较权威的解答!感谢各位
大家在做方法验证的时候,系统实用性溶液怎么配的,浓度多大,遵循什么原则。例如我有个产品活性成分含量大约50%,HPLC就出这一个峰,系统适用性溶液怎么配呢,是不是配置50%的对照品溶液,还是多大浓度的啊!谢谢
本人饲料行业,液相做中间产物的时候,系统适用性可以改为每周做一次,其余天数只走回针考察吗ps:液相不是不停运行的、对照品有效期为一周
[b]高效液相色谱系统适用性试验设计的变化趋势[/b][i]周晓源,李雪茹[/i]高效液相色谱法(HPLC 法)是药物分析中常用的一种定性、定量色谱分析方法。具有较强的专属性,相对较高的检测灵敏度和良好的量化功能。2005版《中国药典》使用 HPLC 法的品种中,色谱系统适用性试验设计有了较大的变化:指标更加细致、周到,检测更重实效,色谱系统适用性的试验用溶液的制备方法也呈现多样化,体现出一些变化趋势。 1. 色谱系统适用性试验的设计与实验目的更加匹配 系统适用性试验的严格细腻程度取决于实验目的。首先应考虑色谱系统被用于何种实验,根据实验目的来设计系统适用性试验。如果一个 HPLC 方法仅用于定性鉴别,就其色谱系统的适用性试验而言可以相对简单宽松,只要可以确保被测成分峰与其他色谱峰有一定的分离度,具有适宜的出峰时间即可达到实验目的。 如果用于定量分析(如含量测定),则除要保证被测成分峰具有适宜的出峰时间外,还需检验系统是否能够保证被测成分峰与其他色谱峰完全分离,分离度一般应在1.5以上,同时还应测试被测成分峰峰面积的重复性是否良好,对照品溶液连续进样5针的峰面积相对标准偏差应不大于2%,被测成分峰的峰型也应基本对称,以保证分离效果和测量精度。对于小峰(如占总面积10%以下的色谱峰)峰面积的定量,或用峰高法定量时,就应对拖尾因子或对称因子加以严格的规定,一般来说,拖尾因子应在 0.95~1.05之间,因为峰的对称性对测量结果影响较大。 如果检查某种药品的有关物质,且还需要分别检查单个杂质和杂质总量,那么系统适用性试验还应有一个重点,就是要有常见杂质难分离物质对分离度的测定指标。此外系统的检测灵敏度试验也就相对比较重要。如盐酸二甲双胍的有关物质检查项下要求: 盐酸二甲双胍与双氰胺的分离度应大于1.5,检测灵敏度要求调节双氰胺峰高为满量程的10%。 如 果色谱系统是一个梯度洗脱系统,有时一个难分离物质对分离度的测试也不能完全达到实验目的。如果在梯度变化的前后均有需要检测的杂质,分离度的测定指标一般应根据需要在梯度变化之前和之后都可加以制订。在梯度洗脱系统中某个成分峰的保留时间也经常用来做系统适用性检测的指标,给出吐峰时间范围,如头孢地尼,主成分头孢地尼峰的保留时间要求22分钟,E-异构体峰保留时间约为33分钟,理论板数按头孢地尼峰计算应不低于 7000。 在2000年版《中国药典》中,有些标准色谱系统适用性试验的要求就与其色谱系统的实验目的不完全匹配。如有些品种含量测定与有关物质共用一套色谱系统,且有关物质还需要分别检查单个杂质和杂质总量,但系统适用性试验指标仅有一个理论板数的要求,或对分离度的设计为“被测成分峰与相邻杂质峰间的分离度应符合规定”这样一个对系统性能缓冲空间很大的一个指标要求。在2005年版《中国药典》中,这种实属很虚的指标开始减少。如2000年版头孢曲松反式异构体(光降解产物)峰的保留时间应为头孢曲松峰保留时间的1.3倍,两峰之间的分离度应不小于3.0,理论板数按头孢曲松峰计算应不低于1500,2005年版修订为头孢曲松峰和头孢曲松反式异构体峰间的分离度应不小于6.0。2.系统适用性试验用溶液的制备更加注重方便性、实用性和可操作性系统适用性试验用溶液的配制方法,最简单的莫过于用主成分对照品与杂质对照品混合配制,但有些杂质对照品不能得到,如性质不稳定或与主成分理化性质太接近,分离提取技术要求太高,成本太大等,但这些杂质峰恰恰又是与主成分峰最难分离的色谱峰,且较常存在于 药 品中需要检查的,在2005年版《中国药典》中,这一问题得到了较好的解决。如喹诺酮类药物中较常出现光降解产物,而此光降解产物是引起这类药物不良反应的主要因素,所以需要在有关物质检查中做为重点检测的杂质之一。 因 此 ,在2005年 版 《 中 国 药 典 》中,这些药物系统适用性试验用溶液的制备就通 过把对照 品溶液进行 光 照 处理,得到能产生明显光降解产物色谱峰的溶液。 3.实验过程、操作步骤趋于严谨规范 色谱系统适用性试验设计、规定的完备、灵敏度检测试验的规范,溶剂的选择、溶解制备方式等各方面均体现出对实验目的的理解更加明确,对实验细节考虑更加严谨周到,标准的书写格式均更 加规范 、统 一 ,如2005年 版《中 国 药典》收载的 β-内酰胺类抗生素中检查高分子聚合物的品种将原来收载的8个品种的色谱条件与系统适用性试验均修订与新增 13个品种项下书写格式相同,系统适用性试验统一为理论板数以蓝色葡聚糖2000峰计算均不低于……。拖尾因子均应小于2.0,在两种流动相系统中蓝色葡聚糖 2000峰保留时间比值均应在0.93~1.07之间,对照品溶液主峰与供试品溶液中聚合物峰与相应色谱系统中蓝色葡聚糖 2000峰保留时间的比值均应在0.93~1.07之间。
采用面积归一化法测定高分子杂质,但系统适用性的对照品溶液峰面积rsd达到9%以上,这样会影响杂质定量么,有人说峰面积归一化法不用对照品峰面积,不用管,有做过的么?(对照品溶液和杂质不是一个物质)
关于液相系统适用性的问题,由于系统适用性需要用到杂质标准品,但如果一直使用杂质标准品,费用又很高。如果公司自己制作内部标准品,有没有这样的科研力量。如何解决?
在做薄层色谱分析时需做系统适用性试验,那么对于已知杂质的薄层分析系统适用性试验该如何做?已知杂质的限度是0.2%,系统适用性试验:1.将杂质与供试品按照0.2%:1制成混合溶液进行系统展开2.将杂质与供试品按照0.2%:0.2%制成混合溶液进行系统展开上述两种系统适用性试验应选择哪种?选择1很容易出现杂质被包裹现象,两种系统适用性试验分离度都能满足要求。求指点~~
如题,在进行方法验证时,系统适用性怎么验证?我最初接受的培训是:因为系统适用性是证明系统对于分析方法是适用的,是每天在分析样品之前进行的,因此在分析方法验证的每一天进行各项验证前,需分析系统适用性,分离度、理论塔板数、拖尾因子或峰面积RSD%(含量计算时对照溶液RSD计算如果定为系统适用性的话,每天必须将系统适用性几样5针)等符合要求才能进行后面的验证;然后将每天的系统适用性数据进行统计。同样的,在进行耐用性验证时,也必须先进系统适用性。后来,我去了其他公司,看到的方式是:系统适用性作为专属性的一部分,在验证专属性时,将各个杂质配制成含约1%或0.1%的混合溶液,分析分离度等,符合要求即认为系统适用性符合要求。各位来讨论讨论,那种方式更为合理?

2010 版中国药典 VIII R 规定TOC分析仪是要定期进行系统适用性测试,GE为客户提供即开即用的液体系统适用性标准品,省心省力。8月15日-9月15日,GE携全国代理商惠馈客户,优惠活动如下:如果了解更多有关TOC的系统适用性测试知识,可以点击此处http://ng1.17img.cn/bbsfiles/images/2012/08/201208221014_385307_2359237_3.jpg
阿奇霉素系统适用性标准图谱谁有?中检所出对照了,可是没标准图谱
有谁做过阿莫西林含量和有关物质,药典中提到色谱图应与标准图谱一致,标准图谱指的是什么,哪里有,含量中提到用系统适用性对照品,哪里可以买到?
SN/T2050-2008 方法测定6种β-内酰胺抗生素的残留量,结果阿莫西林和氨苄西林的回收非常低,约20和10%左右,后来经过排查,发现是加缓冲液(pH8.5)后两组分就会显著降低,大家有没有什么好的建议?方法大概是:5g样→乙腈水(15:2)溶液40ml提取→取20ml在37℃旋蒸至近干→加25ml磷酸盐缓冲液(pH8.5)溶解→上HLB小柱萃取→乙腈洗脱→氮吹→磷酸盐缓冲液(pH7.0)溶样,过滤→上机有哪位专家做过这俩组分的方法,指导下,急盼http://simg.instrument.com.cn/bbs/images/brow/emyc1010.gif
你购买过液相色谱系统适应性专用对照品吗,还是直接以含量用对照品来做,如阿莫西林?
请教各位,阿莫西林和氨苄西林两种药物,HPLC怎么分析,包括前处理过程和液相分析方法,能否简要介绍一下大家的经验
系统适用性的五个项目(塔板数、分离度、灵敏度、拖尾因子、重复性)每次实验都需要做吗?如果实验方法里没有提系统适用性,还用做吗?

作者:汪杰; 刘元瑞; 刘祖德;(武警部队药品检验所; 武警总医院 北京;)摘要:目的 建立顶空毛细管气相色谱法测定注射用氨苄西林钠中的有机溶剂残留量。方法 用DM-624毛细管气相色谱柱,顶空进样法,FID检测器,以甲醇为内标进行测定。结果 二氯甲烷、丙酮、异丙醇的线性范围分别为2.633-1 316.8μg·mL-1(r=0.999 8),1.569-784.4μg·mL-1(r=0.999 9),1.562-781.2μg·mL-1(r=0.999 9);二氯甲烷、丙酮、异丙醇的平均回收率分别为100.8%,98.3%,99.3%;RSD分别为2.0%,2.3%,1.6%(n=5)。结论 本方法简单、准确、灵敏度高、重现性好,适用于注射用氨苄西林钠中有机溶剂残留量的测定。谱图:http://ng1.17img.cn/bbsfiles/images/2012/08/201208271108_386366_1606903_3.jpg
我现在才学习[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]也没什么人教哈!用PGE-1000的柱子照药典2005做氨苄西林钠有关二氯甲烷的检出分离度达的到要求,但是出峰时间太夸张了,二氯甲烷大概在8.8分钟出,二氯乙烷大概在20分钟才出,这种情况正常不?希望高手速度解决
求教各位老师,药品的方法研究中系统适用性和方法的专属性指的是什么,该怎么做?
USP中规定用填充柱检测,现改为用毛细管柱是否违背系统适用性的要求
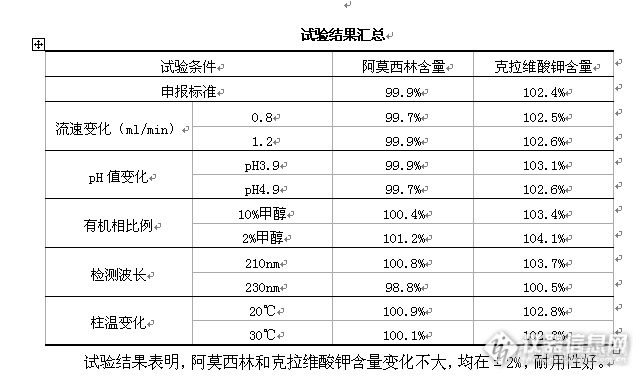
继”阿莫西林克拉维酸钾胶囊有关物质方法学”项目结束,整理的含量测定方法学。项目:含量测定(3.2.P.5.2.9)检查方法:照高效液相色谱法(中国药典2010年版二部附录Ⅴ D)测定试验条件:仪器:LC-2010CHT (SHIMADZU)万分之一电子天平(Sartorius ABS-124S型)工作站(LCsolution色谱工作站)色谱柱(填料:C18,规格:250mm×4.6mm,填料粒径:5μm)Xtimate C18 4.6*250 ,PN:Xt5B18425 ,SN:411101950UV检测器(检测波长:220nm)柱温:室温流动相:0.05mol/L磷酸二氢钠溶液(取磷酸二氢钠7.8g,加水900ml使溶解,用10%磷酸溶液或氢氧化钠试液调节pH值至4.4±0.1,加水稀释至1000ml)-甲醇(95:5)。流速:1.0ml/min。运行时间:约20分钟。系统适用性:取阿莫西林克拉维酸系统适用性试验对照品,加流动相溶解并稀释制成每1ml中含0.8mg的溶液,取20μl注入液相色谱仪,记录的色谱图应与标准图谱一致。具体试验操作:取装量差异项下的内容物适量,精密称取适量,加水适量,超声使溶解并定量稀释制成每1ml中含阿莫西林0.5mg的溶液,滤过,立即精密量取续滤液20μl注入液相色谱仪,记录色谱图;另分别精密称取阿莫西林对照品与克拉维酸对照品各适量,加水溶解并定量稀释制成每1ml中约含阿莫西林0.5mg和每1ml中含克拉维酸0.125mg的混合溶液,同法测定。按外标法以峰面积分别计算供试品中C16H19N3O5S和C8H9NO5的含量。计算公式:标示量百分含量(%)=××100%式中:Cs为对照品的浓度(mg/ml);At为供试液的主峰面积;Nt为供试液的稀释倍数;AS为对照品溶液的主峰面积;W为供试品取样量(mg)。“色”路蹒跚,藤下葡萄,某品种含量测定方法学之耐用性试验部分路蹒跚,藤下葡萄,某品种含量测定方法学之耐用性试验部分http://ng1.17img.cn/bbsfiles/images/2013/06/201306292159_448382_1621890_3.gif3.2.P.5.3.6.1波长选择本品含量测定检测波长参照中国药典2010年版二部收载的阿莫西林克拉维酸钾相关制剂质量标准含量测定项,即220nm。3.2.P.5.3.6.2流动相选择(色谱图见附件1122~1124)参照中国药典2010年版二部收载的阿莫西林克拉维酸钾相关制剂质量标准含量测定项,以0.05mol/L磷酸二氢钠溶液(取磷酸二氢钠7.8g,加水900ml使溶解,用10%磷酸溶液或氢氧化钠试液调节pH值至4.4±0.1,加水稀释至1000ml)-甲醇(95:5)为流动相。试验过程:系统适用性试验供试液:精密称取阿莫西林克拉维酸钾系统适用性对照品4.2mg至5ml量瓶中,加流动相适量超声使溶解并稀释至刻度,摇匀,滤过,取续滤液作为供试液;对照品溶液:精密称取阿莫西林对照品29.1mg和克拉维酸钾对照品7.3mg至50ml量瓶中,加水适量超声使溶解并稀释至刻度,摇匀,滤过,取续滤液作为供试液;精密量取上述供试液各20μl注入高效液相色谱仪,记录色谱图,典型色谱图见下图:http://ng1.17img.cn/bbsfiles/images/2013/06/201306292200_448383_1621890_3.gifhttp://ng1.17img.cn/bbsfiles/images/2013/06/201306292200_448384_1621890_3.gifhttp://ng1.17img.cn/bbsfiles/images/2013/06/201306292201_448385_1621890_3.gif3.2.P.5.3.6.3进样精密度试验(色谱图见附件1125~1130)







 我要推广仪器
我要推广仪器
 下载APP
下载APP