哪位知道国产的异丁醇,异戊醇标准品哪有?
染发剂中对苯二胺等染料的HPLC测定方法采用《化妆品卫生规范》所规定的液相色谱标准检测方法,对氧化型染发剂中包括对苯二胺和邻苯二胺在内的八种染料进行检测。[size=4][b]分析方法[/b]色谱柱:Shimadzu Shim-pack VP-ODS 4.6×150mm 5μm检测波长:280nm 流动相:乙腈:(水:三乙醇胺=98:1)=5:95 流速:1mL/min 柱温:室温进样量:10μL洗脱方式:等度洗脱。[/size][size=4]流动相中使用了三乙醇胺,比较少见,它在分析苯胺类化合物有什么突出的优点吗?可否用三乙胺替代?[/size][img]http://ng1.17img.cn/bbsfiles/images/2010/05/201005082043_217200_1638724_3.jpg[/img][img]http://ng1.17img.cn/bbsfiles/images/2010/05/201005090755_217230_1638724_3.jpg[/img]
请教: 成盐的手性化合物的分析和制备请问如果一个化合物成盐,包括盐酸盐,三氟乙酸盐和有机盐。1. 在手性分析时,该化合物是什么状态在手性柱上分离的?游离态还是成盐的状态?如果化合物是成的有机盐,该有机盐可以由UV检测,那么分析其消旋体时,分析上应该有几根峰?2 . 如果手性液相制备的话,成盐的化合物制备完后是什么状态,游离or成盐?如,一个碱性化合物成盐酸盐,经带有二乙胺的流动相(正己烷:乙醇)制备后还是盐酸盐吗?另外,在手性制备后处理时,二乙胺一般如何除去?旋蒸后还有很多残余~~
急求乙烯中羰基化合物测定的相关标准,多谢!
急求乙烯中羰基化合物测定的相关标准,多谢
化合物纯度的鉴定方法,从快速,便宜,简便的要求出发,主要来之于以下几点:一 通过TLC的纯度的鉴定, 我将自己的心得分述如下1 展开溶剂的选择,不只是至少需要3种不同极性展开系统展开,我的经验是首先要选择三种分子间作用力不同的溶剂系统,如氯仿\甲醇,环己烷\乙酸乙酯,正丁醇\醋酸\水,分别展开来确定组分是否为单一斑点.这样做的好处是很明显的,通过组份间的各种差别将组分分开,有可能几个相似组份在一种溶剂系统中是单一斑点,因为该溶剂系统与这几个组分的分子间力作用无显著的差别,不足以在TLC区分.而换了分子间作用力不同的另一溶剂系统,就有可能分开.这是用3种不同极性展开系统展开所不能达到的.2 对于一种溶剂系统正如wxw0825所言,至少需要3种不同极性展开系统展开,一种极性的展开系统将目标组分的Rf推至0.5,另两种极性的展开系统将目标组分的Rf推至0.8,0.2。其作用是检查有没有极性比目标组分更大或更小的杂质。3 显色方法,光展开是不够的,还要用各种显色方法。一般一定要使用通用型显色剂,如10%硫酸,碘,因为每种显色剂(不论是通用型显色剂,还是专属显色剂在工作中都遇到他们都有一化合物不显色的时候),再根据组分可能含有混杂组份的情况,选用专属显色剂。只有在多个显色剂下均为单一斑点,这时才能下结论样品为薄层纯二 通过熔程,判断纯度。原理很简单,纯化合物,熔程很短,1,2度。混合物熔点下降,熔程变长。三,基于HPLC的纯度鉴定,对于HPLC因为常用的系统较少,加之其分离效果好,我们一般不要求选择三种分子间作用力不同的溶剂系统,只要求选这三种不同极性的溶剂系统,使目标峰在不同的保留时间出峰。四,基于软电离质谱的纯度鉴定。如ESI-MS,APCI-MS。大极性化合物选用ESI-MS,极性很小的化合物选用APCI-MS,这些软电离质谱的特点是只给出化合物的准分子离子峰,通过正负离子的相互沟通来确定分子量。如果样品不纯,就会检出多对准分子离子峰,不但确定了纯度,还能明确混杂物的分子量。五,基于核磁共振的纯度鉴定,从氢谱中如果发现有很多积分不到一的小峰,就有可能是样品是样品中的杂质。利用门控去偶的技术通过对碳谱的定量也能实现纯度鉴定。好了,不能再多写了。这里只是对常见的纯度鉴定方法做了一个小结,从快速,便宜,简便的要求出发,以第一点最合要求,往后次之,所以对第一点详加讲述。当然每种方法多有各自的局限性,如基于氢谱的纯度鉴定,如果发现有很多积分不到一的小峰,还有可能使样品中的活泼质子,基于软电离质谱的纯度鉴定,如果混杂物的分子量与目标物一样就无法检出。等等还有很多。这需要大家在工做中积累,思考。要讲的话,我看好几篇都讲不完。最后说一下对化合物纯度的要求,世界上不存在100%纯的化合物。你希望要多高的纯度应该与你的目的有关,例如,如想测核磁共振鉴定结构,一般要求95%的纯度,如果想测EI-MS,纯度越高越好。99%以上。还有,以上的方法都不能区分对应异构体。

[align=center][b][url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法测定水性木器涂料中挥发性有机化合物、苯系物、乙二醇醚及其酯类含量[/b][/align]摘要:试样经稀释后,通过[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]分析技术使试样中各种挥发性有机化合物分离,定性鉴定被测化合物后,用内标法测试其质量。用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法测定水性木器涂料中挥发性有机化合物、苯系物、乙二醇醚及其酯类含量,可一次测定30中组分含量;方法的回收率范围为84.16%-125.63%,相对标准偏差小于10%,方法检出限为21.20mg/kg。本方法简单、快速、准确,可用于室内装饰装修用水性涂料中有害物质含量的测定。关键词:[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法,水性涂料,挥发性有机物,苯系物,酯类引言[color=#333333]水性木器涂料一般采用丙烯酸乳液制得,因丙烯酸乳液具有固体含量高、干燥速度快、硬度强、耐候性好和成本低等优点。水性木器漆中可能含有的有害物质有三苯 ( 苯、甲苯、二甲苯 ) 、卤代烃、甲醛及甲醛缩聚物,水性木器漆与溶剂型木器漆相比其最大优势在于其挥发性有机化合物总量低,因此此项应严格控制。水性木器涂料在生产过程中为提高其性能,添加了不少润湿剂、表面改性剂、全部或部分消泡剂,增稠剂等化学物质,这些物质或多或少会带来有机挥发物,特别是苯系物,VOC等的残留。本文通过[url=https://insevent.instrument.com.cn/t/Mp]gc[/url]-FID测定其中有害物质含量,为室内装修装饰材料有害物质是否满足标准限值提供有力佐证。[color=#333333][/color][/color]实验部分1.1[b]试剂[/b]甲醇、苯、甲苯、乙苯、二甲苯、乙二醇甲醚、乙二醇甲醚醋酸酯、乙二醇乙醚、乙二醇乙醚醋酸酯、二乙二醇丁醚醋酸酯、丙酮、乙醇、异丙醇、三乙胺、异丁醇(内标物)、1-丁醇、丙二醇单甲醚、二丙二醇单甲醚、乙酸正丁酯、二甲基乙醇胺、甲基异戊基酮、丙二醇正丁醚、乙二醇单丁醚、1,2-丙二醇、乙二醇、N-甲基吡咯烷酮、二丙二醇正丁醚、二乙二醇单丁醚、丙二醇苯醚、二乙二醇、乙二醇苯醚,共计30种。1.4[b]仪器[/b]安捷伦7890B [url=https://insevent.instrument.com.cn/t/Mp]gc[/url]-FID[img=,690,1226]http://ng1.17img.cn/bbsfiles/images/2017/09/201709261539_01_1657564_3.jpg[/img]湘仪离心机[img=,690,1226]http://ng1.17img.cn/bbsfiles/images/2017/09/201709261540_01_1657564_3.jpg[/img]梅特勒-托利多电子天平XSE204(精度0.1mg)[img=,690,388]http://ng1.17img.cn/bbsfiles/images/2017/09/201709261540_02_1657564_3.jpg[/img]色谱柱条件:HP-5 聚乙二醇毛细管柱,30x0.25mmx0.25μm;进样口温度:240°C,检测器(FID)温度:250°C;柱温:程序升温,60°C保持1分钟,然后以20°C/min升至240°C保持20分钟;分流比:10:1;进样量:1.0μL。1.5[b]分析步骤[/b]称取搅拌均匀后的试样1g(精确至0.1mg)以及与被测物质近似相等的内标物于配样瓶中,加入10mL稀释溶剂稀释试样,密封配样瓶并摇匀, 高速离心机离心15min后用0.45um过滤头过滤后上机测试 采用内标的相对校正因子进行计算 目标化合物采用各自的校正因子,未识别峰用异丁醇的相应因子计算。[b]1.试验结果报告[/b]2.1[b]出峰时间的确定[/b]分别称取各标准物质0.5g于32个50mL容量瓶中,用甲醇定容,摇匀;取各标准样品1mL于试样瓶中,进入编辑好方法的[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url]分析,结果见表1。[img=,690,365]http://ng1.17img.cn/bbsfiles/images/2017/09/201709261457_01_1657564_3.png[/img]2[b].[/b]2[b]校正因子的计算[/b] 分别称取2.1鉴定出的各标准物质(异丁醇分开配制)0.5g(精确至0.1mg)于同一个50mL容量瓶中,用甲醇定容。取该溶液20uL及异丁醇20uL于有960uL甲醇的配样瓶中,摇匀(200ppm)上机测试结果见表2。[img=,642,428]http://ng1.17img.cn/bbsfiles/images/2017/09/201709261527_01_1657564_3.png[/img][b]2.3方法的检出限[/b]取20uL已配制好的内标物(异丁醇)加入980uL甲醇中上机测试10次,将峰面积加和作为TVOC结果,数据如表3;[img=,690,134]http://ng1.17img.cn/bbsfiles/images/2017/09/201709261531_01_1657564_3.png[/img]2.4[b]方法的准确度与精密度实验数据[/b]取已配制好的200ppm混标溶液(含内标)上机测试8次结果见表4。[img=,650,374]http://ng1.17img.cn/bbsfiles/images/2017/09/201709261533_01_1657564_3.png[/img][b]3.小结[/b] 用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法测定水性木器涂料中挥发性有机化合物、苯系物、乙二醇醚及其酯类含量,可一次测定30中组分含量;方法的回收率范围为84.16%-125.63%,相对标准偏差小于10%,方法检出限为21.20mg/kg。本方法简单、快速、准确,可用于室内装饰装修用水性涂料中有害物质含量的测定。[b]4.参考资料[/b] GB24410-2009室内装饰装修材料 水性木器涂料中有害物质限量中附录A挥发性有机化合物、苯系物、乙二醇醚及其酯类含量的测试。GB18582-2008 室内装饰装修材料内墙涂料中有害物质限量。[align=center][/align]
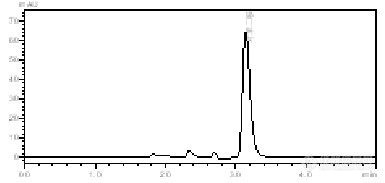
想鉴定某种植物里某种化合物的含量,前期有分离出该种化合物的纯品,但是后期做定量时,不同浓度的标准品的出峰时间有细微差别,且样品中的该化合物的峰要么是出峰时间与标准品不一样,要么就是分不开,是什么原因呢?还有基线总是跑不平(如图所示),流动性是乙腈比水为4:6,
化合物纯度的判定化合物纯度的鉴定方法,从快速,便宜,简便的要求出发,主要来之于以下几点:一 通过TLC的纯度的鉴定, 我将自己的心得分述如下1 展开溶剂的选择,不只是至少需要3种不同极性展开系统展开,我的经验是首先要选择三种分子间作用力不同的溶剂系统,如氯仿\甲醇,环己烷\乙酸乙酯,正丁醇\醋酸\水,分别展开来确定组分是否为单一斑点.这样做的好处是很明显的,通过组份间的各种差别将组分分开,有可能几个相似组份在一种溶剂系统中是单一斑点,因为该溶剂系统与这几个组分的分子间力作用无显著的差别,不足以在TLC区分.而换了分子间作用力不同的另一溶剂系统,就有可能分开.这是用3种不同极性展开系统展开所不能达到的.2 对于一种溶剂系统正如wxw0825所言,至少需要3种不同极性展开系统展开,一种极性的展开系统将目标组分的Rf推至0.5,另两种极性的展开系统将目标组分的Rf推至0.8,0.2。其作用是检查有没有极性比目标组分更大或更小的杂质。3 显色方法,光展开是不够的,还要用各种显色方法。一般一定要使用通用型显色剂,如10%硫酸,碘,因为每种显色剂(不论是通用型显色剂,还是专属显色剂在工作中都遇到他们都有一化合物不显色的时候),再根据组分可能含有混杂组份的情况,选用专属显色剂。只有在多个显色剂下均为单一斑点,这时才能下结论样品为薄层纯二 通过熔程,判断纯度。原理很简单,纯化合物,熔程很短,1,2度。混合物熔点下降,熔程变长。三,基于HPLC的纯度鉴定,对于HPLC因为常用的系统较少,加之其分离效果好,我们一般不要求选择三种分子间作用力不同的溶剂系统,只要求选这三种不同极性的溶剂系统,使目标峰在不同的保留时间出峰。四,基于软电离质谱的纯度鉴定。如ESI-MS,APCI-MS。大极性化合物选用ESI-MS,极性很小的化合物选用APCI-MS,这些软电离质谱的特点是只给出化合物的准分子离子峰,通过正负离子的相互沟通来确定分子量。如果样品不纯,就会检出多对准分子离子峰,不但确定了纯度,还能明确混杂物的分子量。五,基于核磁共振的纯度鉴定,从氢谱中如果发现有很多积分不到一的小峰,就有可能是样品是样品中的杂质。利用门控去偶的技术通过对碳谱的定量也能实现纯度鉴定。好了,不能再多写了。这里只是对常见的纯度鉴定方法做了一个小结,从快速,便宜,简便的要求出发,以第一点最合要求,往后次之,所以对第一点详加讲述。当然每种方法多有各自的局限性,如基于氢谱的纯度鉴定,如果发现有很多积分不到一的小峰,还有可能使样品中的活泼质子,基于软电离质谱的纯度鉴定,如果混杂物的分子量与目标物一样就无法检出。等等还有很多。这需要大家在工做中积累,思考。要讲的话,我看好几篇都讲不完。最后说一下对化合物纯度的要求,世界上不存在100%纯的化合物。你希望要多高的纯度应该与你的目的有关,例如,如想测核磁共振鉴定结构,一般要求95%的纯度,如果想测EI-MS,纯度越高越好。99%以上。还有,以上的方法都不能区分对应异构体。
如题,苯胺类化合物目前国内外标准都有哪些?
有谁做过空[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质[/color][/url]量苯胺类化合物的测定盐酸萘乙二胺分光光度法的,标准曲线的吸光度数据大概是多少?
化合物纯度的鉴定方法,从快速,便宜,简便的要求出发,主要来之于以下几点:一 通过TLC的纯度的鉴定, 我将自己的心得分述如下1 展开溶剂的选择,不只是至少需要3种不同极性展开系统展开,我的经验是首先要选择三种分子间作用力不同的溶剂系统,如氯仿\甲醇,环己烷\乙酸乙酯,正丁醇\醋酸\水,分别展开来确定组分是否为单一斑点.这样做的好处是很明显的,通过组份间的各种差别将组分分开,有可能几个相似组份在一种溶剂系统中是单一斑点,因为该溶剂系统与这几个组分的分子间力作用无显著的差别,不足以在TLC区分.而换了分子间作用力不同的另一溶剂系统,就有可能分开.这是用3种不同极性展开系统展开所不能达到的.2 对于一种溶剂系统正如wxw0825所言,至少需要3种不同极性展开系统展开,一种极性的展开系统将目标组分的Rf推至0.5,另两种极性的展开系统将目标组分的Rf推至0.8,0.2。其作用是检查有没有极性比目标组分更大或更小的杂质。3 显色方法,光展开是不够的,还要用各种显色方法。一般一定要使用通用型显色剂,如10%硫酸,碘,因为每种显色剂(不论是通用型显色剂,还是专属显色剂在工作中都遇到他们都有一化合物不显色的时候),再根据组分可能含有混杂组份的情况,选用专属显色剂。只有在多个显色剂下均为单一斑点,这时才能下结论样品为薄层纯,二 通过熔程,判断纯度。原理很简单,纯化合物,熔程很短,1,2度。混合物熔点下降,熔程变长。三,基于HPLC的纯度鉴定,对于HPLC因为常用的系统较少,加之其分离效果好,我们一般不要求选择三种分子间作用力不同的溶剂系统,只要求选这三种不同极性的溶剂系统,使目标峰在不同的保留时间出峰。四,基于软电离质谱的纯度鉴定。如ESI-MS,APCI-MS。大极性化合物选用ESI-MS,极性很小的化合物选用APCI-MS,这些软电离质谱的特点是只给出化合物的准分子离子峰,通过正负离子的相互沟通来确定分子量。如果样品不纯,就会检出多对准分子离子峰,不但确定了纯度,还能明确混杂物的分子量。五,基于核磁共振的纯度鉴定,从氢谱中如果发现有很多积分不到一的小峰,就有可能是样品是样品中的杂质。利用门控去偶的技术通过对碳谱的定量也能实现纯度鉴定。好了,不能再多写了。这里只是对常见的纯度鉴定方法做了一个小结,从快速,便宜,简便的要求出发,以第一点最合要求,往后次之,所以对第一点详加讲述。当然每种方法多有各自的局限性,如基于氢谱的纯度鉴定,如果发现有很多积分不到一的小峰,还有可能使样品中的活泼质子,基于软电离质谱的纯度鉴定,如果混杂物的分子量与目标物一样就无法检出。等等还有很多。这需要大家在工做中积累,思考。要讲的话,我看好几篇都讲不完。最后说一下对化合物纯度的要求,世界上不存在100%纯的化合物。你希望要多高的纯度应该与你的目的有关,例如,如想测核磁共振鉴定结构,一般要求95%的纯度,如果想测EI-MS,纯度越高越好。99%以上。还有,以上的方法都不能区分对应异构体。
正丁醇部位的分离是天然药物化学专业分离较难的部分,但分到有价值的东西几率较大,所以值得我们为之一搏。 一般来说,正丁醇部位常含有单糖、二糖等小分子糖,多种酚类化合物,以及苷类化合物,极性较大,在硅胶柱上吸附较多,成点性差,分离效果不好。因此,一般说来,对于正丁醇部位可采取以下方法: 1. 大孔树脂柱砍段。 样品水溶解后上样,依次用水,30%、60%、95%乙醇溶液洗脱,分别合并、收集。一般说来目标化合物多在60%段,水,30%段多为一些水溶性单糖、二糖及多酚类化合物,可以考虑弃去。而95%部分多和乙酸乙酯部位大极性段重叠,可以乙酸乙酯部位合并处理。 2. 反相柱分段。 如果正丁醇部位量不是太大,或者课题组有大反相柱,可以考虑用反相柱砍段。本课题组就有一根500g的反相柱,专门用于砍段,效果比正相柱好了许多。唯一需要提醒注意的是,由于我们一般是用正相硅胶板检测化合物分离情况,所以反相柱 砍段后往往各组份在硅胶板上看起来比较混乱,不如硅胶柱砍段后那么直观,一定要小心对比,否则会越分越乱。 3.正相柱分配色谱层析。 如果正丁醇部位量太大,或者课题组没有大的相柱用来分段,那么可以考虑氯仿:甲醇:水 体系来砍段。我一般用8:2:1、7:3:1以及6.5:3.5:1依次洗脱,效果不会很好,但两到三次反复上柱后各部分还是可以看得到点的。这时再会反相柱细分,拿十到二十个点应该没问题的。 总体来说,这部分较难,但等你分到好东西了,难也值得,小小经验和大家分享,祝大家实验顺利!
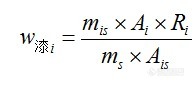
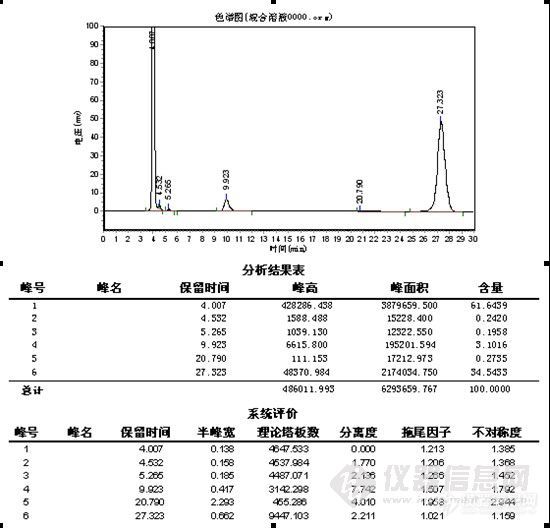
[align=center]涂料挥发性有机化合物方法研究分析 [/align][align=center]西安国联质量检测技术股份有限公司[/align][align=center]化工室+高杰[/align][align=left]一、方法概述[/align]本方法采用《溶剂型木器涂料 挥发性有机化合物[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法》将溶剂型木器涂料按产品明示的施工比制备混合试样,搅拌均匀后,称取2g试样,用适量的稀释剂稀释后加入标记物己二酸二乙酯后通过[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质[/color][/url]联定性己二酸二乙酯(沸点251°C)之前的组分。用内标法测试其含量二、仪器与试剂及标准品 1.仪器 1.1 [url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url]-火焰离子化检测器 1.1 [url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]质谱联用仪2.试剂2.1乙腈:色谱纯,不含任何干扰测试的物质。3.标准品3.1内标:试样中不存在的化合物,且该化合物能够与色谱图上其他成分完全分离,纯度至少为99%;如,邻苯二甲酸二甲酯;领苯二甲酸二乙酯等。3.2标记物:己二酸二乙酯,纯度大于99%3.3校准化合物:甲醇、乙醇、正丙醇、异丙醇、正丁醇、异丁醇、苯、甲苯、乙苯、二甲苯、三乙胺、二甲基乙醇胺、2-氨基-2-甲基-1-丙醇、1,2-丙二醇、1,3-丙二醇、二乙二醇、乙二醇单丁醚、二乙二醇单丁醚、二乙二醇乙醚醋酸酯、二乙二醇丁醚醋酸酯、2,2,4-三甲基-1,3-戊二醇。纯度至少为99%,或已知纯度。 4.[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]条件色谱柱:聚二甲基硅氧烷毛细管柱,30m×0.25mm×0.25μm 进样口温度:300℃;检测器:FID,温度300℃:柱温:起始温度45℃保持4min,然后10℃/min升至230℃保持15min 载气流速:1.0ml/min 分流比:分流进样,分流比可调:进样量:1.0ul。三、测试步骤 1 密度 按照产品明示的施工比例制备混合试样,搅拌均匀后,按照GB/T6750-2007的规定测定试样的密度。 2 水分含量 按照GB18582-2009 附录B进行。 3 挥发性有机化合物(VOC)含量四、分析步骤1.校准1.1如果校准中用到的化合物都可以买到,应使用下列方法测定其相对校正因子。分别称取一定量(精确至0.1mg)经过鉴定出的各种校准化合物于配样瓶中,称取的质量与试样中各自化合物的含量应在同一数量级。再称取与待测化合物相同数量级的内标物(3.1)于同一配样瓶中,用适量的稀释剂稀释混合物,密封配样并摇匀。1.2相对校正因子的测试:在与测试试样相同的[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]条件下,优化仪器条件,将适量的校准混合物注入[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url]中,记录峰面积,计算每种化合物的相对校正因子;若出现未能定性的色谱峰或者校准用的化合物未商品化,假设其相对于邻苯二甲酸二甲酯的校正因子为1.0。2试样的测试2.1试样的配制:按产品明示的施工比制备混合试样,搅拌均匀后,称取1g(精确至0.1mg)以及被测物相同数量级的内标物于配样瓶中,加入适量稀释剂溶剂10mL(2.1)于同一配样瓶中,密封并摇匀。五、结果处理1将1.0ul按照2.1配置的试样注入[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url],记录色谱图,并计算各种保留时间低于标记物化合物峰面积,然后按式1的计算公式分别计算试样中所含的有机化合物质量分数。[align=center][img=,195,87]http://ng1.17img.cn/bbsfiles/images/2017/09/201709071647_01_2904018_3.png[/img][/align][align=left]式中:w漆i,式中挥发性有机化合物i的质量分数,单位克每克(g/g) [/align] Ri,被测化合物i的相对校正因子; mis,内标物的质量,单位为克(g) ms,试样的质量,单位为克(g); Ai,被测化合物i的峰面积; Ais,内标物的峰面积。涂料产品按式(VOC)含量按下列公式计算[align=center][img=,325,103]http://ng1.17img.cn/bbsfiles/images/2017/09/201709071648_01_2904018_3.png[/img].[/align][align=left]式中,ρ(VOC)涂料产品的VOC含量,单位为克每升(g/L) [/align] wi,试样中被测化合物的质量分数,单位为克每克(g/g) ωw,试样中水的质量分数,单位为克每克(g/g); ρs,试样的密度,单位为克每毫升(g/ml) ρw,水的密度,单位为克每毫升(g/ml) 1000,转换因子。六、方法检出限 用10ppm浓度的混合标准品重复10次分别计算每个组分的标准偏差,以三倍的标准偏差作为每个物质的检出限,以综合作为VOC方法的检出限。[align=center][img=,690,304]http://ng1.17img.cn/bbsfiles/images/2017/09/201709071649_01_2904018_3.png[/img][/align][align=center][img=,690,359]http://ng1.17img.cn/bbsfiles/images/2017/09/201709071651_01_2904018_3.png[/img][/align][align=center][img=,690,419]http://ng1.17img.cn/bbsfiles/images/2017/09/201709071653_01_2904018_3.png[/img][/align][align=center][img=,690,222]http://ng1.17img.cn/bbsfiles/images/2017/09/201709071708_01_2904018_3.png[/img][/align][align=center][img=,690,91]http://ng1.17img.cn/bbsfiles/images/2017/09/201709071709_01_2904018_3.png[/img][/align]七、回收率 用不含VOC的空白溶剂,如乙腈,做空白加标回收率,回收率试验值应在80%——110%之间。[align=center][img=,303,401]http://ng1.17img.cn/bbsfiles/images/2017/09/201709071711_01_2904018_3.png[/img][/align][align=center][img=,305,194]http://ng1.17img.cn/bbsfiles/images/2017/09/201709071712_01_2904018_3.png[/img][/align]八、总结本方法中VOC检出限分别为0.319ug/L,0.003 ug/L,0.001 ug/L,0.003 ug/L;本方法的精密度分别为1.35%,0.00,1.41%,2.25%,符合《室内内墙涂料 挥发性有机化合物方法》(GB 18582-2008)中给出卤代烃统一样品的精密度要求.对引用水中卤代烃含量测定方法的检出限、精密度和准确度的评价,本方法测定水中卤代烃数据准确,结果可信。此方法的准确好,测定结果真实可靠,可用于水中卤代烃的测定。
有幸参加极限色谱柱的试用活动,分享一下使用结果,希望对大家在色谱柱使用上有点帮助。言归正传。本次检测目的是监测有机合成反应进程,待测产品本为一个侧链接乌洛托品的化合物(B),原料为含氯代乙酮侧链的物质(A),B在酸性条件下极易水解,在100mmol/L磷酸二氢钠溶液-甲醇(1:1)(用三乙胺调PH=7.0)条件下,一个小时内几乎完全水解成伯胺化合物(C),C由B上的乌洛托品开环所得。本化合物主体为较大的非极性基团,所以色谱柱选择上首选C18柱,分析条件选择有分析结果如下:仪器:岛津LC-10AT泵、SPD-10A紫外检测器、检测波长:224nm 色谱柱:C18(250*4.6 5um),一根为极限色谱柱,一根为其它品牌色谱柱。 (1)甲醇-水(1:1) 由于易水解,没有调节PH,峰形很差,像一个山包,重复性也差,有时什么峰也不出来,以为是样品分解了,结果临用新配的样品结果也是如此,所以放弃些流动相。把甲醇换成相似洗脱能力的乙腈,结果也是一样。(2)甲醇-水(1:1)(含2%三乙胺,用磷酸调节PH=7.0) 实验结果与(1)没有明显差别,常常出现的是B与C共存的现象,5分钟内用流动相溶解样品后进样,结果也是一样。(3)100mmol/L磷酸二氢钠溶液-甲醇(1:1)(用三乙胺调PH=7.0)这个是没有办法的情况下,大大加大了缓冲盐的深度,结果还是存在样品水解的情况。直到现在还没有找到一个重复性好,能很好反应实验进程的流动相,最后经过与合成人员的探讨,得知B完全水解仅会产生C,几乎不存在其它副反应,且高压可能会加速B水解,得知B完全水解仅会产生C,几乎不存在其它副反应,最后决定直接使B转化为C后再检测也不失为一个好办法,C性质稳定,容易检测。以下是在流动相(3)下得出的谱图。极限色谱柱:http://ng1.17img.cn/bbsfiles/images/2009/11/200911040041_180792_1638724_3.jpg其它品牌:http://ng1.17img.cn/bbsfiles/images/2009/11/200911040041_180793_1638724_3.jpg总结:对比中,在纯甲醇 1.0ml/min下,极限色谱柱压为5.8MPa,其它品牌色谱柱压为6.7MPa,加有保护柱,可能会略高于其它用户。4.0min为物质C,要完全水解下,未见B色谱峰,24.2min\27.3min为A,理论塔板数、分离度明显极限色谱柱较高,分析时间较短,但在峰形上,特别是A物质来看,极限色谱柱没有体现出优势,不过从A物质的峰形没有托尾上看,极限色谱柱在封端技术上有其独到之处。本实验试用的三种流动相非是最佳方案,后述会继续了解极限色谱柱在酸性条件下的表达,其使用寿命也是一个重要的考察项目。前几年也恰好参与过极限色谱柱的试用活动,检测的物质为18种氨基酸注射液,方法为柱前衍生,使用效果还不错,有做这一方面的版油可以一试。
亚硝胺的致癌作用已经清楚地在动物试验中得以证明,同时这类化合物被一些国际组织怀疑对人体有致癌作用,这些组织包括国际癌症研究总署,加拿大卫生署和美国国家毒理学规划处。这些化合物可能存在于环境空气中,这源于直接排放或者源于仲胺间或叔胺间在大气层中发生反应。在橡胶工业区内所检测出的亚硝胺浓度为人类环境中最高 这类化合物还存在于烟草烟雾中。美国国家职业安全和健康研究所(NIOSH) 已经开发出2522 方法,用于捕集和分析空气中的亚硝胺类化合物。然而,该方法使用的空气采样器需要较大的样品体积且采样时间长,亚硝胺还必须用溶剂再次提取,从而导致整个方法缓慢而冗长。[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url](GC) 和[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]/质谱联用仪(GC/MS) 经常被用于测定亚硝胺。然而亚硝胺热稳定性差,所以检测灵敏度受限于[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]进样器的高温。本应用简报描述了利用创新型安捷伦微吸附采样器(CTS)、热分离进样杆(TSP) 和Agilent 5975T 低热容[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质联用仪[/color][/url]建立起一种灵敏的空气亚硝胺检测方法。该方法快速,经济且易于使用,能够从大体积空气中捕获分析物,因此推测方法检测限(MDL) 低于1 ng。实验部分试剂和标准品亚硝胺类化合物标准品从Supelco公司获得,EPA 8270 亚硝胺混合物,浓度为2000 ng/μL(货号48489),六种亚硝胺化合物被检测(表1)。[img]http://img.vogel.com.cn/2014/0227/0944313156.png[/img]仪器这个方法是利用配有分流/不分流进样口的Agilent 5975T GC/MS系统开发而成,CTS(部件号G1181A)用于空气中样品的收集,TSP(部件号G4381A)安装在分流/不分流进样器上用于样品脱附。表2 列出了仪器使用条件。[img]http://img.vogel.com.cn/2014/0227/0947357839.png[/img]样品前处理标准品在空气中进行稀释,利用静态稀释瓶技术将其贮存在5 L 的玻璃瓶内,玻璃瓶事先已用纯氮吹扫5 分钟。将10 μL的2000 ng/μL的亚硝胺混合物注入瓶中后,让该样品在室温平衡4 小时使其充分气化,最终浓度为4000 ng/L。稀释样品是由贮存物制备而成,通过分别抽取2、60、100 和200 mL 蒸气到不同的1L 瓶中制成系列工作标准品,其浓度分别为8.0、240、400 和800 ng/L。结果与讨论CTS 操作CTS 包含一个气泵(流速范围10-300 mL/min)、一个手持式采样头和一个适配器。采样头可以容纳六根相同或不同类型的捕集柱。该方法使用了六根内径为530 μm的PoraPLOT Q 柱。TSP用于直接脱附CTS 捕集的样品。每根捕集毛细管置于一次性微量瓶中,然后放入TSP。TSP 然后被插入5975T LTM GC/MS 一个加热的分流/不分流进样口中。捕集柱被迅速而有效地脱附到GC进样器中。采集样品是通过将CTS 头直接插入瓶嘴完成的,同时捕集柱戳穿密封膜而不容许外部气体进入瓶内。校准标准品的制备是通过使用CTS 分别从240、400 和800 ng/L 的工作标准品的瓶中泵取200 mL 气体。另外,通过CTS 从4000 ng/L 的工作标准品中泵取100、200和400 mL 的气体制备成三个独立的样品。最后将这些制备好后的校准水平为48、80、160、400、800 和1600 ng的亚硝胺标准品混合物用于GC/MS 分析。CTS 性能对CTS 收集的400 ng亚硝胺标准样品进行分析,其色谱图上仅呈现了尖锐的亚硝胺色谱峰,而没有其他主要成分信息。该实验证明CTS 具有浓缩和定量转移亚硝胺而不引入污染物或伪迹的能力(图1)。这是一个收效显著的结果,因为考虑到亚硝胺化合物的高活性,其能在样品前处理过程中产生相互作用。定量准确性利用CTS 制备的校准样品用于建立校准曲线来分析混合标样中的六种亚硝胺。六种亚硝胺的所有相关系数(R2) ? 0.990,线性范围48 至1600 ng(表3)。图2 为所有六种化合物的校准曲线。当高于2000 ng或低于48 ng时校准曲线将不成线性。[img]http://img.vogel.com.cn/2014/0227/0949198654.png[/img][img]http://img.vogel.com.cn/2014/0227/0950015814.png[/img][img]http://img.vogel.com.cn/2014/0227/0951022218.png[/img]灵敏度和选择性这个方法能够用于检测低至8 ng/L 的气体浓度,通过泵取500 mL工作标准品,使注入GC/MS 的每种亚硝胺具有4 ng。所有的化合物呈现较好的信噪比并且没有显著性干扰(图3)。CTS 在采样过程中能够去除空气中的一些干扰物质,使后续分析具有一定的选择性。CTS 也可以通过泵取125 mL 的8 ng/L 工作标准品来捕集1.0 ng的亚硝胺混合标样。在提取离子流色谱图中,所有化合物的信噪比范围为3 到9,使方法检出限低于1 ng变为可能。因为CTS 能有效从大体积样品中捕集这类化合物,因此能够用于分析痕量水平的亚硝胺。方法回收率通过液体进样校准,在200 ng水平的NDBA 回收率为100%。由于采用TSP 的液体进样方式会导致由于溶剂的瞬间气化而引起高挥发性化合物的微量损失,因此其他5 种化合物的回收率会高于100%。其他亚硝胺的回收率范围为105.0% 至125.5%。[img]http://img.vogel.com.cn/2014/0227/0951381337.png[/img]结论这款创新型安捷伦微吸附采样器(CTS) 能够提供有用、经济有效的数据用于挥发性亚硝胺的筛查、日常监测和在一个很宽浓度范围内的定量分析。这个采样装置使用非常简单和灵活,对高浓度空气样品允许小体积采样,对低浓度空气样品允许大体积采样。现场采样能在数秒到数分钟内完成。CTS 与TSP 和车载式5975TLTM GC/MS 搭配提供了一个灵敏的分析系统用于空气样品的分析。这个系统非常适合各种应用领域,包括在筛查性调查中快速得出分析结果,到为定量研究提供准确和精确的数据

如题,原料药合成中,溶剂用到三乙胺,那么三乙胺作为溶剂残留,质量标准应该定多少合适?查了ICH,把三乙胺归到了3类溶剂里,标准推算如下图:[img=,690,504]https://ng1.17img.cn/bbsfiles/images/2019/05/201905091239303097_6773_2700892_3.png!w690x504.jpg[/img]6250ppm是根据毒理学数据计算的结果。但查了相关欧盟方面的规定,三乙胺定了320ppm,结合以上信息,那三乙胺作为溶残到底应该定多少比较合理?
最近在看GBT 24800.12-2009 化妆品中对苯二胺、邻苯二胺和间苯二胺的测定有一事不明:标准在流动相中用到了三乙醇胺请问它的作用是什么?我只知道为了改善碱性化合物的峰形经常会用到三乙胺那三乙醇胺的作用是什么呢?请各位指教!谢谢~
各位大佬你们好,我有一个疑问,我师兄之前做的是80%甲醇超声提取的植物提取液,拿去跑的[font='Times New Roman']UPLC-Q-Orbitrap HRMS 通过比对确认化合物结构,很多结构别人的文章已经发现过了,然而我老师让我做70%甲醇提取过后正丁醇萃取部位的东西。那我就很好奇70%甲醇提取之后萃取部位怎么说都是80%甲醇提取液总成分的子集,那能出什么新化合物? 我有些不理解 [/font]
有哪位大侠有,三聚氰胺甲醛模制化合物的标准规范 标准号:ASTM D 704-1999。中文,英文的都可以。 谢谢
做硝基呋喃类化合物检测用的标准品大家都是用的什么呀?有标准中说的是用对照品,有标准中用的是代谢物,不知道这其中有没有什么区别呀?大家都根据哪个标准做的呢?
请问各位高手有没有叔丁醇锂的国标,行业标准也行,谢谢!
请问大家,萜烯类化合物标准品哪里有买?
我采用岛津的气质联用仪测定挥发物,检测结果采用NIST系统确定化合物名称。现在投稿返回意见是核对化合物的中英文名称。我想知道有没有网站可以查询化合物,这些化合物没有错误,就是中英文名称让重新核对下,写其标准名称。如乙酸叶醇酯 3-hexenyl acetate这样的。谢谢!
[align=right][b]SGLC-GC/MS-016[/b][/align][b]摘要:[/b]建立了9种N-亚硝胺化合物同时测定的[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]GC-MS[/color][/url]方法。本应用按照国标GB5009.26-2016方法,采用SH-PolarWax色谱柱对N-二甲基亚硝胺(NDMA)、N-二乙基亚硝胺(NDEA)、N-二丙基亚硝胺(NDPA)、N-二丁基亚硝胺(NDBA)、N-亚硝基吡咯烷(NPYR)、N-亚硝基哌啶(NPIP)、N-甲基乙基亚硝基(NMEA)、N-亚硝基吗啉(NMorPh)、N-二苯基亚硝胺(NDPhA)等9种N-亚硝胺化合物进行分析,峰形对称,RSD小于3%,重现性好,满足国标要求,本方法适用于N-亚硝胺化合物的同时测定。[b]关键词:[/b]N-亚硝胺 SH-PolarWax [url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]GC-MS[/color][/url][b]1. 实验部分1.1 实验仪器及耗材[/b]仪器配置:岛津[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]GCMS[/color][/url]-QP2020 [url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]-质谱联用仪;色谱柱:SH-PolarWax(30 m* 0.25 mm *0.25 μm;P/N:227-36305-02);SHIMSEN Arc Disc HPTFE针式过滤器(P/N:380-00341-05);[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]GC-MS[/color][/url]认证样品瓶LabTotal Vial(P/N:227-34002-01);SHIMSEN Pipet[url=https://insevent.instrument.com.cn/t/9p][color=#3333ff]移液枪[/color][/url]:SHIMSEN Pipet PMII-10(P/N:380-00751-02);SHIMSEN Pipet PMII-100(P/N:380-00751-04);SHIMSEN Pipet PMII-1000(P/N:380-00751-06)。[b]1.2 分析条件1.2.1 色谱条件:[/b]毛细管柱: SH-PolarWax(30 m* 0.25 mm *0.25 μm;P/N:227-36305-02)程序升温:初始温度40℃, 以10℃/min升温到80℃,再以1℃/min升温到100℃,以20℃/min升温到240℃, 保持4 min;载气:He流速:1.0ml/min进样口温度:220 ℃进样量:1μL 进样方式:不分流进样[b]1.2.2 质谱条件:[/b]电离模式:电子轰击电离(EI);电子轰击能量:70 eV离子源温度: 230 ℃接口温度:230 ℃溶剂延迟:4 min数据采集模式: SIM 各化合物SIM参数见下表[img=9种N-亚硝胺化合物的测定]https://img.shimadzumall.com/Storage//userfiles/images/Img_articles/SGLC-[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]GCMS[/color][/url]-016_01.png[/img][font=arial, &][size=12px][/size][/font][b]1.3 标准品溶液配制[/b]精密量取9种N-亚硝胺混合标准品母液适量,用二氯甲烷稀释,使最终浓度为200 μg/L,备用。[b]2. 结果及讨论2.1标准品的SIM色谱图[/b]按照上述色谱条件(1.3)进行采集,系统适用性溶液色谱图如下:[img=9种N-亚硝胺化合物的测定]https://img.shimadzumall.com/Storage//userfiles/images/Img_articles/SGLC-[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]GCMS[/color][/url]-016_02.png[/img][font=arial, &][size=12px][/size][/font][align=center]9种N-亚硝胺化合物混合标准品的SIM色谱图(200 μg/L)[/align][b]2.2 重现性数据[/b][img=9种N-亚硝胺化合物的测定]https://img.shimadzumall.com/Storage//userfiles/images/Img_articles/SGLC-[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]GCMS[/color][/url]-016_03.png[/img][font=arial, &][size=12px] [/size][/font][b]3. 结论[/b]建立了9种N-亚硝胺同时测定的[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]GC-MS[/color][/url]方法。本应用按照新国标方法,采用SH-PolarWax色谱柱对N-二甲基亚硝胺(NDMA)、N-二乙基亚硝胺(NDEA)、N-二丙基亚硝胺(NDPA)、N-二丁基亚硝胺(NDBA)、N-亚硝基吡咯烷(NPYR)、N-亚硝基哌啶(NPIP)、N-甲基乙基亚硝基(NMEA)、N-亚硝基吗啉(NMorPh)、N-二苯基亚硝胺(NDPhA)等9种N-亚硝胺化合物进行分析,峰形对称,RSD小于3%,重现性好,满足国标要求,本方法适用于N-亚硝胺化合物的同时测定。
GB 5009.26-2016 食品中亚硝胺类化合物的测定,第一法中是只测一种亚硝胺化合物的含量吗?
歐盟官方於2013年6月29日公佈通報2013/C 187/09,指定歐盟玩具安全標準《EN 71-12:2013 亞硝胺與亞硝胺化合物》將成為新玩具指令(2009/48/EC)的協調標準。主要针对以下材料或部件: ——打算为3岁以下儿童使用的由“弹性体”所组成的玩具或玩具部件; ——打算放入口中的由“弹性体”所组成的玩具或玩具部件; ——3岁以下儿童使用的手指油漆。举例:由“弹性体”组成的玩具如气球和出牙器等产品。欧盟委员会要求各成员国将这个标准强制转化为本国标准或要求的最后期限为2013年12月,同时,相冲突的国内标准最迟将于2013年12月被废除。为方便客户的使用,安谱定制了相应的2套混标MIX1-目标分析物CDGG-116965-01-1ml 品牌:美国o2si13种亚硝胺混标《EN 71-12:2013 亚硝胺与亚硝胺化合物》 标准品1000mg/L于甲醇,1 mlName of the substance CAS number AbbreviationN-nitrosodiethanolamine 1116-54-7 NDELAN-nitrosodimethylamine 62-75-9 NDMAN-nitrosodiethylamie 55-18-5 NDEAN-nitrosodipropylamine 621-64-7 NDPAN-nitrosodiisopropylamine 601-77-4 NDiPAN-nitrosodibutylamine 924-16-3 NDBAN-nitrosodiisobutylamine 997-95-5 NDiBAN-nitrosodiisononylamine 1207995-62-7 NDiNAN-nitrosomorpholine 59-89-2 NMORN-nitrosopiperidine 100-75-4 NPIPN-nitrosodibenzylamine 5336-53-8 NDBzAN-nitroso-N-methyl-N-phenylamine 614-00-6 NMPhAN-nitroso-N-ethyl-N-phenylamine 612-64-6 NEPhAMIX2-内标CDGG-116960-10-1ml 品牌:美国o2si2种亚硝胺内标混标《EN 71-12:2013 亚硝胺与亚硝胺化合物》 标准品1000mg/L于甲醇,1 mlN-Nitrosodiethan-d8-olamine (d8-NDELA) 1173019-53-8N-nitrosodimethyl-d6-amine (d6-NDMA) 17829-05-9欢迎来电详询。
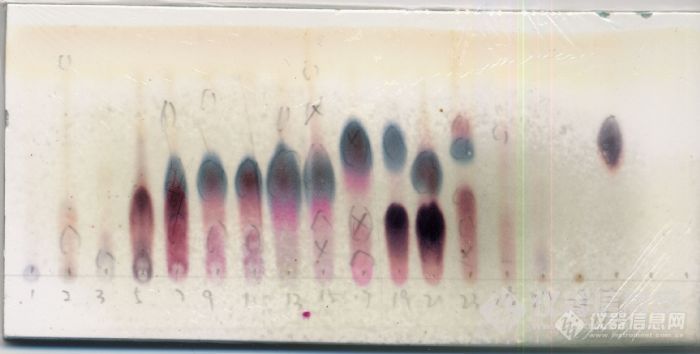
维权声明:本文为环烯醚萜原创作品,本作者与仪器信息网是该作品合法使用者,该作品暂不对外授权转载。其他任何网站、组织、单位或个人等将该作品在本站以外的任何媒体任何形式出现均属侵权违法行为,我们将追究法律责任。环烯醚萜类化合物分离纯化心得体会基本介绍 环烯醚萜(iridoids)为臭蚁二醛(iridodial)的缩醛衍生物。臭蚁二醛是由伊蚊(Iridomyrmex detectus)的防收性分泌物中分得的物质。自1958年的Halpem和Schmid确定的环烯醚萜的基本骨架以来,各国学者对该类化合物作了大量深入的研究。环烯醚萜类化合物具有多种生物活性,近来受到极大关注,发展也很迅速。 环烯醚萜类主要分为:环烯醚萜类、裂环环烯醚萜类、3,4-位无取代的环烯醚萜类、聚合环烯醚萜类等等。本人做的是普通类的环烯醚萜类化合物,且以其苷居多,做的比较浅,下面斗胆一谈,各位看官莫要见笑。提取部分 环烯醚萜苷类化合物在醇(甲醇、乙醇)中溶解度较好,部分苷类在水中溶解度也很好。本人在对某植物进行提取的时候,实际上并非针对这类化合物,采用的是60%的乙醇/水,是为了兼顾各类成分。后来在实验过程中发现,60%的乙醇/水条件下,这类化合物的提取率是很高的。 注释:其实如果要针对性分离,可以将提取液简单处理后进行D101大孔柱色谱,对环烯醚萜类化合物进行富集。萃取部分 提取之后,将药液进行浓缩,至无醇味混悬于水中,然后进行萃取。萃取的过程为:等体积的环己烷、乙酸乙酯、正丁醇分别萃取三次,合并各层提取液浓缩得各层浸膏。就目前实验进展情况来看,环烯醚萜苷类化合物主要集中在正丁醇层,水层也有一部分(我目前还没开始这部分工作)。 注释1:萃取的过程,涉及到溶剂的回收,由于这类化合物在高温下不太稳定,所以用旋转蒸发仪进行减压回收溶剂的时候,温度不能过高,我采用的温度是60度(其实60度已经很高了,但是没办法,不设60度,正丁醇回收不了)。 硅胶柱色谱 正丁醇层进行硅胶柱色谱分离,采用氯仿/甲醇梯度洗脱,样品500g,拌样硅胶1500g,柱床硅胶500g,洗脱梯度为50:1→20:1→10:1→5:1→2:1→1:1→0:1。实验的过程中发现,环烯醚萜类化合物,主要集中在氯仿:甲醇=10:1和5:1部分。 注释1:选择硅胶柱色谱,也是人之常情,此处也可选择D101大孔柱色谱,对这类化合物进行富集; 注释2:选择氯仿/甲醇系统,是因为经过小试,此系统对样品分离较好,最重要的,样品在此系统中成点性很好; 注释3:拌样1500g,柱床500g,你没有看错,我没有写错。书上说,拌样:柱床在1:1-1:10甚至1:20或者1:50,而我这里却是3:1。我可以很负责任地告诉你,没有必要按书上的说法,柱床500g足够了,分离效果一点也不差。以前做另外一个植物,拌样用了3000g,柱床才600g,分离效果也不差,一点问题都没有; 注释4:梯度的选择,建议6-8个。梯度太少,各流分成分可能过于复杂;梯度太多,后续分离麻烦。这种大型硅胶柱色谱,属于平常所说的“粗分”,不宜太多,不宜太少,不然就是给自己找麻烦。http://ng1.17img.cn/bbsfiles/images/2010/10/201010061143_249374_1745326_3.jpgODS柱色谱 包括开放型ODS柱色谱和中低压型ODS柱色谱,其实原理一样,只是规模大小不同而已。 我将5:1洗脱的样品进行中低压ODS柱色谱,采用甲醇/水梯度洗脱,水→10%甲醇/水→20%甲醇/水→30%甲醇/水→50%甲醇/水→甲醇,实验结果表明,ODS柱色谱对此类化合物具有良好的分离效果。 注释1:环烯醚萜苷类化合物一般极性较大,一般集中在10%甲醇/水、20%甲醇/水、30%甲醇/水部分; 注释2:水洗下来的,一般为糖苷类化合物,我从此流分中分离得到几个糖类化合物(题外话); 注释3:ODS柱色谱可以多次进行,反复纯化,利用ODS柱色谱可以得到部分单体化合物;http://ng1.17img.cn/bbsfiles/images/2010/10/201010061144_249375_1745326_3.jpg(注:此图为未合并相同流分前的点板情况)制备液相(反相) 从ODS柱色谱上洗脱的样品,经过分析,可以考虑进行制备液相,半制备液相等等。 事实上,一般而言,PHPLC也是获得环烯醚萜类单体化合物最重要的手段之一。 流动相可采用甲醇/水,若峰形不好,可加入少量乙酸改善(这一招屡试不爽)。 波长的选择,可以使用230nm、240nm都可以(我们有PAD检测器,验证过)。其它一些化合物,由于连上芥子酰基,对羟基香豆酰基,还可以选择320nm的吸收。 色谱柱一般是C18的居多(目前未使用过其它柱子),品牌好像都还行(我们主要使用YMC)。凝胶其它填料 由于分离原理的缘故,使用凝胶对环烯醚萜这一类化合物(分子量相差不大,且结构极为类似)进行分离,前景似乎并不明朗。但是,用凝胶将这一类化合物与其他类型的化合物分离,效果还是很不错的。 其它如大孔树脂,前面提过,用来富集是个不错的选择;聚酰胺,我没有使用过,不过从其分离原理来看,对这类化合物不会很敏感。显色剂的选择 部分环烯醚萜类在254nm紫外下有暗斑,这个很实用; 最常用的是浓硫酸-香草醛显色剂:母核上有羟基取代的显蓝色;母核上无羟基取代显紫红色(非绝对); 其它显色剂如硫酸乙醇、碘等等都可以,我个人偏爱浓-香显色剂。结构测定与解析(简) 环烯醚萜类化合物进行NMR测试,首先氘代甲醇,这个没有任何疑问。 我在一些文献中,也看到一些特例,比如使用重水、氘代DMSO,这些基本可以忽略。 关于结构解析部分,此处不再赘述。
DMA做溶剂,却有醇峰,定位炔丁醇和混合溶剂跑出来的峰,混合溶剂里炔丁醇附近有两个峰,是反应了吗
有一个亚胺类化合物(标样)用甲醇和水作流动相,出现了两个色谱峰,而用乙腈和水却只有一个峰。请问各位这是为什么?难道是甲醇与亚胺反应?谢谢!







 我要推广仪器
我要推广仪器
 下载APP
下载APP