配1%三乙胺溶液加磷酸调ph值至7.0,这样会生成磷酸铵吗?三乙胺的氨基不是饱和了吗应该不会,是吗?可是为什么加完磷酸后,那股三乙胺的味就没有了呢?标准流动相是上述溶液-甲醇 50-50 要是生成磷酸铵的话不就和甲醇析出结晶。

什么是氢化可的松呢?氢化可的松是人工合成也是天然存在的糖皮质激素。氢化可的松的结构和性质[img=,360,263]https://ng1.17img.cn/bbsfiles/images/2019/10/201910260900022643_4962_4009900_3.jpg!w360x263.jpg[/img]1、结构中有共轭体系,具有紫外吸收。2、氢化可的松松3号位上有羰基,可以和生物碱发生沉淀反应。3、4号位上有双键,可以被高锰酸钾氧化,也可以与溴水发生加成反应。4、11号位上的羟基可与酸酐或酰氯成酯,利用酯具有一定的熔点,可进行熔点测定,其酯在碱性条件下再与羟胺作用,生成异羟肟酸,并在高铁离子在酸性条件下络合,显紫红色。5、此结构与硫酸、磷酸、高氯酸等作用呈色。6、Α-醇酮基与碱性酒石酸铜试液反应成砖红色的氧化亚铜沉淀。
三乙胺和磷酸反应吗,如果反应那生成物遇到三氟乙酸会出现什么情况?谢谢!
[color=#444444]求问:测紫外光谱在文献中,配pH=2.5的缓冲溶液用的是磷酸三乙胺缓冲溶液,为什么不用磷酸和胺的缓冲溶液或者其他pH可以达到2.5的缓冲溶液,想问问磷酸三乙胺有什么好处呢?[/color]
我要配置20 mM的三乙胺-磷酸缓冲液,始终不知道这个20 mM是谁的浓度在网上查到的都是这样:“取磷酸约4ml与三乙胺约7ml,加50%甲醇 ..”但是这样 怎么能知道到底三乙胺是多大浓度呢?
有这个标准:取样品0.5g,精密加浓氨试液-甲醇(1:20),冷浸1小时,加热回流1小时。按这个方法处理出来的样品峰形很难看,而且他离度也不好。它所用的流动相是甲醇:0.1%磷酸溶液(用三乙胺调PH至6.0)。同样是含碱性,为什么处理样品不用三乙胺-甲醇(1:20)?
关于印发化妆品中氢化可的松等禁用物质或限用物质检测方法的通知 国食药监保化13号 2012年01月18日 发布 各省、自治区、直辖市食品药品监督管理局(药品监督管理局): 为规范化妆品中禁用物质和限用物质检测技术要求,提高化妆品质量安全,化妆品中氢化可的松等禁用物质或限用物质的检测方法已经国家食品药品监督管理局化妆品标准专家委员会审议通过,现予印发。 附件:1.化妆品中氢化可的松等7种禁限用物质的检测方法 2.化妆品中水杨酸的检测方法 3.化妆品中酮麝香的检测方法 4.化妆品中巯基乙酸的检测方法 5.化妆品中8种邻苯二甲酸酯的检测方法 6.化妆品中4-氨基偶氮苯和联苯胺的检测方法 7.化妆品中苯并芘的检测方法 8.化妆品中4-氨基联苯及其盐的检测方法 9.化妆品中间苯二酚的检测方法 10.化妆品中32种禁限用染料成分的检测方法 11.化妆品中苯扎氯铵的检测方法 12.化妆品中羟基喹啉的检测方法 13.化妆品中过氧化氢的检测方法 14.化妆品中苄索氯铵、劳拉氯铵和西他氯铵的检测方法 15.化妆品中颜料橙5等5种禁用着色剂检测方法 16.化妆品中呋喃香豆素类(三甲沙林、8-甲氧基补骨脂素、5-甲氧基补骨脂素)和欧前胡内酯的检测方法 17.化妆品中补骨脂特征成分补骨脂素、异补骨脂素、新补骨脂异黄酮和补骨脂二氢黄酮的检测方法 国家食品药品监督管理局 二○一二年一月十六日

如题,原料药合成中,溶剂用到三乙胺,那么三乙胺作为溶剂残留,质量标准应该定多少合适?查了ICH,把三乙胺归到了3类溶剂里,标准推算如下图:[img=,690,504]https://ng1.17img.cn/bbsfiles/images/2019/05/201905091239303097_6773_2700892_3.png!w690x504.jpg[/img]6250ppm是根据毒理学数据计算的结果。但查了相关欧盟方面的规定,三乙胺定了320ppm,结合以上信息,那三乙胺作为溶残到底应该定多少比较合理?
问题描述: 四烷基季铵盐(如四丁基硫酸氢铵、四丁基溴化铵、四丁基氢氧化铵等)在水中电离后,也形成了类似N+H(CH2CH3)3的结构N+(CH2CH2CH2CH3)4 ,这种结构也能有效的与Si-O-产生较强的静电作用,此类离子对用的比较少,但它的作用仅是掩藏Si-O-吗,它对物质的保留性能有何影响,实验中发现检测胺化物时,流动相中加入磷酸缓冲盐有增加物质保留的趋势,而加入三乙胺则会降低物质的保留能力? 解答: 四丁基氢氧化铵等离子对试剂确实不如辛磺酸钠等极性基阴离子的离子对试剂用得普遍,原因是酸类极性物质很容易通过降低pH值的方法提高在反相色谱中的保留能力,降低pH可抑制酸的电离,使酸处于中性状态而与疏水碳链的作用力增强。而碱类分析物则受硅胶基质pH上限的严重影响(以前上限是8,后来抬到10,直到最近杂化硅胶才将pH上限提到12左右)。 铵盐类离子对试剂确实有辛磺酸钠等所不具有的屏蔽硅醇基作用,但其主要作用还是疏水端和疏水固定相结合,外露的阳离子亲水端和酸阴离子作用,从而提高其保留能力。当然同样还有第二种解释机理,离子对试剂先和分析物结合,掩藏极性基,从而提高极性分析物在反相色谱中的保留能力。而且丁基比辛基疏水性差,这种情况下,认为后面的结合机理占上风的可能性更大。 检测胺类化合物,加入三乙胺预先和硅醇基结合,胺和硅醇基作用被三乙胺取代,保留下降是肯定的。
看到氢化可的松注射液的辅料里有乙醇和注射用水,想扣除辅料干扰就得配制相应浓度的辅料,谁能透露下,这处方里头的乙醇大概浓度多少啊? 叩谢各位大大。
最近在做水质三乙胺的检测,发现校准曲线很难做直,请问主要有哪些影响因素,还有三乙胺的标样何处可以买到?
如题,最近要做氢化可的松的检测,按照的是欧洲标准,文献上的梯度洗脱的要求是:0~18min,76%水,26%乙腈;18~32min,76%~55%水,26%~45%乙腈;32~48min,45%~55%乙腈。但是设定时间的时候,流动相还是停留在26%的乙腈,不能换流动相。各位大侠,能不能帮帮设定一个时间流程呢?谢了哈
谁有三乙胺三氟化氢的分析方法?请共享好么?
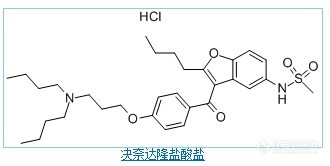
盐酸决奈达隆 结构式我做这个样品的时候存在严重的拖尾现象1、乙腈:水=55:45 峰分布出来,大概需要10分钟左右才能出一个趴着的峰2、乙腈:水(0.02mol/L 磷酸二氢钠)=55:45 拖尾3.33、乙腈:水(0.02mol/L磷酸二氢钠):三乙胺=550:450:1 拖尾2.1 此时PH=7.284、乙腈:水(0.04mol/L磷酸二氢钠):三乙胺=550:450:1 拖尾2.1 此时PH=7.12我觉得这两个基团(甲基磺酰胺 另外一个氨基 )会和ODS填料的硅羟基形成氢键,所以加入了三乙胺扫尾,效果是稍微好了一点,但是拖尾因子到2.1时候不再改善,我先加入磷酸将PH调整到2.5,后又用三乙胺将PH调整到7,峰型又有所改善,感觉是因为加大了三乙胺的量,可是网上查的三乙胺最大添加量为1ml/L,也没有见到有先将PH调低再加入大量三乙胺的做法求教各位大大,这种基团应该怎么处理,有什么思路吗,在线等,万分感谢http://ng1.17img.cn/bbsfiles/images/2011/04/201104072058_287643_1638724_3.jpg
谁有三乙胺的GB标准吗?
北京康农兴牧,他家有售,美国进口,氢化可的松试剂盒(猪尿)通常情况下都是检测人体的氢化可的松,用在兽药方面的试剂盒还不多见,有需要的朋友可以联系他们电话:010-65919921我们买了,用了还是不错的,方便一些,是进口
做肉中环丙沙星残留量的测定,国标方法中,流动相是这样配制的:0.05mol/L磷酸-三乙胺溶液:取浓磷酸3.4mL,用水稀释至1000mL。用三乙胺调pH至2.4。本人用的是85%磷酸(色谱纯),取了3.4mL,稀释至1000mL。搅匀后,测pH是3.6。这是不可能再往里面加三乙胺了,三乙胺是碱性的,越加pH只会越大。但这时我往溶液中起码再加入了十几毫升的磷酸,pH也才是3.0左右。我应该继续往溶液中加磷酸,直到pH=2.4吗? 这样流动相中磷酸是不是过多了?国标中这个溶液的配制方法是不是有问题呢?磷酸是中强酸,才取3.4mL配成1000mL,pH怎么可能低于2.4?这时如果还往里面加三乙胺,pH只会越来越大,永远也不可能调至2.4了。望大家帮我分析下,到底哪里出错了?感激不尽!
[color=#444444]三乙胺-丙酮-盐的混合液,三乙胺的含量在5%以下,请问有什么好的方法可以测定三乙胺的含量,在实验室用氢火焰毛细柱打可以出峰,但是如果含量特别低时担心[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]打不出,导致微量三乙胺检测不出来。查文献有溴酚蓝分光光度法、红外色谱,[url=https://insevent.instrument.com.cn/t/3p][color=#3333ff]离子色谱[/color][/url]、电位返滴定法,以及混合液放入盐酸,用过量NaOH滴定未反应酸,溴酚蓝做指示剂等方法。请问这几种方法哪个比较准确,并且好操作,谢谢各位了![/color]
染发剂中对苯二胺等染料的HPLC测定方法采用《化妆品卫生规范》所规定的液相色谱标准检测方法,对氧化型染发剂中包括对苯二胺和邻苯二胺在内的八种染料进行检测。[size=4][b]分析方法[/b]色谱柱:Shimadzu Shim-pack VP-ODS 4.6×150mm 5μm检测波长:280nm 流动相:乙腈:(水:三乙醇胺=98:1)=5:95 流速:1mL/min 柱温:室温进样量:10μL洗脱方式:等度洗脱。[/size][size=4]流动相中使用了三乙醇胺,比较少见,它在分析苯胺类化合物有什么突出的优点吗?可否用三乙胺替代?[/size][img]http://ng1.17img.cn/bbsfiles/images/2010/05/201005082043_217200_1638724_3.jpg[/img][img]http://ng1.17img.cn/bbsfiles/images/2010/05/201005090755_217230_1638724_3.jpg[/img]
我按照标准在水相中加了1%三乙胺用的岛津的走的梯度结果却是在9分钟的时候一直向下漂直到检测器OVER,现考察结果如下:三乙胺肯定是影响漂移的但这又是国外标准的浓度,柱子影响基本可以排除,用安捷伦的做是7分钟和24分钟的时候向上漂可以考虑设备也有影响,还有什么影响及怎么才能尽量让漂移小到10-20mAU啊请大家帮帮忙图如下:[size=3][font=Times New Roman] [/font][/size]
刚接触分析没多长时间,公司领导要求我做一个车间回收三乙胺定量的方法,面积百分比不准,然后就尝试做一个外标法,因为试样当中有邻二氯苯和三乙胺两种物质,所以我就配置了不同浓度的三乙胺溶液,5%.10%.15%.20%.25%.的溶液,用自动进样器进样,发现峰面积相差很大,这是为什么?到底该怎么做呢?求助前辈~我的气谱方法是这样的,进样0.2ul,分流比100:1,进样口气化温度250,检测器300,柱箱150保持5分钟。HP-5中性柱,安捷伦7820A,FID检测器

头孢曲松钠的测定和泼尼松龙和氢化可的松的测定http://ng1.17img.cn/bbsfiles/images/2009/11/200911021846_180184_1896702_3.jpg
三乙胺我们在生产中用作缚酸剂,现在得到一个三乙胺邻二氯苯混合溶液,配置了不同浓度的三乙胺溶液,但是打出来同样溶度的三乙胺峰面积相差挺大的,这个是为什么啊?
药典规定流动相含0.5%磷酸,考虑到ph过低,我用3ml三乙胺中和至ph3左右,现发现压力变大,保留时间变久,请教各位有没有错误?
顶空[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]测三乙胺,谁有相关经验吗?设置条件大概是多少?三乙胺溶液里面需要添加碱溶液吗?求回复
就是这样,三乙胺加多了,pH到7了,然后我又用磷酸把PH调到4.6,有什么影响吗??我的流动相中有磷酸氢二钾。。求各位师傅
各位老师。我最近在做一个化合的异构体分离,流动相中需要加如三乙胺扫尾,但是发现采用三乙胺扫尾后,色谱峰比不加三乙胺的保留时间要延迟很多,这是什么原因呢?我一直认为三乙胺是不会影响手性保留的!大家有什么高见?
小弟分析生物碱,用的流动相为甲醇/水(含0.14%H3PO4和0.5%三乙胺),防拖尾效果不错。 但0.5%三乙胺是不是太高了?会对柱子不好吗?望论坛高手释疑
职业卫生的标准测定三乙胺,重复性特别差,质控浓度80μg/ml,有时测出来是30,有时测出来是70.岛津GC-2014C型[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]胺分析专用柱进样口210℃,柱流量3.0ml/min,分流比10:1柱温:150℃检测器:230℃出峰时间:0.705min
要测一个原料药中三乙胺的残留。仪器是安捷伦的6890N,7694E,条件如下:色谱柱HP-1,柱温100,进样口250,分流比1:1,柱压7.7psiFID250顶空平衡30min,温度70/100/110三乙胺浓度16ug/ml(用水溶),取2ml至10ml顶空瓶我的问题是我进了三乙胺后跟了一针空白,三乙胺的位置有出峰,再跟一针空白就没有了,我试着做进样精密度,连续进了三针峰面积从29涨到了40,RSD非常差。而且换了DB-624的柱子,这个问题还是存在,哪位大侠指点一下!







 我要推广仪器
我要推广仪器
 下载APP
下载APP